Computational and Statistical Analyses of Insertional Polymorphic Endogenous Retroviruses in a Non-Model Organism


 ,
,
Abstract
:1. Introduction
2. Experimental Section
2.1. Fluorescent In Situ Hybridization (FISH)
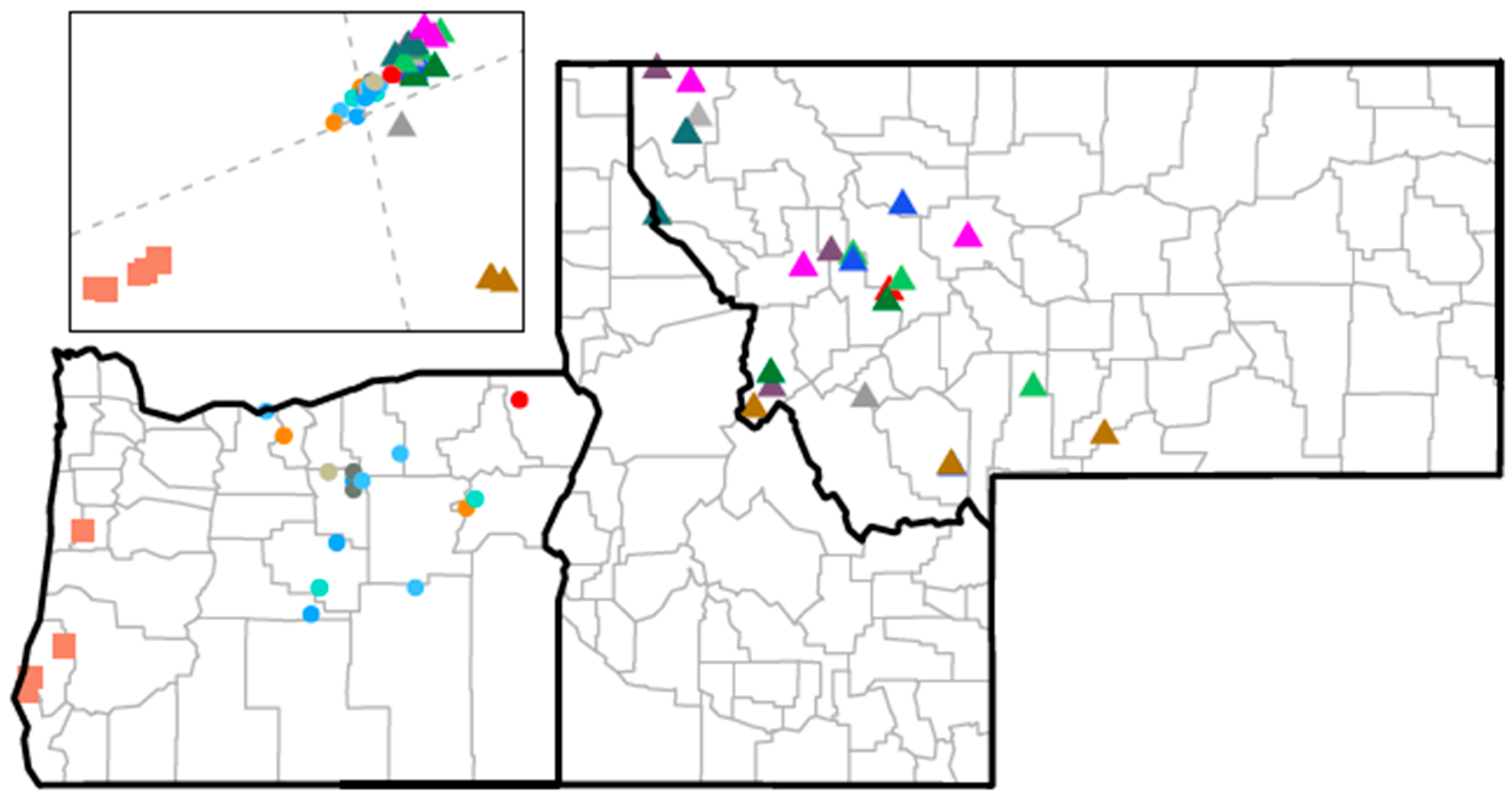
2.2. Animal Samples
2.3. Junction Fragment Analysis
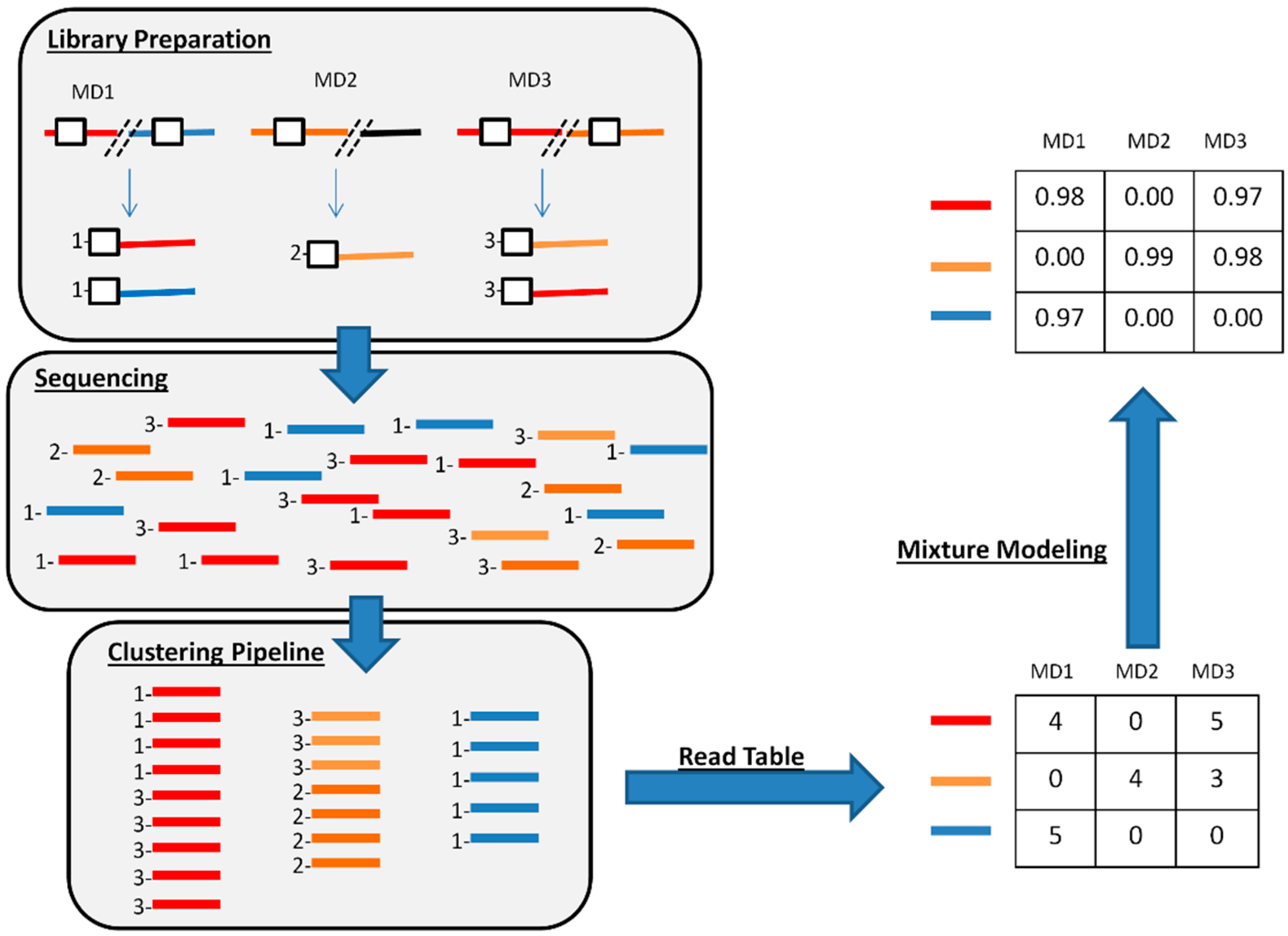
2.4. De Novo Clustering of Host-Virus Junction Fragments
2.5. Mixture Model
2.6. Validation of Mixture Model via Replicated Individuals
2.7. Principal Component Analysis
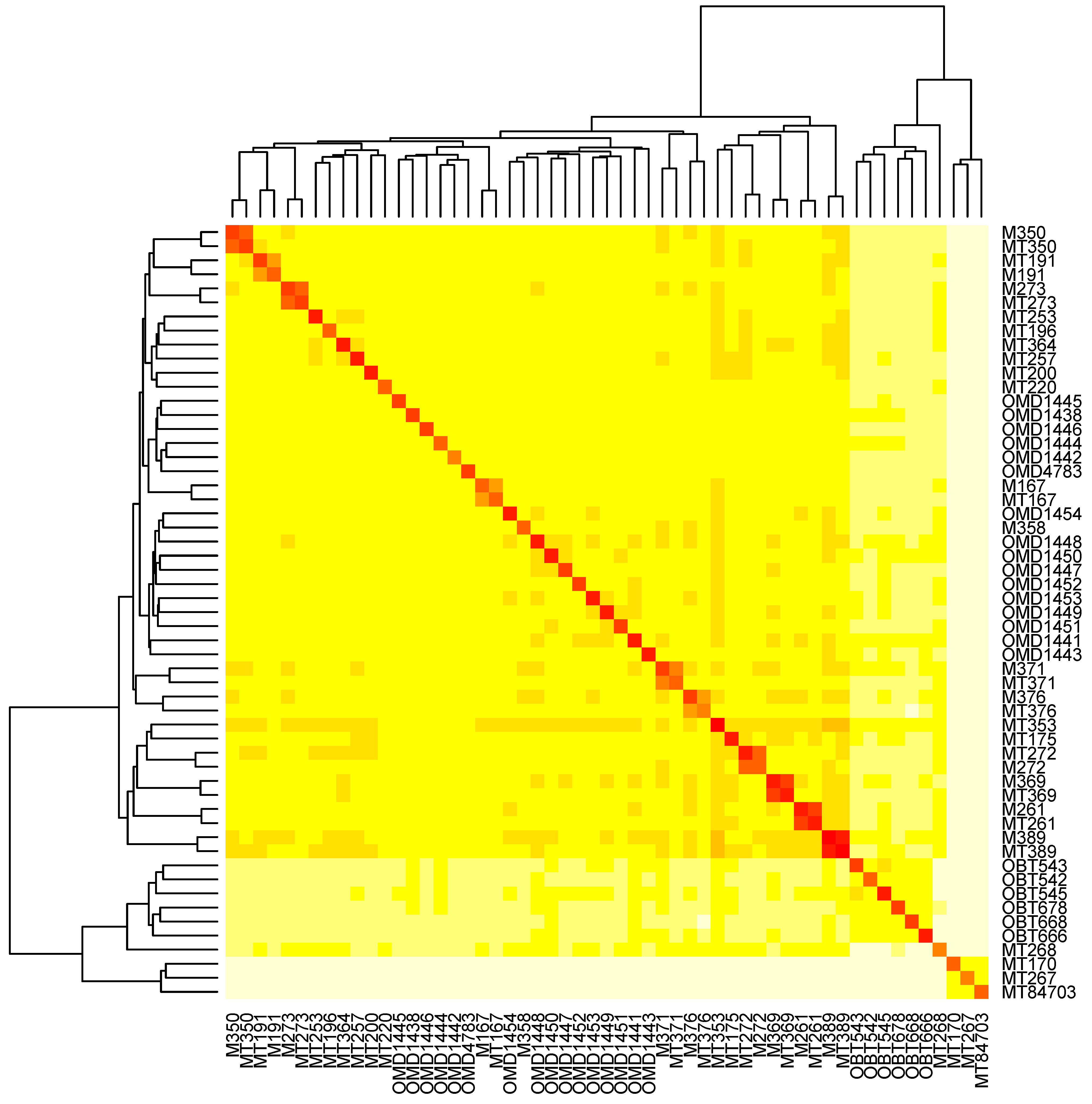
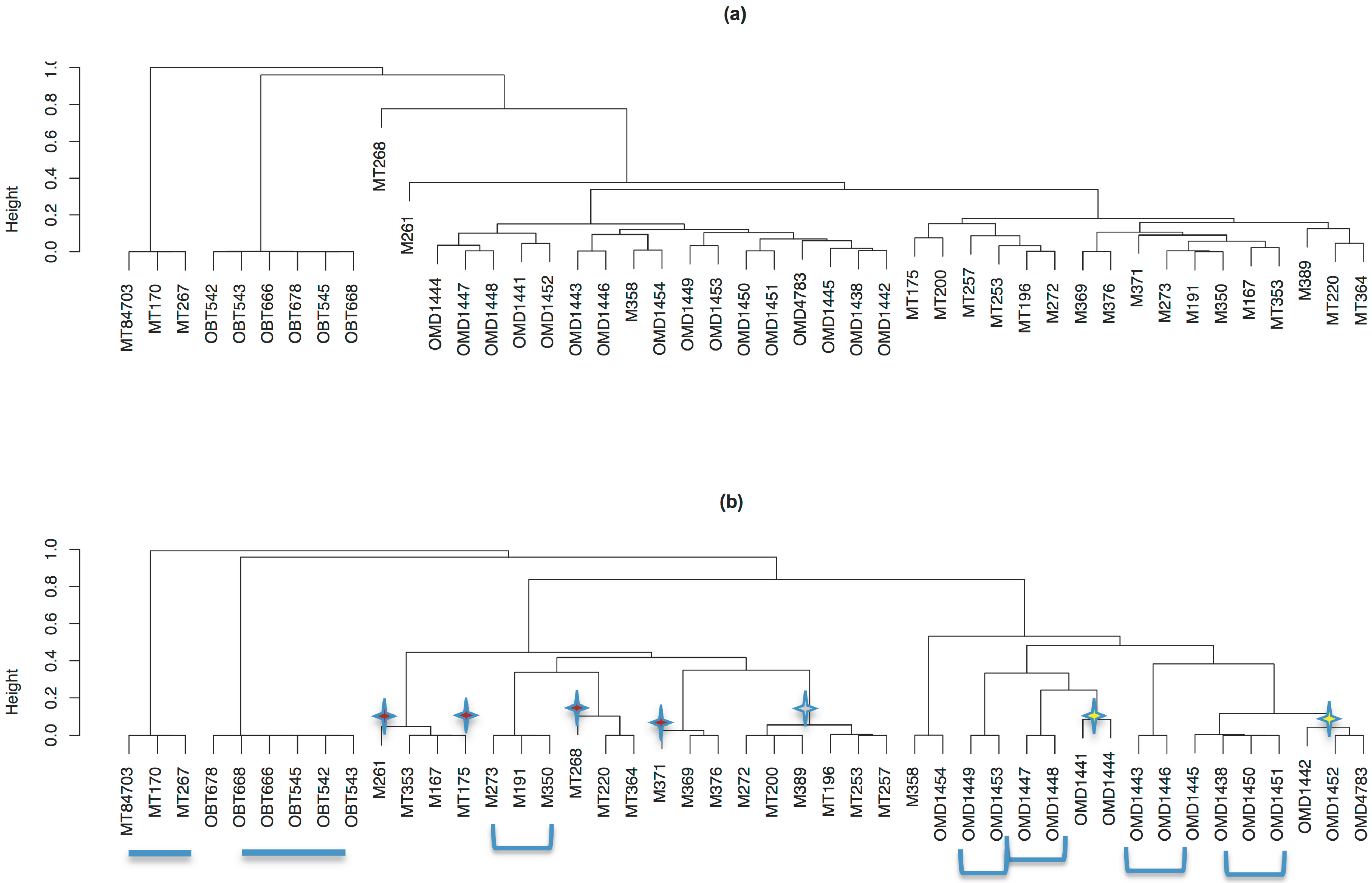
2.8. Determine the Relationship of Animals via Ensemble Cluster
2.9. Visualization of Animal Relatedness by Hierarchical Clustering
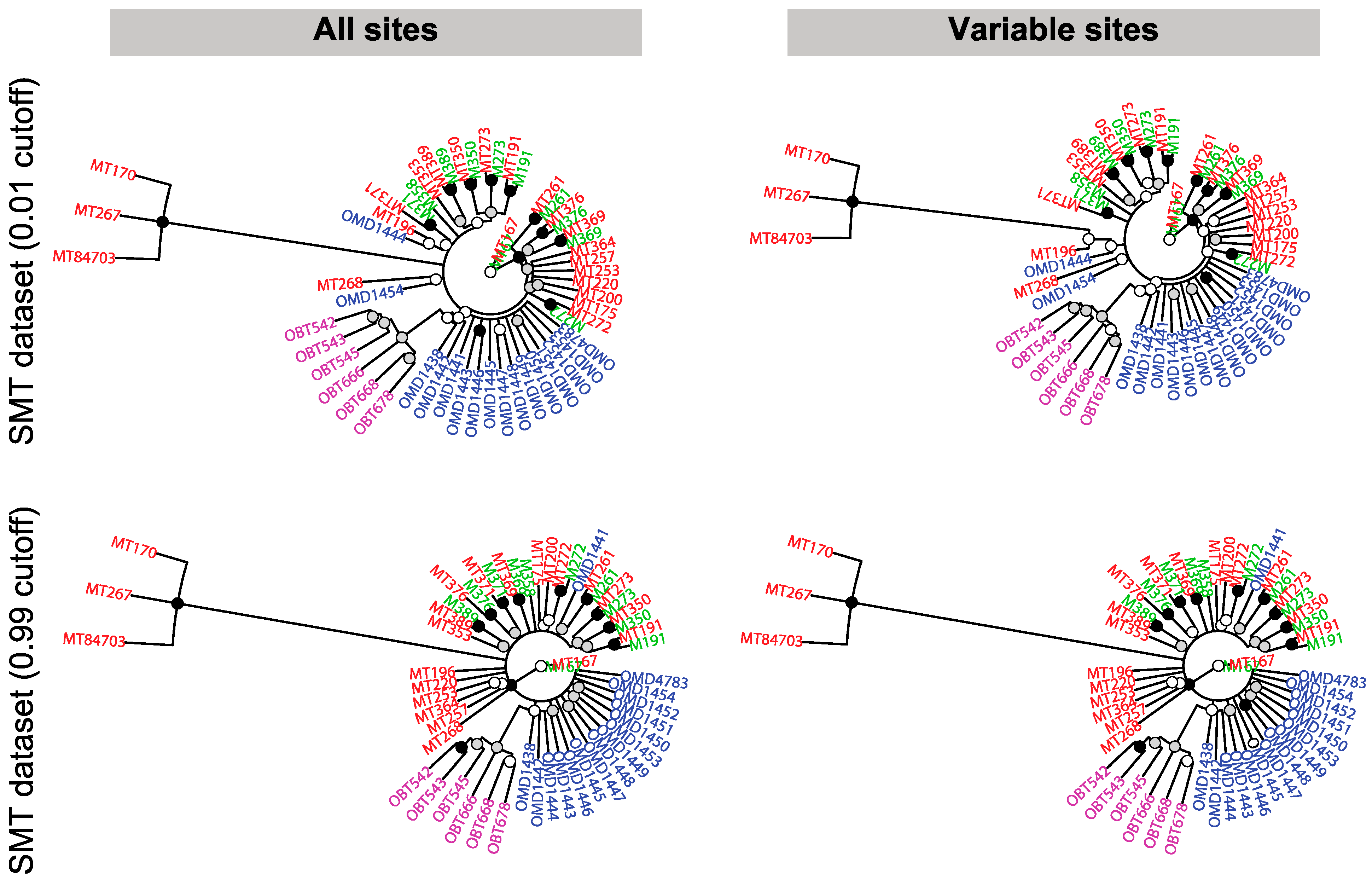
2.10. Phylogenetic Analysis
3. Results and Discussion
3.1. Overview of Research Objectives and Experimental Design
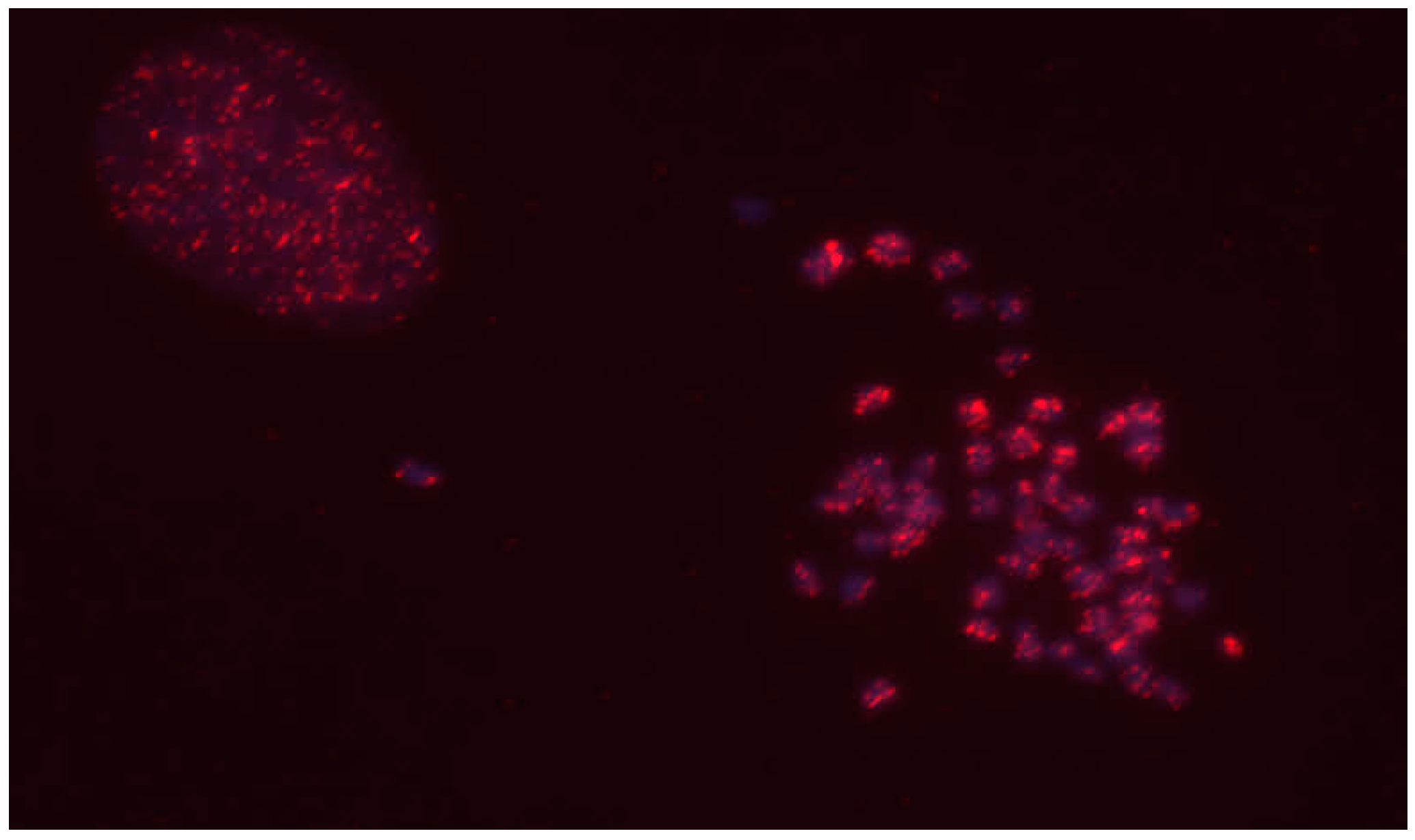
3.2. Fluorescent In Situ Hybridization Analysis of CrERV Locations
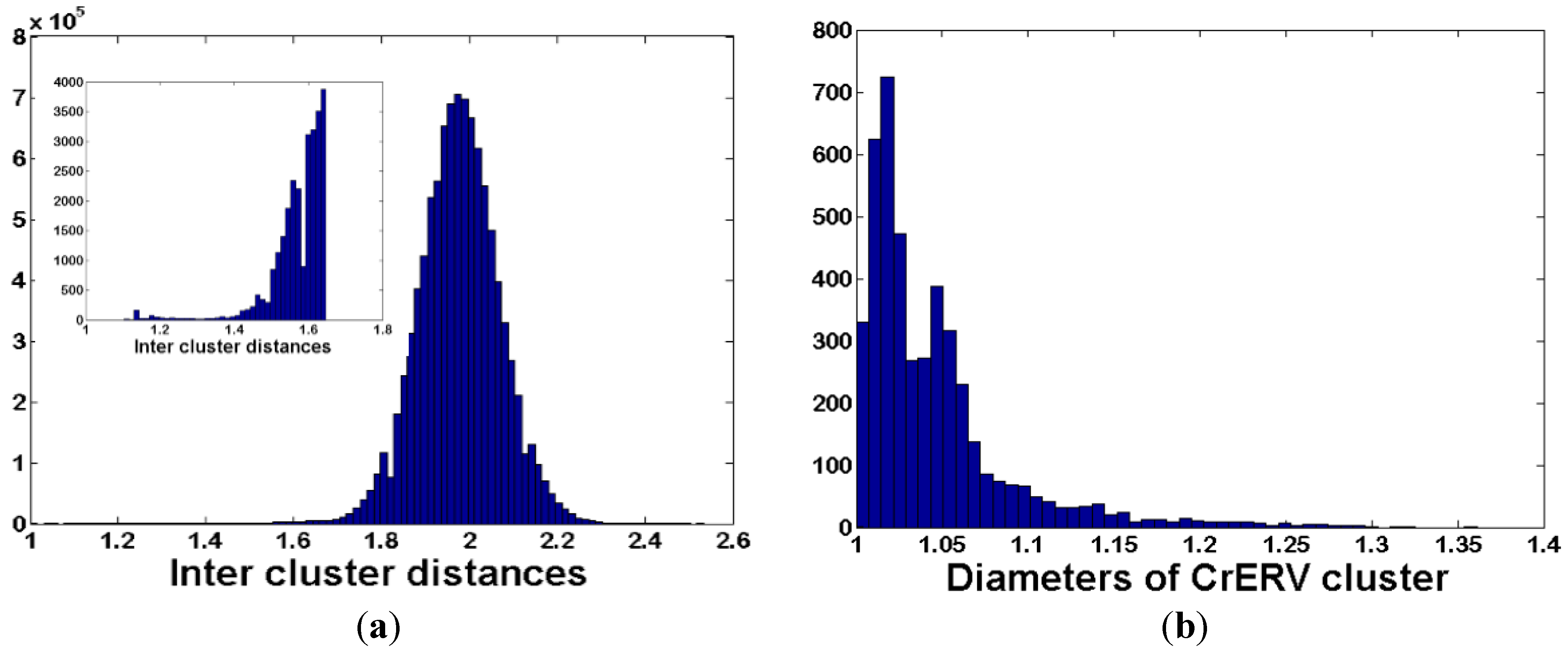
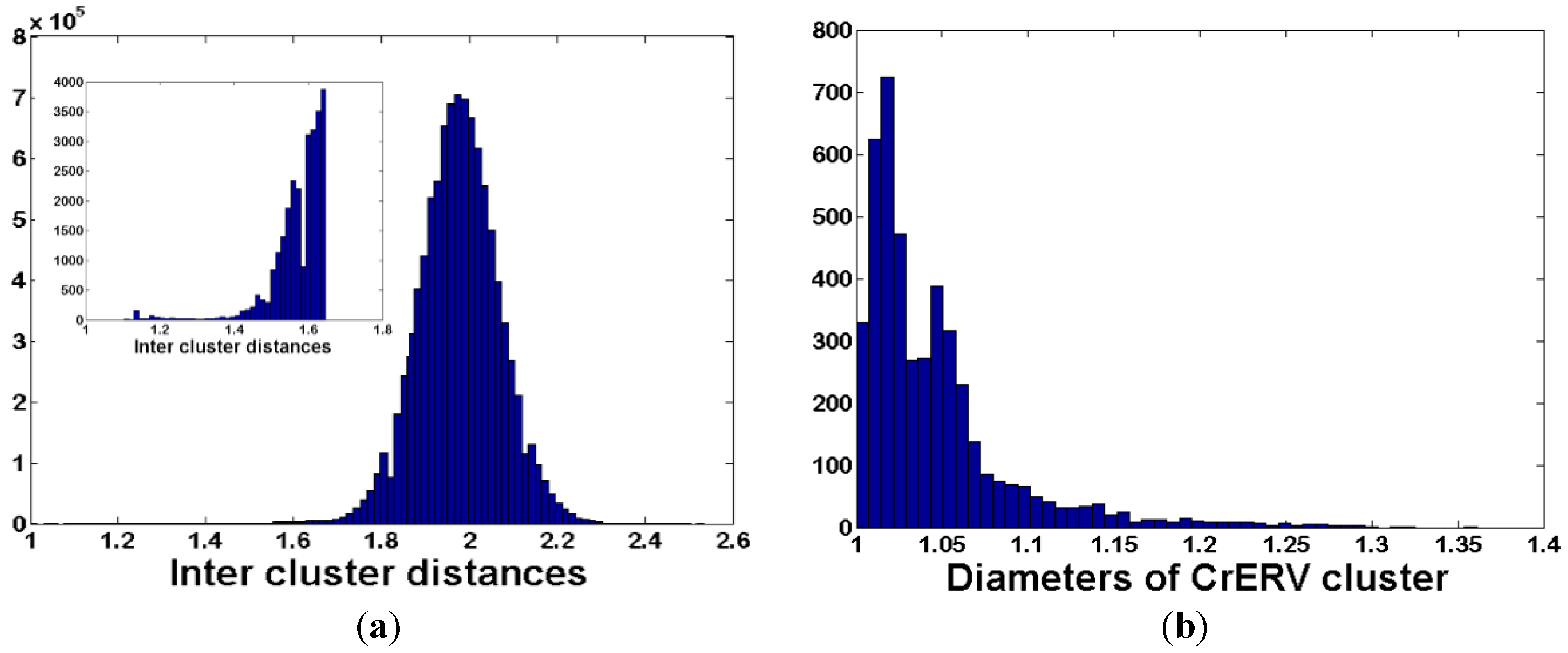
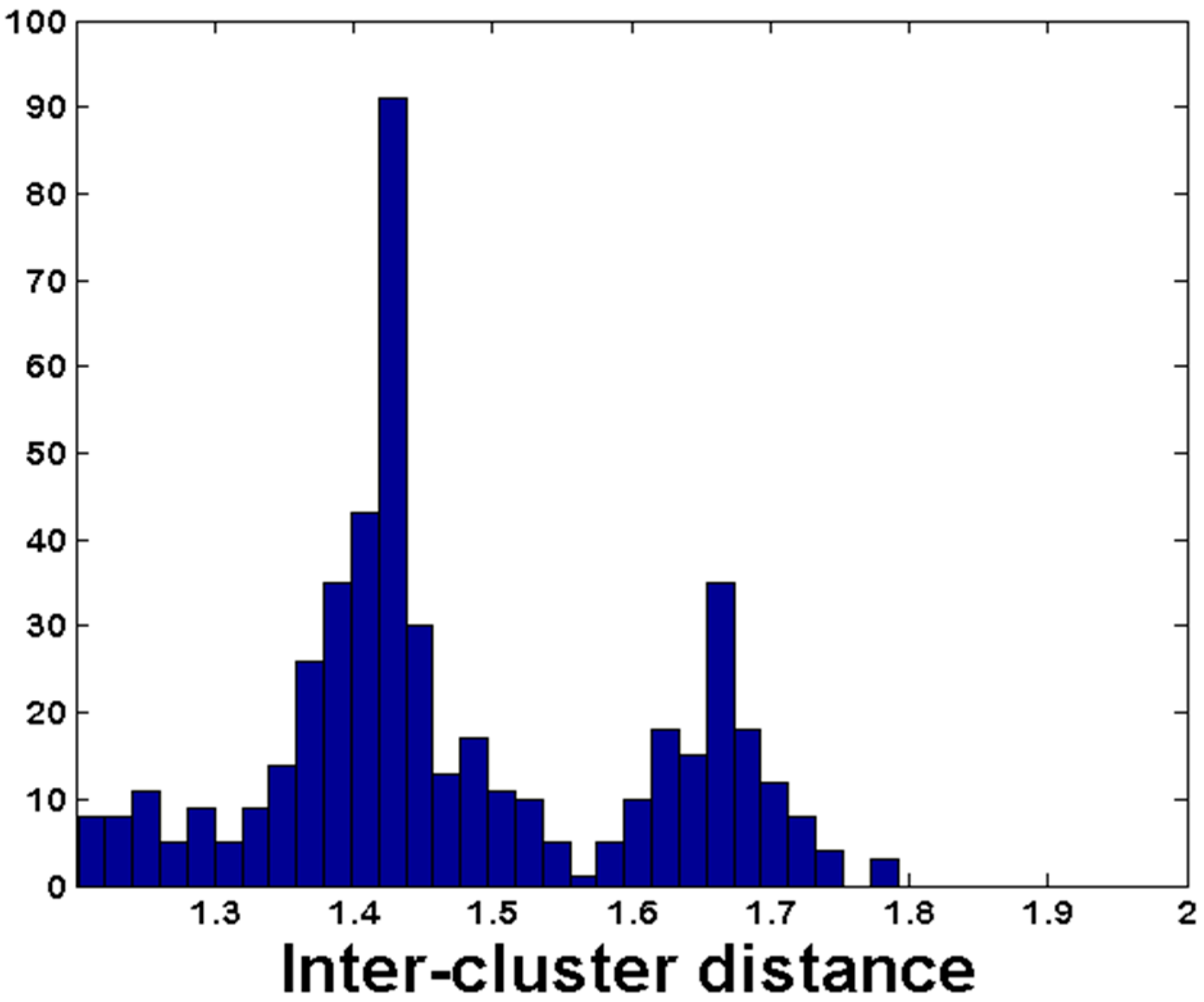
3.3. De Novo Clustering Analyses of CrERV-Host Junction Fragments
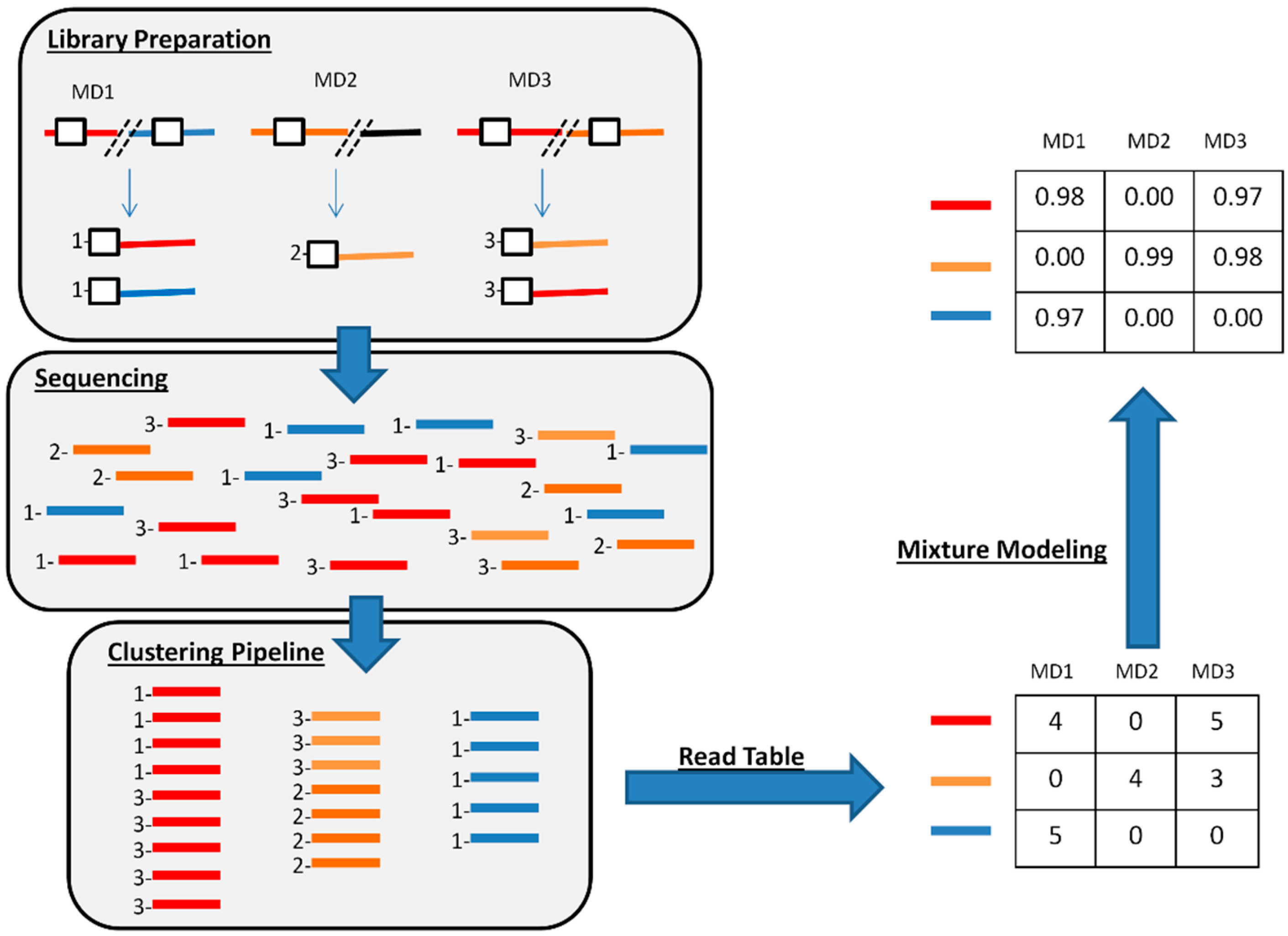

3.4. Estimating the Probability of a CrERV Assignment Using a Mixture Model



{kind=link}
{kind=link}
{kind=link}
{kind=link}
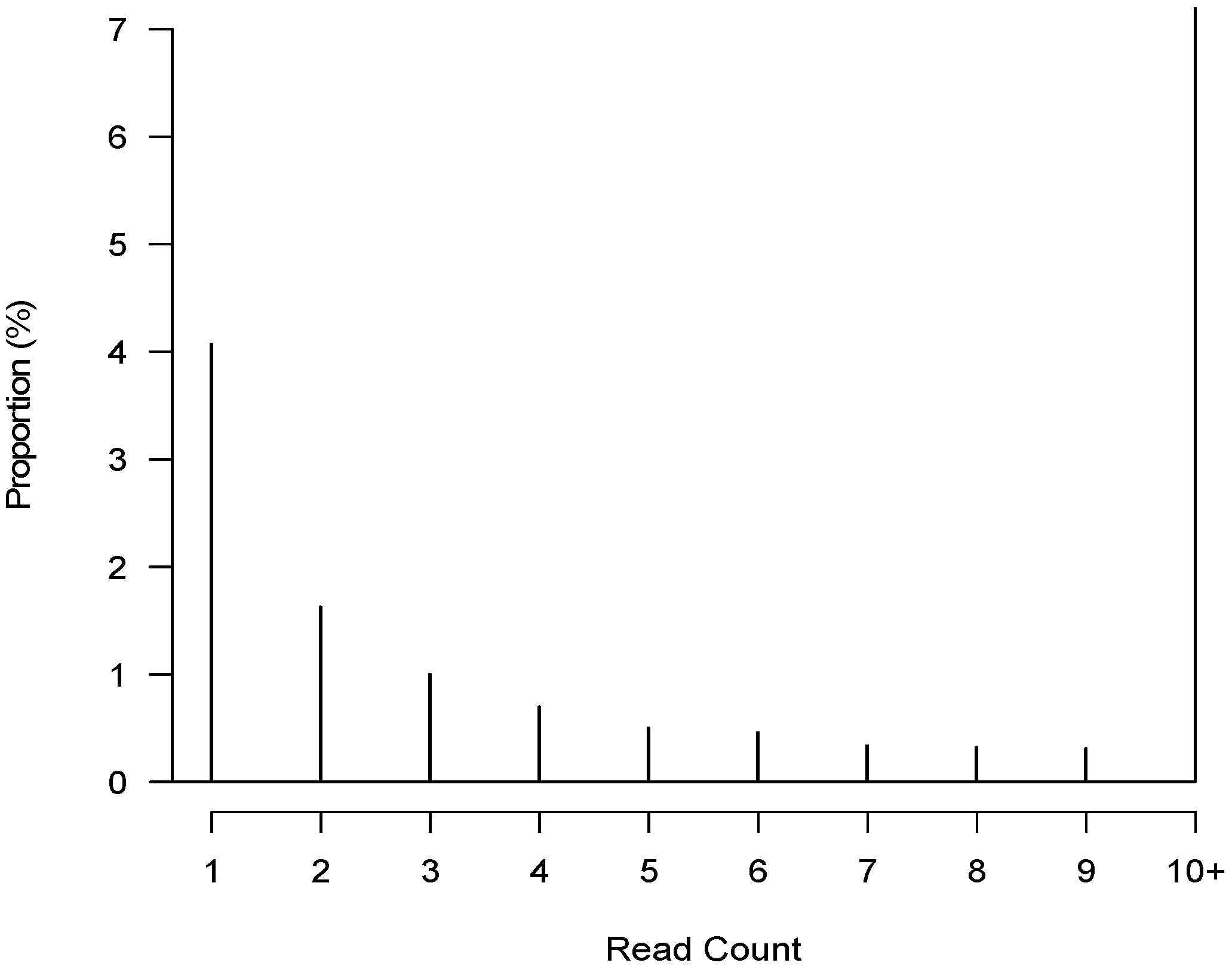{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Animal ID | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | Mixture Model |
|---|---|---|---|---|---|---|---|---|---|---|---|
| M191 | 0.083 | 0.048 | 0.039 | 0.044 | 0.05 | 0.053 | 0.059 | 0.065 | 0.068 | 0.076 | 0.049 |
| M389 | 0.174 | 0.101 | 0.067 | 0.047 | 0.038 | 0.033 | 0.034 | 0.034 | 0.037 | 0.034 | 0.034 |
| M350 | 0.196 | 0.106 | 0.061 | 0.041 | 0.035 | 0.034 | 0.029 | 0.029 | 0.027 | 0.025 | 0.025 |
| M261 | 0.061 | 0.032 | 0.033 | 0.033 | 0.04 | 0.039 | 0.04 | 0.043 | 0.042 | 0.041 | 0.033 |
| M369 | 0.11 | 0.04 | 0.027 | 0.025 | 0.028 | 0.027 | 0.026 | 0.021 | 0.023 | 0.024 | 0.028 |
| M167 | 0.157 | 0.079 | 0.053 | 0.047 | 0.04 | 0.035 | 0.035 | 0.034 | 0.035 | 0.035 | 0.047 |
| M371 | 0.208 | 0.094 | 0.057 | 0.047 | 0.041 | 0.037 | 0.035 | 0.034 | 0.038 | 0.039 | 0.051 |
| M376 | 0.07 | 0.055 | 0.06 | 0.064 | 0.066 | 0.071 | 0.072 | 0.075 | 0.08 | 0.081 | 0.062 |
| M272 | 0.249 | 0.127 | 0.077 | 0.064 | 0.061 | 0.059 | 0.056 | 0.06 | 0.063 | 0.064 | 0.038 |
| M273 | 0.103 | 0.057 | 0.042 | 0.034 | 0.030 | 0.027 | 0.028 | 0.030 | 0.028 | 0.027 | 0.027 |
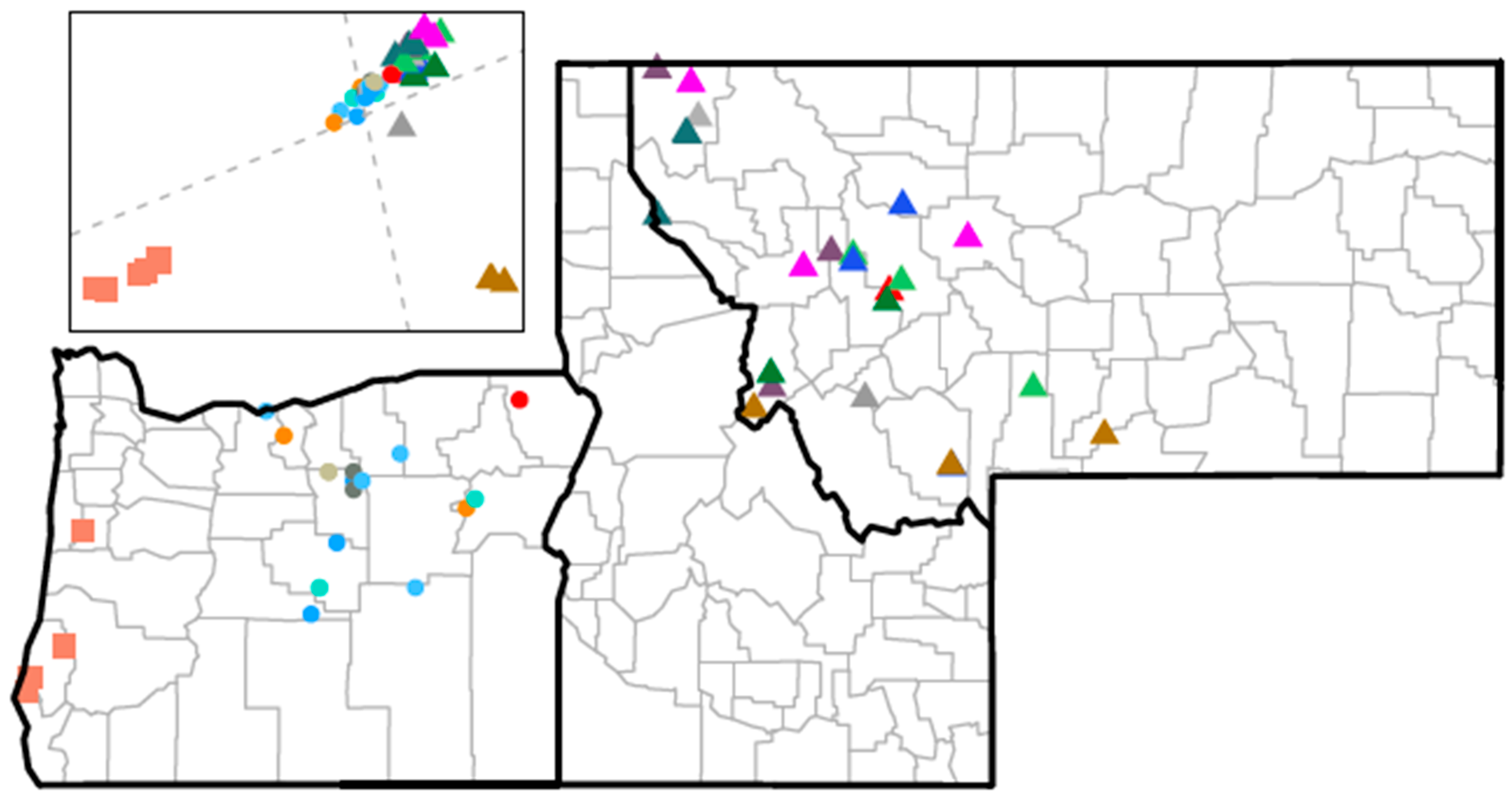
3.5. CrERV Distribution in Mule Deer
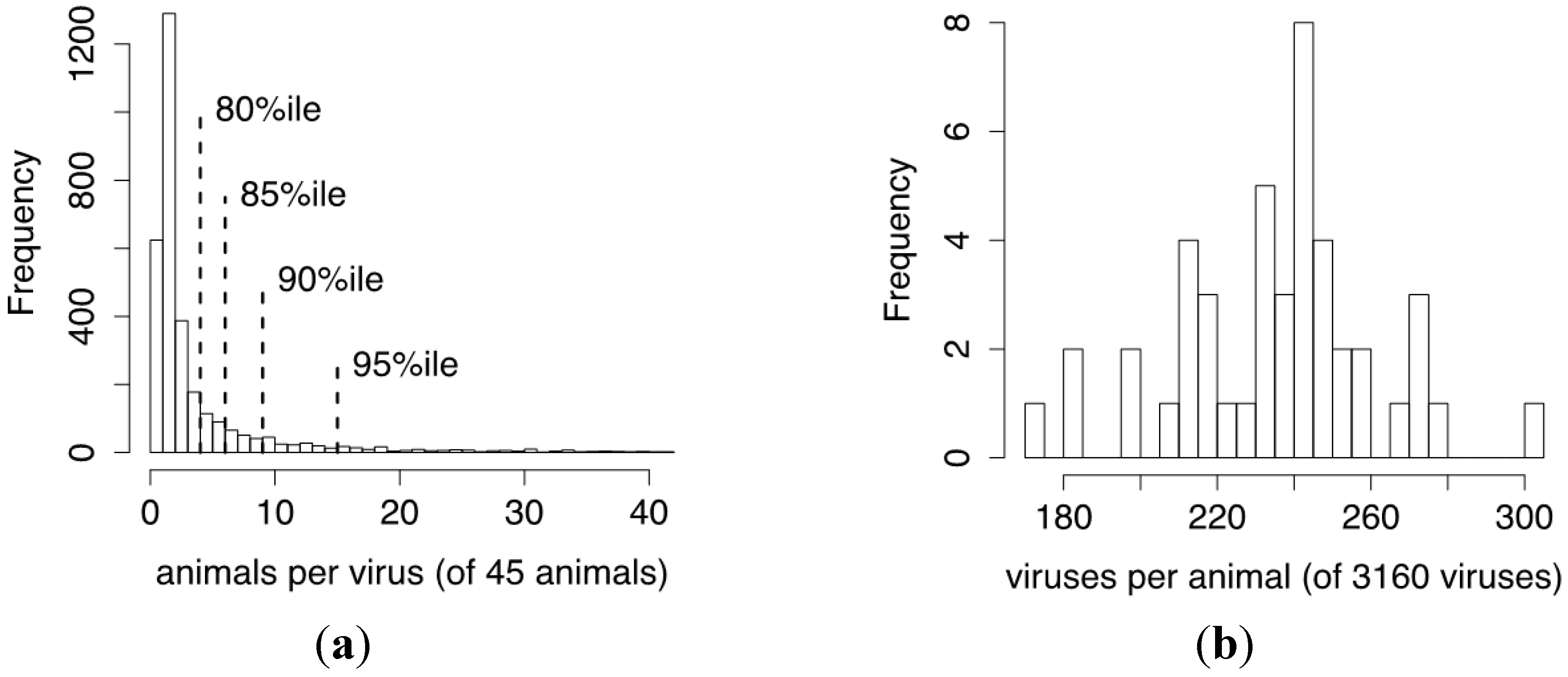
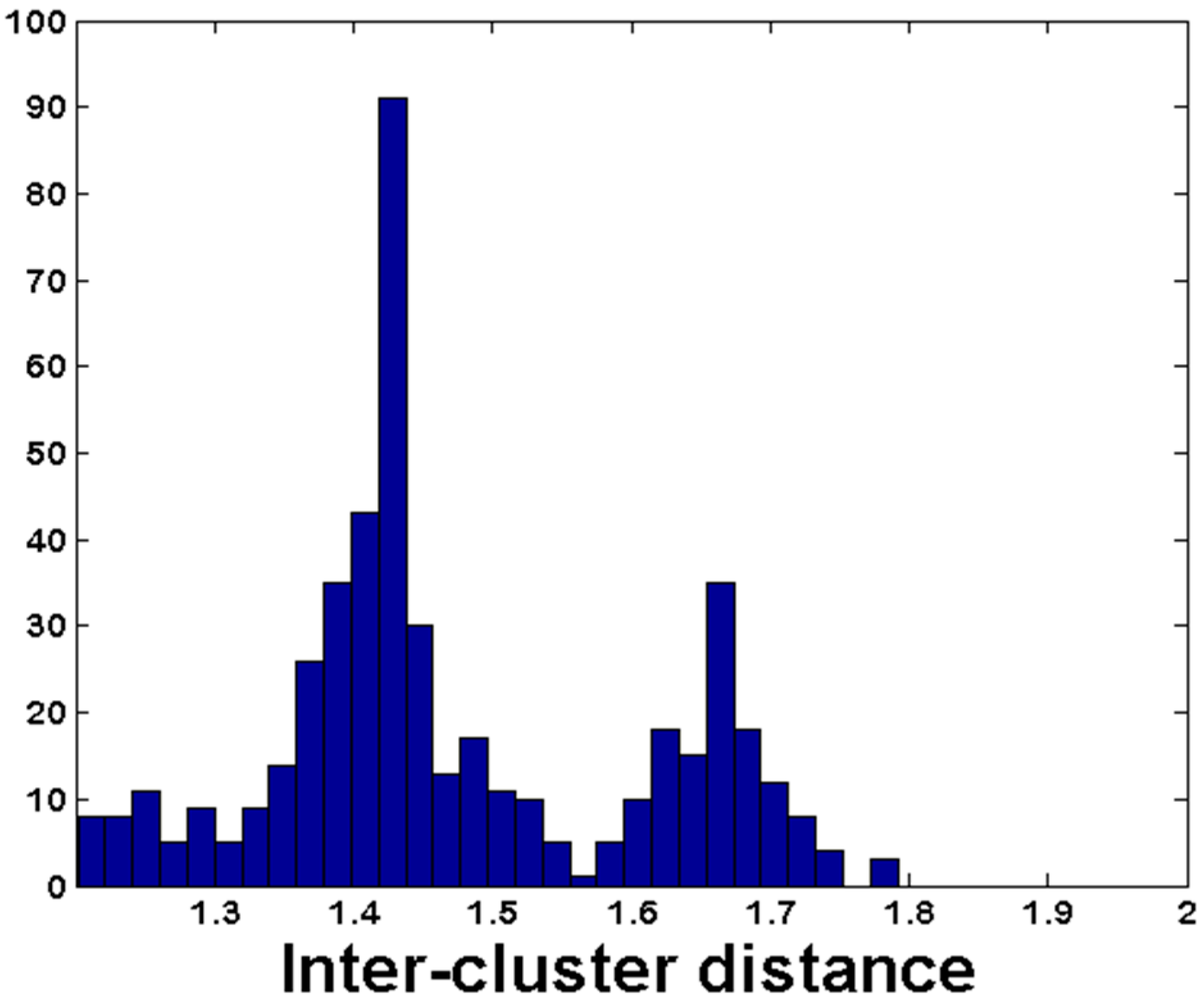
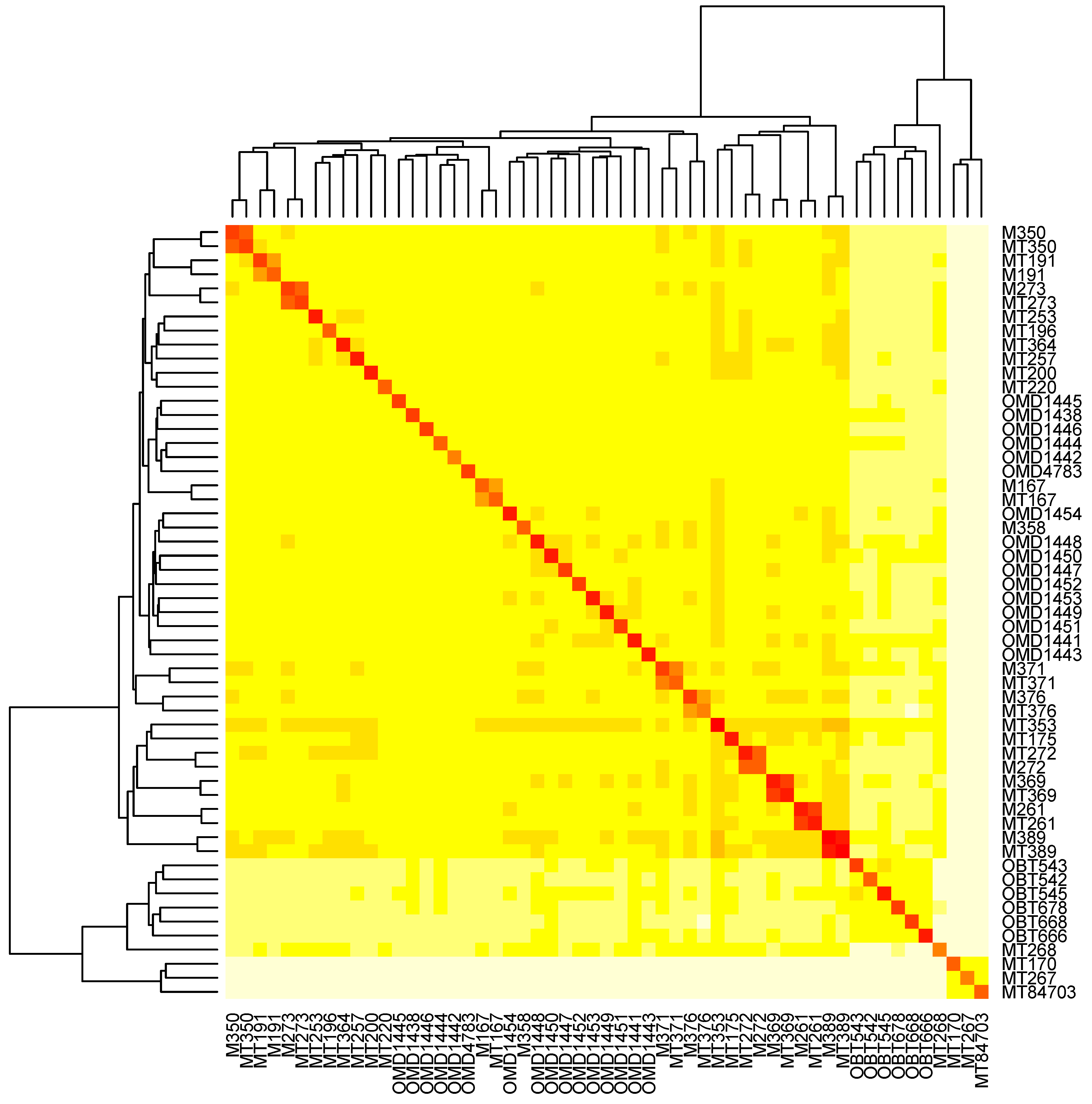
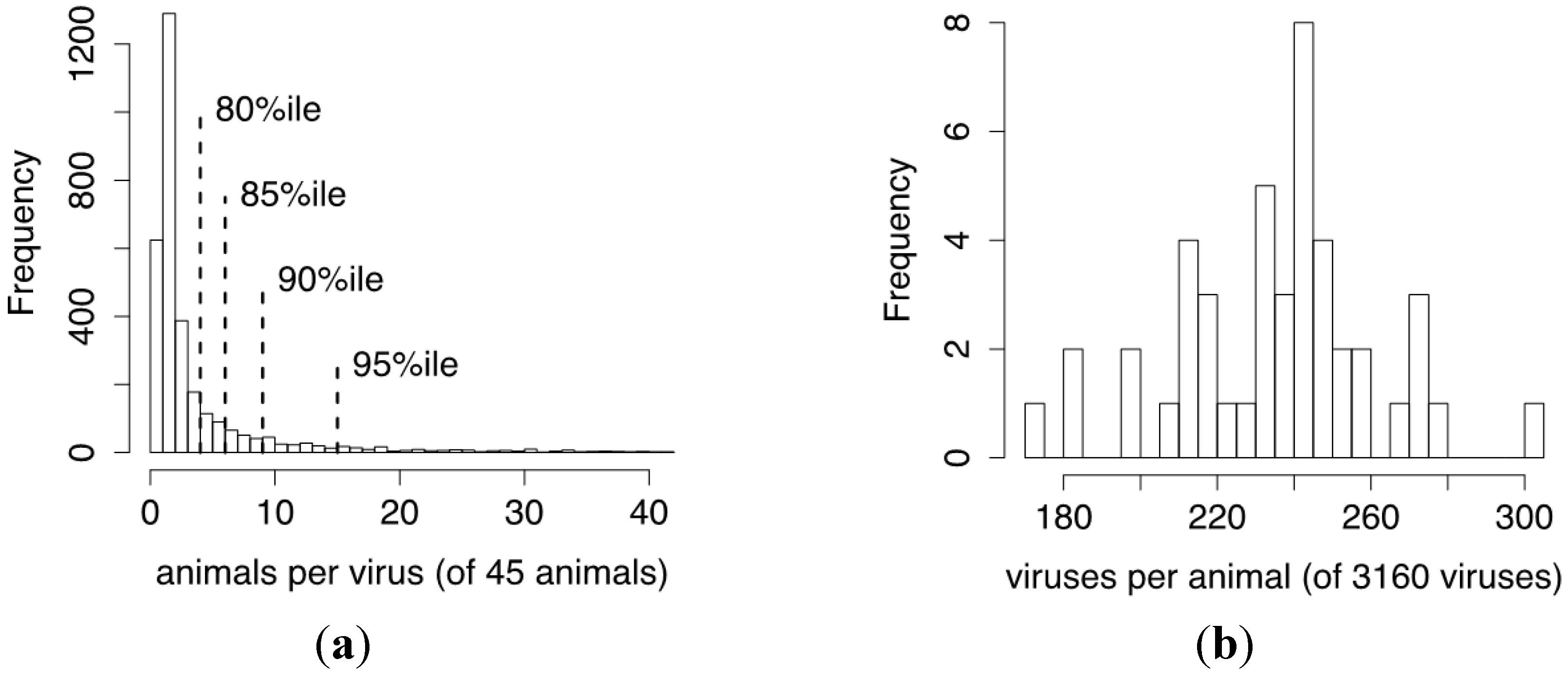
3.6. The Relatedness of Mule Deer Based on Shared CrERVs
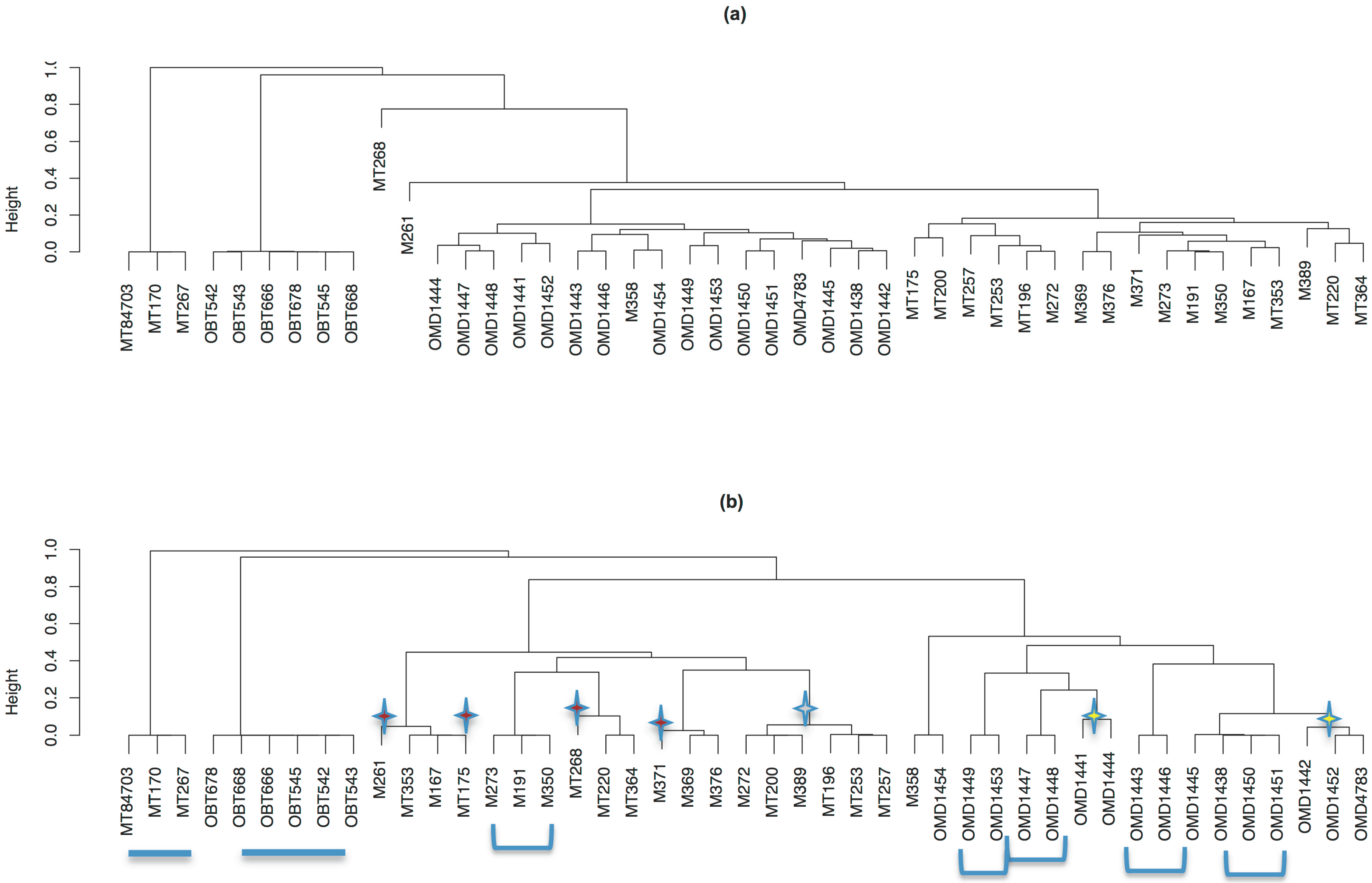
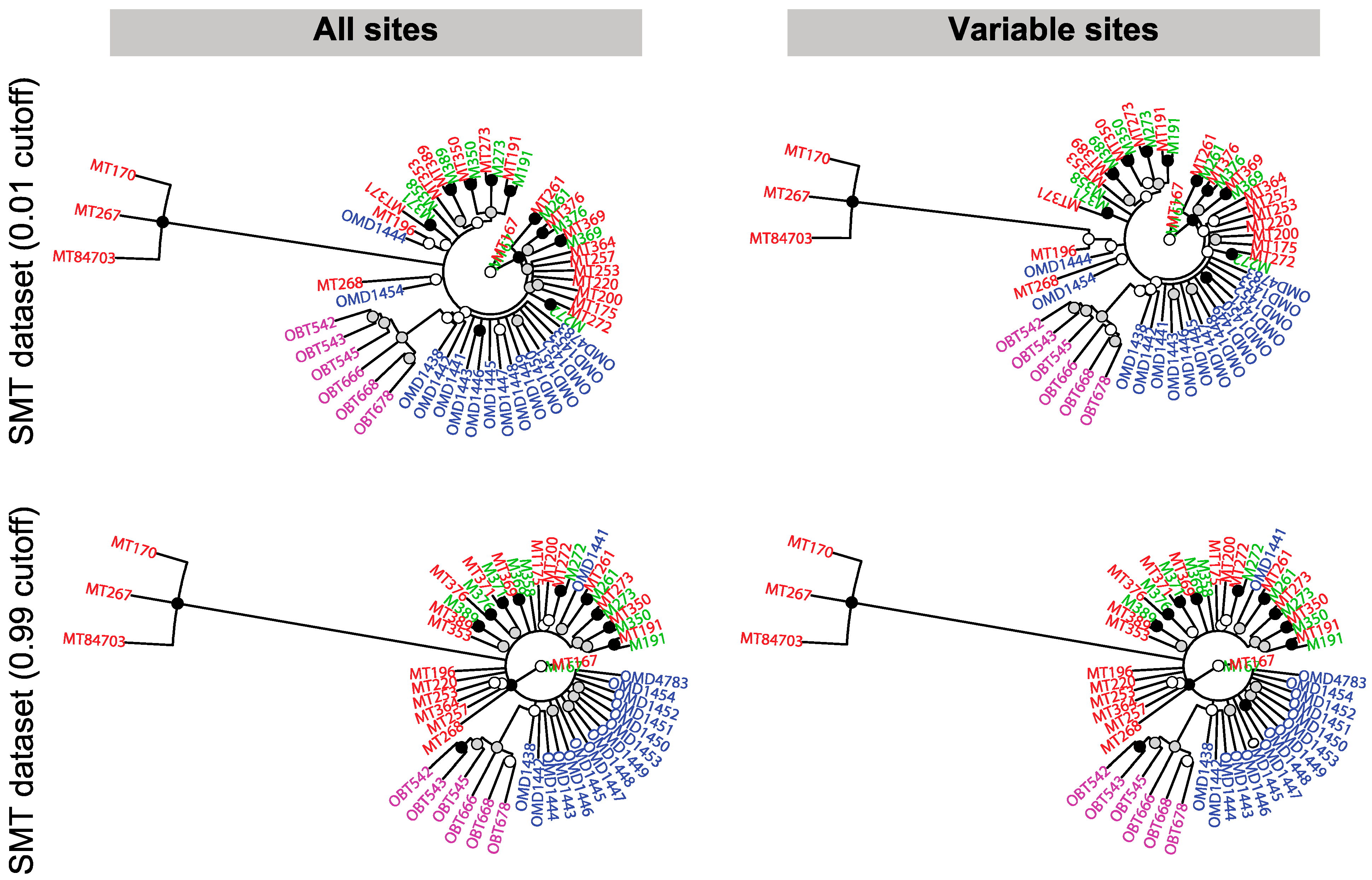
3.7. Discussion
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Schnable, P.S.; Ware, D.; Fulton, R.S.; Stein, J.C.; Wei, F.; Pasternak, S.; Liang, C.; Zhang, J.; Fulton, L.; Graves, T.A.; et al. The B73 maize genome: Complexity, diversity, and dynamics. Science 2009, 326, 1112–1115. [Google Scholar] [CrossRef] [PubMed]
- De Koning, A.P.J.; Gu, W.; Castoe, T.A.; Batzer, M.A.; Pollock, D.D. Repetitive elements may comprise over two-thirds of the human genome. PLoS Genet. 2011, 7. [Google Scholar] [CrossRef]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [PubMed]
- Kazazian, H.H. Mobile elements: Drivers of genome evolution. Science 2004, 303, 1626–1632. [Google Scholar] [CrossRef] [PubMed]
- Bourque, G.; Leong, B.; Vega, V.B.; Chen, X.; Lee, Y.L.; Srinivasan, K.G.; Chew, J.L.; Ruan, Y.; Wei, C.L.; Ng, H.H.; et al. Evolution of the mammalian transcription factor binding repertoire via transposable elements. Genome Res. 2008, 18, 1752–1762. [Google Scholar] [PubMed]
- Feschotte, C. Transposable elements and the evolution of regulatory networks. Nat. Rev. Genet. 2008, 9, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Jern, P.; Coffin, J.M. Effects of retroviruses on host genome function. Annu. Rev. Genet. 2008, 42, 709–732. [Google Scholar] [CrossRef]
- Feschotte, C.; Gilbert, C. Endogenous viruses: Insights into viral evolution and impact on host biology. Nat. Rev. Genet. 2012, 13, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Stoye, J.P. Studies of endogenous retroviruses reveal a continuing evolutionary saga. Nat. Rev. Microbiol. 2012, 10, 395–406. [Google Scholar] [PubMed]
- Marchi, E.; Kanapin, A.; Magiorkinis, G.; Belshaw, R. Unfixed endogenous retroviral insertions in the human population. J. Virol. 2014, 148. [Google Scholar] [CrossRef]
- Belshaw, R.; Dawson, A.L.A.; Woolven, A.J.; Redding, J.; Burt, A.; Tristem, M. Genomewide screening reveals high levels of insertional polymorphism in the human endogenous retrovirus family HERV-K(HML2): Implications for present-day activity. J. Virol. 2005, 79, 12507–12514. [Google Scholar] [CrossRef] [PubMed]
- Elleder, D.; Kim, O.; Padhi, A.; Bankert, J.G.; Simeonov, I.; Schuster, S.C.; Wittekindt, N.E.; Motameny, S.; Poss, M. Polymorphic integrations of an endogenous gammaretrovirus in the mule deer genome. J. Virol. 2012, 86, 2787–2796. [Google Scholar] [CrossRef] [PubMed]
- Ávila-Arcos, M.C.; Ho, S.Y.W.; Ishida, Y.; Nikolaidis, N.; Tsangaras, K.; Hönig, K.; Medina, R.; Rasmussen, M.; Fordyce, S.L.; Calvignac-Spencer, S.; et al. One hundred twenty years of koala retrovirus evolution determined from museum skins. Mol. Biol. Evol. 2013, 30, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, C.; Ropiquet, A.; Hassanin, A. Mitochondrial and nuclear phylogenies of Cervidae (Mammalia, Ruminantia): Systematics, morphology, and biogeography. Mol. Phylogenet. Evol. 2006, 40, 101–117. [Google Scholar] [CrossRef] [PubMed]
- Hedges, S.B.; Dudley, J.; Kumar, S. TimeTree: A public knowledge-base of divergence times among organisms. Bioinformatics 2006, 22, 2971–2972. [Google Scholar] [PubMed]
- Slotkin, R.K.; Martienssen, R. Transposable elements and the epigenetic regulation of the genome. Nat. Rev. Genet. 2007, 8, 272–285. [Google Scholar] [CrossRef] [PubMed]
- Contreras-Galindo, R.; Kaplan, M.H.; Leissner, P.; Verjat, T.; Ferlenghi, I.; Bagnoli, F.; Giusti, F.; Dosik, M.H.; Hayes, D.F.; Gitlin, S.D.; et al. Human endogenous retrovirus K (HML-2) elements in the plasma of people with lymphoma and breast cancer. J. Virol. 2008, 82, 9329–9336. [Google Scholar] [CrossRef] [PubMed]
- Kewitz, S.; Staege, M.S. Expression and Regulation of the Endogenous Retrovirus 3 in Hodgkin’s Lymphoma Cells. Front. Oncol. 2013, 3. [Google Scholar] [CrossRef]
- Huang, G.; Li, Z.; Wan, X.; Wang, Y.; Dong, J. Human endogenous retroviral K element encodes fusogenic activity in melanoma cells. J Carcinog 2013, 12. [Google Scholar] [CrossRef]
- Takeuchi, K.; Katsumata, K.; Ikeda, H.; Minami, M.; Wakisaka, A.; Yoshiki, T. Expression of endogenous retroviruses, ERV3 and lambda 4-1, in synovial tissues from patients with rheumatoid arthritis. Clin. Exp. Immunol. 1995, 99, 338–344. [Google Scholar] [PubMed]
- García-Montojo, M.; de la Hera, B.; Varadé, J.; de la Encarnación, A.; Camacho, I.; Domínguez-Mozo, M.; Arias-Leal, A.; García-Martínez, Á.; Casanova, I.; Izquierdo, G.; et al. HERV-W polymorphism in chromosome X is associated with multiple sclerosis risk and with differential expression of MSRV. Retrovirology 2014, 11. [Google Scholar] [CrossRef]
- Treangen, T.J.; Salzberg, S.L. Repetitive DNA and next-generation sequencing: Computational challenges and solutions. Nat Rev Genet. 2013, 13, 36–46. [Google Scholar]
- Contreras-Galindo, R.; Kaplan, M.H.; He, S.; Contreras-Galindo, A.C.; Gonzalez-Hernandez, M.J.; Kappes, F.; Dube, D.; Chan, S.M.; Robinson, D.; Meng, F.; et al. HIV infection reveals widespread expansion of novel centromeric human endogenous retroviruses. Genome Res. 2013, 23, 1505–1513. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Akagi, K.; Hu, Y.; Trivett, A.L.; Hlynialuk, C.J.W.; Swing, D.A.; Volfovsky, N.; Morgan, T.C.; Golubeva, Y.; Stephens, R.M.; et al. Mouse endogenous retroviruses can trigger premature transcriptional termination at a distance. Genome Res. 2012, 22, 870–884. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Carrel, L. Escape from X chromosome inactivation is an intrinsic property of the Jarid1c locus. Proc. Natl. Acad. Sci. USA 2008, 105, 17055–17060. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.; Gustashaw, K.; Wolff, D.J.; Rider, S.H.; Monaco, A.P.; Eble, B.; Schlessinger, D.; Gorski, J.L.; van Ommen, G.J.; Weissenbach, J. Three genes that escape X chromosome inactivation are clustered within a 6 Mb YAC contig and STS map in Xp11.21–p11.22. Hum. Mol. Genet. 1995, 4, 731–739. [Google Scholar] [CrossRef] [PubMed]
- Iskow, R.C.; McCabe, M.T.; Mills, R.E.; Torene, S.; Pittard, W.S.; Neuwald, A.F.; van Meir, E.G.; Vertino, P.M.; Devine, S.E. Natural mutagenesis of human genomes by endogenous retrotransposons. Cell 2010, 141, 1253–1261. [Google Scholar] [CrossRef] [PubMed]
- Witherspoon, D.J.; Xing, J.; Zhang, Y.; Watkins, W.S.; Batzer, M.A.; Jorde, L.B. Mobile element scanning (ME-Scan) by targeted high-throughput sequencing. BMC Genomics 2010, 11. [Google Scholar] [CrossRef] [PubMed]
- Ray, A.; Rahbari, R.; Badge, R.M. IAP Display: A Simple Method to Identify Mouse Strain Specific IAP Insertions. Mol. Biotechnol. 2011, 47, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Ciuffi, A.; Ronen, K.; Brady, T.; Malani, N.; Wang, G.; Berry, C.C.; Bushman, F.D. Methods for integration site distribution analyses in animal cell genomes. Methods 2009, 47, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Kamath, P.; Elleder, D.; Bao, L. The Population History of Endogenous Retroviruses in Mule Deer (Odocoileus hemionus). J. Hered. 2014, 105, 173–187. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, R.; Elleder, D.; Bao, L.; Hunter, D.; Acharya, R.; Poss, M. Clustering Pipeline for Determining Consensus Sequences in Targeted Next-Generation Sequencing. ArXiv E-Prints 2014. arXiv:1410.1608. [Google Scholar]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed]
- Dempster, A.; Laird, N.; Rubin, D. Maximum Likelihood from Incomplete Data via the EM Algorithm. J. R. Stat. Soc. B 1977, 39, 1–38. [Google Scholar]
- Strehl, A.; Ghosh, J. Cluster Ensembles—A Knowledge Reuse Framework for Combining Multiple Partitions. J. Mach. Learn. Res. 2002, 3, 583–617. [Google Scholar]
- Raymond, G.; Olsen, E.; Lee, K. Inhibition of protease-resistant prion protein formation in a transformed deer cell line infected with chronic wasting disease. J.Virol. 2006, 80, 596–604. [Google Scholar] [CrossRef] [PubMed]
- Dunn, J.C.A. Fuzzy Relative of the ISODATA Process and Its Use in Detecting Compact Well-Separated Clusters. J. Cybern. 1973, 3, 32–57. [Google Scholar] [CrossRef]
- Davies, D.L.; Bouldin, D.W. A Cluster Separation Measure. IEEE Trans. Pattern Anal. Mach. Intell. 1979, PAMI-1, 224–227. [Google Scholar]
- Latch, E.K.; Heffelfinger, J.R.; Fike, J.A.; Rhodes, O.E. Species-wide phylogeography of North American mule deer (Odocoileus hemionus): Cryptic glacial refugia and postglacial recolonization. Mol. Ecol. 2009, 18, 1730–1745. [Google Scholar] [CrossRef] [PubMed]
- Ilie, L.; Fazayeli, F.; Ilie, S. HiTEC: Accurate error correction in high-throughput sequencing data. Bioinformatics 2011, 27, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Kelley, D.R.; Schatz, M.C.; Salzberg, S.L. Quake: Quality-aware detection and correction of sequencing errors. Genome Biol. 2010, 11. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Schröder, J.; Schmidt, B. Musket: A multistage k-mer spectrum-based error corrector for Illumina sequence data. Bioinformatics 2013, 29, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Schmidt, B.; Maskell, D.L. DecGPU: Distributed error correction on massively parallel graphics processing units using CUDA and MPI. BMC Bioinformatics 2011, 12. [Google Scholar] [CrossRef] [PubMed]
- Medvedev, P.; Scott, E.; Kakaradov, B.; Pevzner, P. Error correction of high-throughput sequencing datasets with non-uniform coverage. Bioinformatics 2011, 27, i137–i141. [Google Scholar] [CrossRef] [PubMed]
- Lindsay, B.G. Mixture Models: Theory, Geometry and Applications; Institute of Mathematical Statistics and American Statistical Association: Philadelphia, PA, USA, 1996. [Google Scholar]
- McLachlan, J.G.; Krishnan, T. EM Algorithm Extensions. In Wiley Series in Probability and Statistics; John Wiley & Sons, Inc.: New York, NY, USA, 1997. [Google Scholar]
- De Queiroz, A.; Gatesy, J. The supermatrix approach to systematics. Trends Ecol. Evol. 2007, 22, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Rokas, A.; Williams, B.L.; King, N.; Carroll, S.B. Genome-scale approaches to resolving incongruence in molecular phylogenies. Nature 2003, 425, 798–804. [Google Scholar] [CrossRef] [PubMed]
- Kolaczkowski, B.; Thornton, J.W. Performance of maximum parsimony and likelihood phylogenetics when evolution is heterogeneous. Nature 2004, 431, 461–463. [Google Scholar] [CrossRef]
- Gadagkar, S.R.; Rosenberg, M.S.; Kumar, S. Inferring species phylogenies from multiple genes: Concatenated sequence tree versus consensus gene tree. J. Exp. Zool. B. Mol. Dev. Evol. 2005, 304, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Mossel, E.; Vigoda, E. Phylogenetic MCMC algorithms are misleading on mixtures of trees. Science 2005, 309, 2207–2209. [Google Scholar] [CrossRef] [PubMed]
- Edwards, S.V.; Liu, L.; Pearl, D.K. High-resolution species trees without concatenation. Proc. Natl. Acad. Sci. USA 2007, 104, 5936–5941. [Google Scholar] [CrossRef] [PubMed]
- Kubatko, L.S.; Degnan, J.H. Inconsistency of phylogenetic estimates from concatenated data under coalescence. Syst. Biol. 2007, 56, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Degnan, J.H.; Rosenberg, N.A. Gene tree discordance, phylogenetic inference and the multispecies coalescent. Trends Ecol. Evol. 2014, 24, 332–340. [Google Scholar] [CrossRef]
- Rannala, B.; Yang, Z. Phylogenetic inference using whole genomes. Annu. Rev. Genomics Hum. Genet. 2008, 9, 217–231. [Google Scholar] [CrossRef] [PubMed]
- Degnan, J.H.; Rosenberg, N.A. Discordance of species trees with their most likely gene trees. PLoS Genet. 2006, 2. [Google Scholar] [CrossRef] [PubMed]
- Degnan, J.H. Anomalous unrooted gene trees. Syst. Biol. 2013, 62, 574–590. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, N.A.; Tao, R. Discordance of species trees with their most likely gene trees: The case of five taxa. Syst. Biol. 2008, 57, 131–140. [Google Scholar] [CrossRef]
- Rosenberg, N.A. Discordance of species trees with their most likely gene trees: A unifying principle. Mol. Biol. Evol. 2013, 30, 2709–2713. [Google Scholar] [CrossRef] [PubMed]
- Heled, J.; Drummond, A.J. Bayesian inference of species trees from multilocus data. Mol. Biol. Evol. 2010, 27, 570–580. [Google Scholar] [CrossRef] [PubMed]
- Liu, L. BEST: Bayesian estimation of species trees under the coalescent model. Bioinformatics 2008, 24, 2542–2543. [Google Scholar] [CrossRef] [PubMed]
- Jewett, E.M.; Rosenberg, N.A. iGLASS: An improvement to the GLASS method for estimating species trees from gene trees. J. Comput. Biol. 2012, 19, 293–315. [Google Scholar] [CrossRef] [PubMed]
- Pickrell, J.K.; Pritchard, J.K. Inference of population splits and mixtures from genome-wide allele frequency data. PLoS Genet. 2012, 8. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bao, L.; Elleder, D.; Malhotra, R.; DeGiorgio, M.; Maravegias, T.; Horvath, L.; Carrel, L.; Gillin, C.; Hron, T.; Fábryová, H.; et al. Computational and Statistical Analyses of Insertional Polymorphic Endogenous Retroviruses in a Non-Model Organism. Computation 2014, 2, 221-245. https://doi.org/10.3390/computation2040221
Bao L, Elleder D, Malhotra R, DeGiorgio M, Maravegias T, Horvath L, Carrel L, Gillin C, Hron T, Fábryová H, et al. Computational and Statistical Analyses of Insertional Polymorphic Endogenous Retroviruses in a Non-Model Organism. Computation. 2014; 2(4):221-245. https://doi.org/10.3390/computation2040221
Chicago/Turabian StyleBao, Le, Daniel Elleder, Raunaq Malhotra, Michael DeGiorgio, Theodora Maravegias, Lindsay Horvath, Laura Carrel, Colin Gillin, Tomáš Hron, Helena Fábryová, and et al. 2014. "Computational and Statistical Analyses of Insertional Polymorphic Endogenous Retroviruses in a Non-Model Organism" Computation 2, no. 4: 221-245. https://doi.org/10.3390/computation2040221
APA StyleBao, L., Elleder, D., Malhotra, R., DeGiorgio, M., Maravegias, T., Horvath, L., Carrel, L., Gillin, C., Hron, T., Fábryová, H., Hunter, D. R., & Poss, M. (2014). Computational and Statistical Analyses of Insertional Polymorphic Endogenous Retroviruses in a Non-Model Organism. Computation, 2(4), 221-245. https://doi.org/10.3390/computation2040221