Abstract
Over the years, as more complete poxvirus genomes have been sequenced, phylogenetic studies of these viruses have become more prevalent. In general, the results show similar relationships between the poxvirus species; however, some inconsistencies are notable. Previous analyses of the viral genomes contained within the vaccinia virus (VACV)-Dryvax vaccine revealed that their phylogenetic relationships were sometimes clouded by low bootstrapping confidence. To analyze the VACV-Dryvax genomes in detail, a new tool-set was developed and integrated into the Base-By-Base bioinformatics software package. Analyses showed that fewer unique positions were present in each VACV-Dryvax genome than expected. A series of patterns, each containing several single nucleotide polymorphisms (SNPs) were identified that were counter to the results of the phylogenetic analysis. The VACV genomes were found to contain short DNA sequence blocks that matched more distantly related clades. Additionally, similar non-conforming SNP patterns were observed in (1) the variola virus clade; (2) some cowpox clades; and (3) VACV-CVA, the direct ancestor of VACV-MVA. Thus, traces of past recombination events are common in the various orthopoxvirus clades, including those associated with smallpox and cowpox viruses.
1. Introduction
The orthopoxviruses comprise one genus of the family Poxviridae; all poxviruses have a linear dsDNA genome, ranging in size from 140 to 350 kb. The central portion of the poxvirus genome tends to be conserved, encoding proteins required for genome replication, mRNA transcription, and building the virion. Eighty-nine genes are conserved in all Chordopoxviruses and 49 in all poxviruses; genus and species-specific genes are often located near the ends of the genome [1,2]. The ends of the genome have inverted terminal repeats (ITRs), which may also contain several genes, and hairpin loops create one covalently closed genomic DNA circle [3].
A variety of animal and human pathogens are present in other poxvirus genera, but the orthopoxviruses have been most studied because they include variola virus (VARV), the causative agent of the smallpox disease, and its vaccine, vaccinia virus (VACV) [4]. Although natural smallpox was eradicated in the 1970s, through a worldwide vaccination program [4], VACV is still intensively studied because: (1) the vaccine remains important due to potential use of smallpox as a bioterrorist weapon; (2) it serves as an excellent laboratory model for all poxviruses; and (3) it has been repurposed as a recombinant vaccine and anti-cancer therapeutics [5,6]. The first smallpox vaccine, as used by Jenner in the 1790s, is thought to have been a poxvirus isolated from cows/milkmaids; such cowpox viruses (CPXV) appear to be endemic in Eurasia, likely with small rodents as their natural reservoir. Recent genomic sequencing of a series of CPXV isolates has distinguished several clades among these viruses and suggests that VACV is more similar to CPXV strains with European ancestry [7,8]. In addition this work reported that several very similar CPXV isolates grouped with VARV and its closest relatives.
At the micro-scale, poxvirus genome evolution proceeds through single nucleotide substitutions; these may be observed as genetic drift with amino acid sequence substitutions in encoded proteins, and also as the dramatic variation in genome nucleotide composition (e.g., A + T%) that exists between some of the poxvirus genera [9]. Genome evolution also proceeds through macro-scale events, such as the creation of small or large insertions (often duplication events) and deletions (indels), as well as rearrangements, including transpositions or inversions of DNA [10,11], which includes homologous recombination between genomes and horizontal gene transfers [12]. The problems that these recombination events create for evolutionary genomics studies are well known [13].
Phylogenetic studies form part of most poxvirus sequencing projects; generally the trees are similar but some inconsistencies have been observed. For example, different phylogenetic relationships have been observed between the orthopoxviruses depending on which region of the genome is analyzed [9,14]. The use of different genes or proteins may also result in apparent differences in evolutionary relationships due to insufficient variation in the sequences. Therefore large data sets, such as concatenated conserved proteins or the central region of the poxvirus genomes, have been used in an attempt to create more reliable phylogeny, but puzzling inconsistencies still exist. An interesting example is the comparison of genomic sequences of multiple VACV isolates selected from a single vial of the Dryvax vaccine. Numerous VACV isolates were sequenced to examine their relationship and a detailed analysis of the Dryvax vaccine [15], which was derived from the New York City Board of Health (NYCBH) strain of VACV, revealed that their phylogenetic relationships were clouded by low bootstrapping confidence. Some sequence comparisons also suggested recombination between the VACV-Dryvax ancestors.
In the present study, VACV-Dryvax genome core sequences and a selection of other orthopoxvirus genomes were analyzed at the level of single nucleotides to identify blocks of sequence that show patterns of recombination. To perform this analysis, it was necessary to develop a series of software tools, installed in the Base-By-Base bioinformatics software [16,17].
2. Experimental Section
2.1. Retrieval of Genome Sequences and Alignment
The core alignment was constructed using the following genome sequences (listed as strain name, GenBank accession number): VACV-Acam3, AY313848; VACV-Acam2000, AY313847; VACV-Dryvax-DPP15, JN654981; VACV-Dryvax-DPP17, JN654983; VACV-Dryvax-DPP13, JN654980; VACV-Dryvax-DPP11, JN654978; VACV-Dryvax-DPP16, JN654982; VACV-Dryvax-DPP9, JN654976; VACV-Dryvax-DPP10, JN654977; VACV-Dryvax-DPP20, JN654985; VACV-Dryvax-DPP19, JN654984; VACV-DUKE, DQ439815; VACV-Dryvax-DPP12, JN654979; VACV-3737, DQ377945; VACV-Dryvax-DPP21, JN654986; HSPV-MNR76, DQ792504; VACV-CVA, AM501482; VACV-MVA, U94848; VACV-Acam3000, AY603355; VACV-WR, NC_006998; VACV-LC16mO, AY678277; VACV-Lister_VACV107, DQ121394; VACV-Lister, AY678276; VACV-Cop, M35027; VACV-TP5, KC207811; RPXV-Utr, AY484669; CPXV-AUS_1999, HQ407377; CPXV-GRI, X94355; CPXV-FIN_2000_MAN, HQ420893; CPXV-GER_1980_EP4, HQ420895; CPXV-GER_2002_MKY, HQ420898; CPXV-GER91, DQ437593; CPXV-GER_1998_2, HQ420897; CPXV-FRA_2001_Nancy, HQ420894; CPXV-NOR_1994_MAN, HQ420899; CPXV-BR, NC_003663; CPXV-UK2000_K2984, HQ420900; CPXV-HumLue09/1, KC813494; CPXV-MarLei07/1, KC813499; CPXV-HumPad07/1, KC813496; CPXV-HumGri07/1, KC813511; CPXV-HumMag07/1, KC813495; CPXV-HumLan08/1, KC813492; CPXV-HumGra07/1, KC813510; CPXV-HumLit08/1, KC813493; TATV-DAH68, NC_008291; CMLV-CMS, AY009089; VARV-GBR44_harv, DQ441444; VARV-YUG72, DQ441448; MPXV-ZAR, NC_003310; ECTV-Mos, NC_004105.
Sequences were aligned using MAFFT [18] and both ends of the genome alignments were removed leaving a 98 kb region spanning the VACV-WR genome nt 36,459–134,689, from gene VACV-WR-049 (ser/thr kinase) to VACV-WR-144 (RNA polymerase (RPO132). The sequences were then manually edited using Base-By-Base to fix alignment errors. For some analyses, all alignment columns containing gap-characters were removed prior to construction of the phylogenetic trees.
2.2. Visual Examination of the Multiple Sequence Alignment
Base-By-Base highlights single nucleotide polymorphisms (SNPs) in various ways [16,17]. The user can choose to compare sequences against the top sequence, against the consensus sequence, or in a pairwise fashion. Differences are highlighted by blue blocks; insertions and deletions are shown by green and red blocks, respectively. This highlighting makes otherwise unrecognizable short patterns of SNPs easily visible to the user. We were thus able to scan the sequence alignment visually for small and imperfect patterns of SNPs and indels.
2.3. Phylogenetic Tree Construction
Maximum likelihood (ML) and Neighbor-joining trees were constructed with MEGA5 [19] and T-REX with 1000 bootstrap replicates; generally tree topology had only small variances in the branching of the Dryvax clade were seen, but this was expected because of low bootstrap values in the ML tree.
2.4. Generation of Artificial Multiple Sequence Alignments
The sequence simulator indel-Seq-Gen (iSG) [20] was used to generate a set of artificial sequences in which the relationships between sequences was based on the organization and branch lengths of the input tree, using the HKY method of evolution [21]. Since Indel-Seq-Gen does not generate gaps in the artificial MSA we constructed the tree from the selected orthopoxvirus core genomes after removing all gap-containing columns. Due to limitations of the program, ECTV-Mos was excluded from the artificial alignment.
2.5. Use of Python Scripts for Further MSA Manipulation and Analysis
The python script snip.py scans a MSA for SNPs that differ from the consensus nucleotide. This SNIP function has also been built into Base-By-Base. The user sets a threshold value that dictates how many sequences can differ from the consensus at a given position before the SNP is changed to be the same as the consensus nucleotide. The SNPs are filtered if there are two types of nucleotides in a column and the number of SNPs in the column is less than or equal to the specified threshold value. If more than two types of nucleotides exist in a column, it will be skipped. For example, if a C and G are both present in a column in which the consensus nucleotide is a T, nothing will be changed. However, if a column has two Cs and the rest are Ts, the two Cs will be changed to Ts, provided that the threshold value is 2 or greater. For our purposes, the threshold was set to one in order to eliminate only unique SNPs and then constructed phylogenetic trees using the “snipped” MSAs in order to observe the effect of removing unique SNPs on terminal branch length.
3. Results and Discussion
Although there are a wide variety of bioinformatics tools for comparative genomics analyses, often the presentation of the results shields the researcher from the raw data; this makes the researcher reliant on the software for correct interpretation of the data. For example, although the percent identity matrices and phylogenetic trees derived from many multiple alignment tools are very good summaries of the data, most users never examine the details of the relationships between the genomes being compared; sometimes, the scale of a project (length or number of sequences) imposes the use of such summaries. In order to solve this problem for the analysis of poxvirus genomes, several new features were incorporated into Base-By-Base, our multiple sequence alignment (MSA) editor; they allow more detailed comparisons of multiple sequences and, by providing both numerical and visual output, permit a researcher to understand the exact nature of the differences between the sequences. These tools function with dozens of sequences, each of which can be hundreds of kilobase-pairs long.
3.1. Development of New Sequence Analysis Tools
The new tools are of two types. The first set provides basic, but detailed information about the aligned sequences, and the second set performs a variety of user-controlled quantitative comparisons for groups of sequences within an alignment. For these types of analyses, the quality of the starting multiple sequence alignment is very important and it must be appreciated that (1) the alignment algorithms that are optimized for large DNA sequences often do not perform well when multiple gaps must be positioned in a short region of DNA; (2) rearranged (e.g., transposed) segments of DNA cannot normally be aligned and visualized in a standard MSA; and (3) regions of DNA with differing numbers of repeats are usually arbitrarily aligned by the algorithms such that gaps may be placed differently in otherwise identical sequences [22].
In Base-By-Base, under the Reports menu, the Get Counts function reports the: (1) total number of columns (includes gaps) in a MSA; (2) number of columns that contain a gap character; (3) number of columns that contain one, two, three, or four different nucleotides; and (4) number of columns with each of the six combinations of two different nucleotides. The Get Unique Positions reports the number of SNPs in each sequence that are unique to it and not a gap, i.e., not shared by any other sequence in the MSA. It is the number of pairwise differences that relates to the length of the terminal branches of a phylogenetic tree. Under the Advanced menu, via the Advanced/Experimental tools selection, there are two new tools: The Pairwise Comparisons function generates a matrix that shows the number of nucleotide differences between each pair of sequences in the MSA; values including and excluding gapped positions are provided, which provide an appreciation of the actual divergence between the sequences that is represented by a phylogenetic tree. The Find Differences function is an interactive tool used to compare multiple, user-selected, sub-sets of the population of sequences comprising the MSA. It can identify those columns (nucleotide positions) in the MSA that satisfy the following conditions: Nucleotides must be identical in all of the sequences in group 1, different from all sequences in group 2, and, optionally, the same in (or different in) at least one sequence in other groups. Examples of its use include: (1) find SNPs common to all VACV-Dryvax genomes and absent from all other orthopoxvirus genomes in an alignment; or (2) find SNPs common to all VACV-Dryvax genomes and absent from all other VACV species, but present in at least one CPXV genome in an alignment. Importantly, the tool outputs nucleotide positions that are found as: (1) a list (the Log) of nucleotide positions that allows the user to quickly estimate the number and distribution of SNPs even while the analysis is still running; and (2) colored highlights within the Base-By-Base MSA viewer that are functionally sequence comments associated with the nucleotides; these can be manipulated as sequence comments and displayed in the Base-By-Base Visual Summary to show complete genomes in a single graphical window, which also has zoom capabilities. The development of these new sequence comparison tools was essential to this project. By adding these search features to Base-By-Base, we were able to compose a series of complex multi-genome SNP searches, each using different user-specified sub-sets of the genomes in a single large MSA, and then take advantage of the unique display features of Base-By-Base to view graphical representations of the results. Base-By-Base is Open Source software and available at www.virology.ca [16,17].
3.2. Genomic Comparison of VACV Strains
The precise origin of VACV, used as the smallpox vaccine in the 20th century, is unclear. Over 200 years have passed since Jenner used material isolated from lesions on the teats of cows to vaccinate against smallpox, since then virus stocks have been crudely passaged in both humans and animals [23,24]. The VACVs form a distinct phylogenetic clade that includes rabbitpox (a VACV with unusually high virulence in rabbits) [25] and horsepox (HSPV) [26], which may be a wild “escaped” VACV analogous to the Brazilian VACVs that currently circulate in cattle [27,28]. When Qin et al. [15] examined the variation among a series of VACV-Dryvax genomes isolated from a vial of vaccine, they found that the phylogenetic branching within the VACV-Dryvax cluster was poorly supported by bootstrap values and because they observed some SNP and deletion patterns that did not conform to the phylogenetic tree it was suggested that perhaps multiple recombination events were obscuring the true relationships between these viruses [15]. In order to focus on the differences between these VACV-Dryvax genomes, and allow comparison to other poxviruses, we aligned all sequenced VACV genomes together with the available CPXV genomes (near identical genomes were omitted) and a selection of other orthopoxvirus genomes using the MAFFT tool [18,29]. Since these genomes vary considerably in length, primarily at the genome ends, this investigation was restricted to the central core section of the alignment (VACV-WR genome nt 48,321–134,689, from gene VACV-WR-061 (virosome component) to VACV-WR-144 (RNA polymerase (RPO132)). The initial alignment was reviewed and then corrected manually using the MSA editing features of Base-By-Base; this step is critical because the MAFFT alignment parameters for complete genomes do not correctly align regions that have multiple small gaps or short repeats. For counting SNPs, we used a MSA from which any columns of nucleotides in the alignment that contained one or more gap characters had been removed.
From this primary MSA of 97,138 nt, the group of 15 VACV-Dryvax sequences was extracted from the MSA and compared to each other independent of other sequences. Within the group of VACV-Dryvax sequences there are 96,343 nucleotide positions where all DNA sequences have the same nucleotide; this leaves 795 nucleotide positions that contain SNPs, of which 793 contain only two different nucleotides, and two nucleotide positions that are occupied by three different nucleotides. Counting the total number of differences between those Dryvax isolates arranged in pairs on the tree and therefore assumed to have evolved from a recent common ancestor gave a range of 178–281 SNPs (Figure 1). Since the terminal branches of the Dryvax isolates are similar in length, it was expected that the number of unique SNPs present in each virus sequence would be approximately half of these totals. However, the number of unique SNPs present in each Dryvax sequence ranged from 8 to 34, which is 5–10-fold lower than expected based on the observed pairwise differences (179–281 SNPs). Thus, most of the SNPs that contribute to the terminal branch length are not unique, but also found in other Dryvax sequences. Although a small number of these SNPs are random coincidental matches with SNPs in the other sequences, it is clear that SNPs unique to a particular isolate account for less than 20% of the observed terminal branch lengths. When this analysis was extended to use all VACV sequences from the primary MSA, we discovered the number of unique SNPs associated with a particular Dryvax isolate dropped even further, to 0–11 per sequence (Table 1). A review of the data revealed that the majority of the SNPs previously found to be unique to a Dryvax isolate were in fact actually common to several other of the more distantly related VACV sequences. Thus, the evolutionary distance between the Dryvax isolates suggested by the length of the terminal branches of the tree appears to be greatly exaggerated.
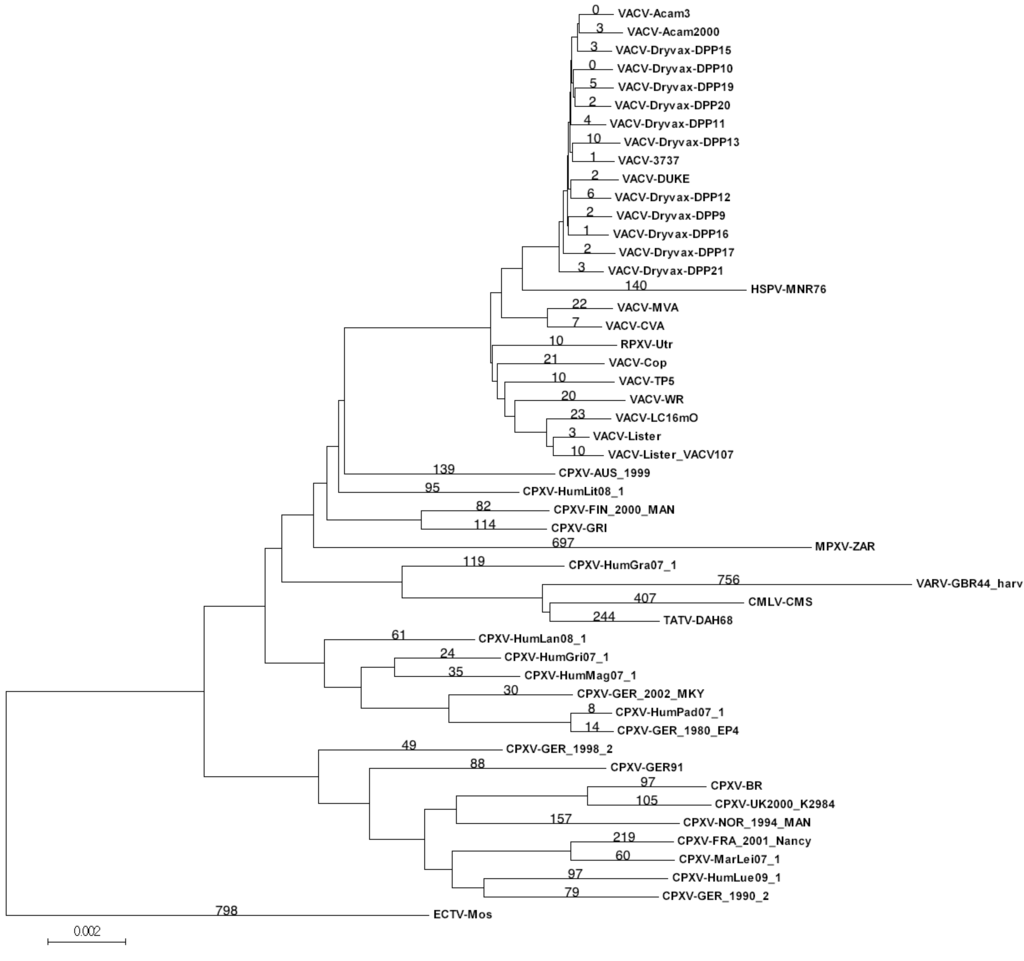
Figure 1.
Orthopoxvirus phylogenetic tree. A Neighbor-joining tree was created based on the MaximumComposite Likeihood method with 1000 bootstrap replicates. All bootstrap values were >90 except those associated with vaccinia virus (VACV). Numbers on branches indicate the number of unique SNPs associated with virus core sequence.
Figure 1.
Orthopoxvirus phylogenetic tree. A Neighbor-joining tree was created based on the MaximumComposite Likeihood method with 1000 bootstrap replicates. All bootstrap values were >90 except those associated with vaccinia virus (VACV). Numbers on branches indicate the number of unique SNPs associated with virus core sequence.
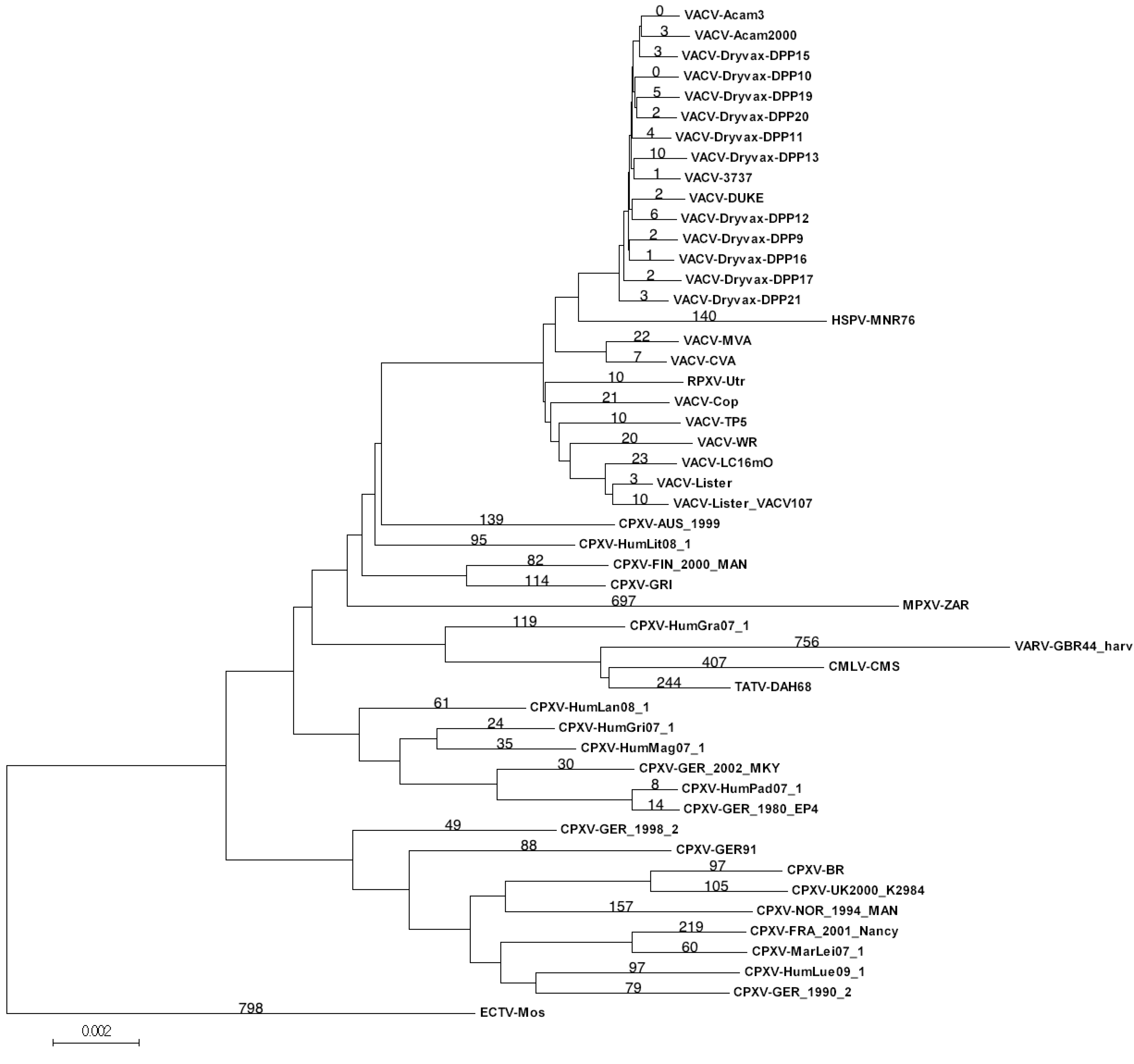

Table 1.
Number of unique single nucleotide polymorphisms (SNPs) in the core region of the VACV-Dryvax viruses calculated within the Dryvax set and full VACV set of sequences.
| Virus: VACV-Dryvax- | Unique positions: Dryvax set | Unique positions: VACV set |
|---|---|---|
| DPP9 | 17 | 2 |
| DPP10 | 16 | 1 |
| DPP11 | 8 | 4 |
| DPP12 | 17 | 6 |
| DPP13 | 29 | 11 |
| DPP15 | 14 | 5 |
| DPP16 | 12 | 1 |
| DPP17 | 23 | 4 |
| DPP19 | 22 | 7 |
| DPP20 | 8 | 2 |
| DPP21 | 27 | 3 |
| Acam3 | 14 | 0 |
| Acam2000 | 34 | 3 |
| 3737 | 29 | 4 |
| DUKE | 25 | 4 |
To provide a control phylogenetic tree, the indel-Seq-Gen software [20] was used to generate a set of artificial DNA sequences with the same phylogenetic relationships (the original tree was used as a guide) as these orthopoxvirus genome segments. In this artificial MSA, those sequences representing the Dryvax genomes were found to have 176–336 differences in the set of pairwise comparisons, similar to the values measured for the actual Dryvax sequences; however, the number of unique SNPs among the artificial sequences (66–173) was much higher than in the real Dryvax sequences (8–34). Thus, the phylogenetic tree does not appear to be an accurate reflection of the relationship between these Dryvax genomes. Since researchers view phylogenetic trees rather than count unique SNPs, we performed an experiment that is the reciprocal of the one above. A custom Python script (snip.py) was written to change each of the unique SNPs in the MSA sequences to the corresponding consensus nucleotide. Both the orthopoxvirus MSA and the counterpart with artificial sequences were modified with this script and then the phylogenetic trees were regenerated. As expected, the removal of unique SNPs from the alignment of artificial sequences is reflected in a substantial change to the structure of the tree; all of the terminal branches are drastically shortened (compare Figure 2A,B with Figure 1). However, the same branches are almost unchanged for the actual VACV-Dryvax sequences. It is noteworthy that (1) the terminal branch lengths of the other VACVs, with the exception of HSPV, also change very little after removal of the unique SNPs; (2) the terminal branch lengths of the genomes in the variola clade change dramatically; and (3) the terminal branch lengths of the CPXV genomes change an intermediate amount. Thus, for the most part, the core regions of the VARV sequences behaved normally, whereas the VACV-Dryvax sequences, which have SNPs that are shared with other non-sister isolates, behaved as though they have been jumbled by a series of recombination events. Base-By-Base was used to count the number of unique SNPs for both the orthopoxvirus sequences and the artificial sequences created to match the same orthopoxvirus phylogenetic tree. These counts revealed that the variance from the expected number of SNPs is greatest for the VACV sequences and least for the VARV sequences. Using a Chi Square statistical test, the numbers of unique SNPs in the VACV genomes were significantly less than the numbers of unique SNPs in the VACV-matched artificial genomes (p < 0.001).
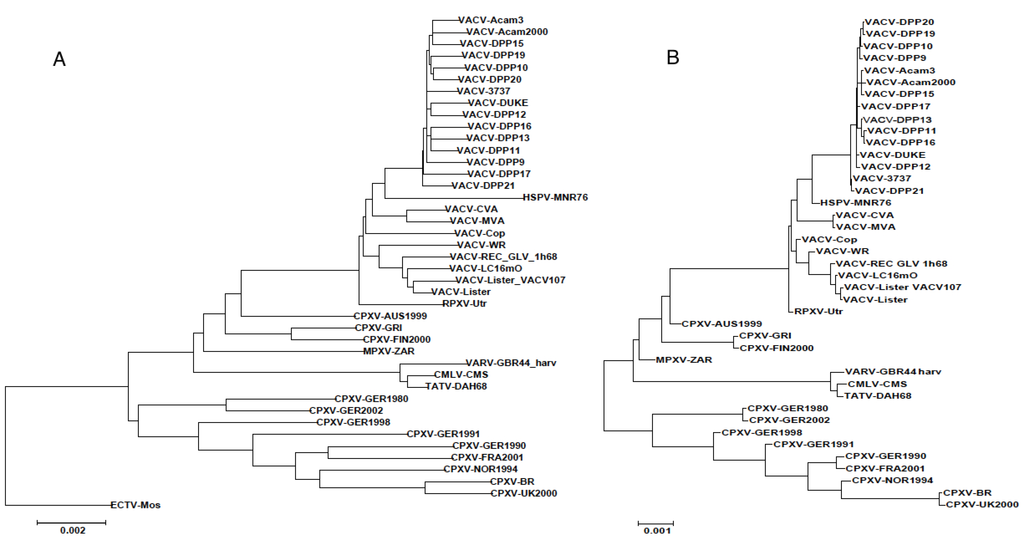
Figure 2.
Modified orthopoxvirus phylogenetic trees. Maximum-likelihood trees were created based on the Tamura-Nei method. (A) “snipped” tree; unique SNPs were removed from the multiple sequence alignment (MSA) prior to creating the tree; (B) Synthetic sequences, “snipped” tree. ECTV-Mos was not included due to limitations of the program. Scale bars refer to nucleotide substitutions per site.
Figure 2.
Modified orthopoxvirus phylogenetic trees. Maximum-likelihood trees were created based on the Tamura-Nei method. (A) “snipped” tree; unique SNPs were removed from the multiple sequence alignment (MSA) prior to creating the tree; (B) Synthetic sequences, “snipped” tree. ECTV-Mos was not included due to limitations of the program. Scale bars refer to nucleotide substitutions per site.
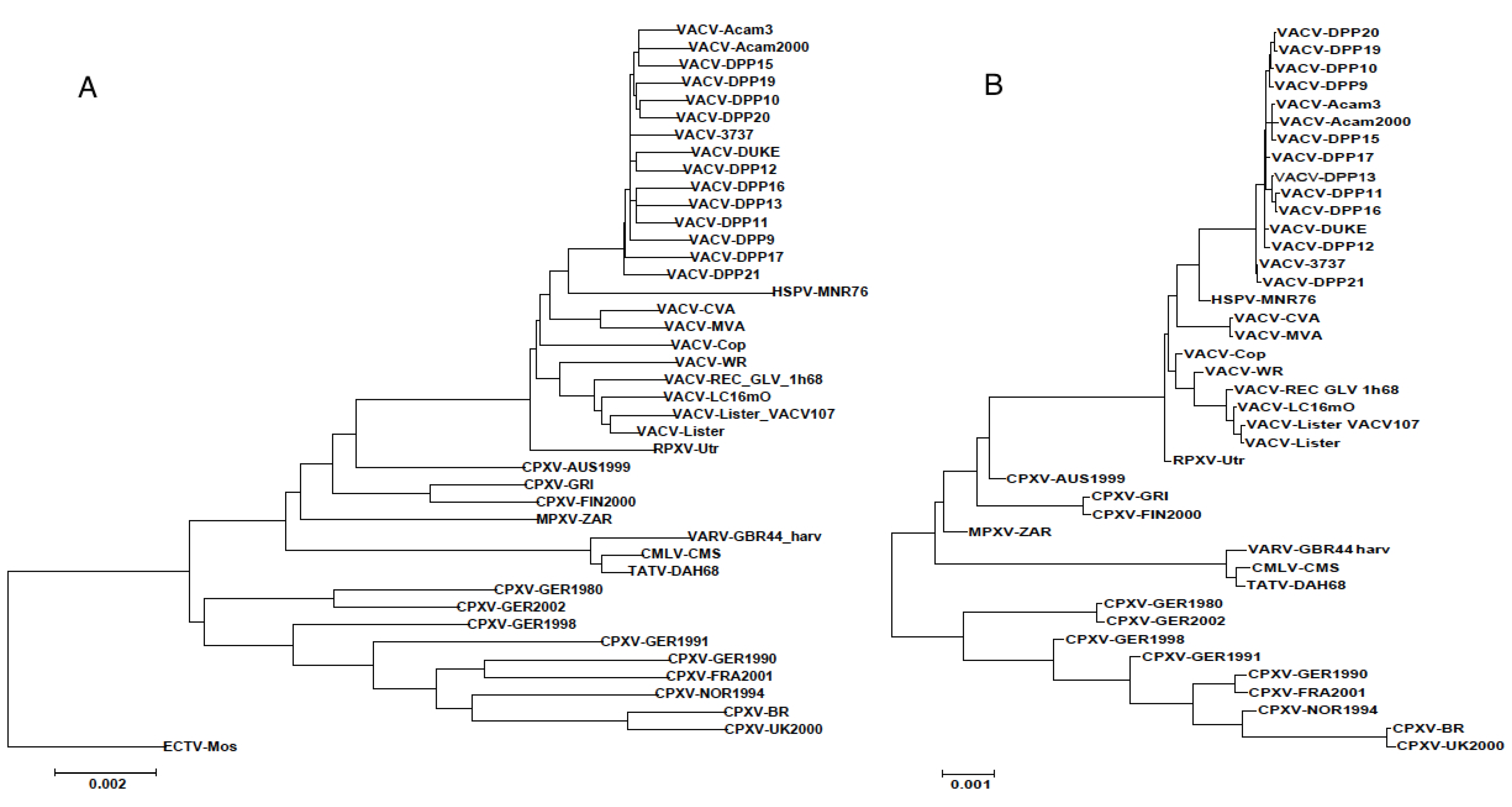
3.3. Sequence Variation in VACV Core Region
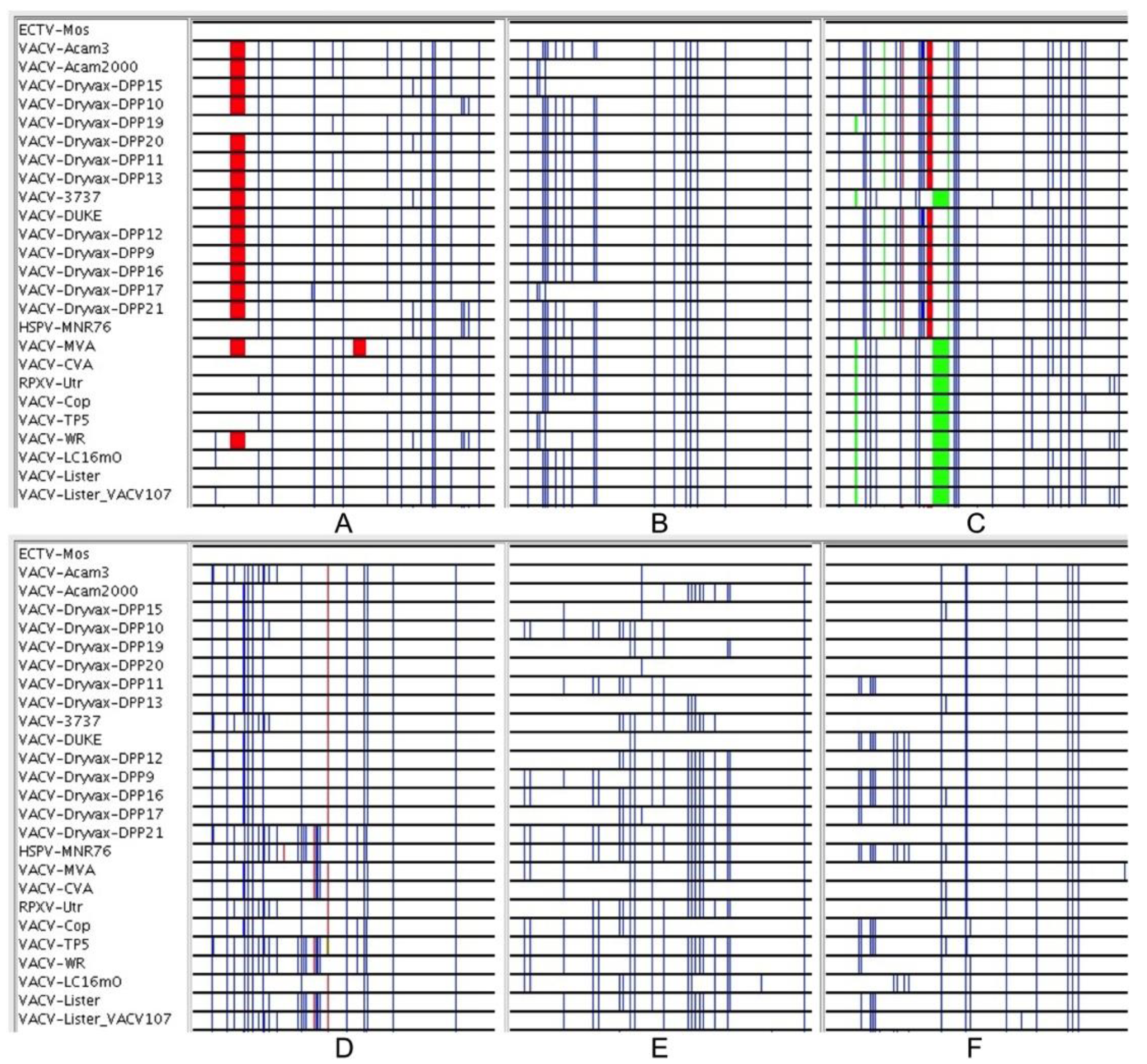
The above results confirm that the relationships portrayed by the terminal branches of orthopoxvirus phylogenetic trees are misleading. The sequencing of VACV-Dryvax isolates previously revealed some discrepancies in the phylogenetic trees; recombination was suggested as an explanation [15]. High levels of VACV recombination in in vitro co-infection experiments has also been confirmed by complete genome sequencing of progeny viruses [11] and the large blocks of sequence exchanged in these experiments were easily observable with the Visual Summary tool of Base-By-Base that displays the SNPs from genome comparisons [11]. However, since this type of large segment exchange cannot be observed in alignments of the natural VACV-Dryvax genomes, we looked at the arrangement of SNPs in these genomes at a much finer scale using the Global Zoom feature of the Base-By-Base Visual Summary tool that allows the user to distinguish SNPs patterns at the nucleotide level. Using an alignment of the core section, we arranged the order of the genomes to match that in the phylogenetic tree and visualized the differences between each of the VACV core segment and the ECTV sequence. Our goal was not to test whether every SNP supported the phylogenetic tree (the low boot-strap values indicate they do not), but to examine the patterns of the SNPs and to search for consistency in the size and arrangement of the blocks of SNPs that are incongruent to the phylogenetic tree. Figure 3 displays six 350 bp segments sampled from the alignment; these panels contain some SNPs that: (1) are unique to a particular VACV sequence or the distantly related ECTV that do not contribute information to the phylogeny; (2) are fully supportive of the tree; or (3) are arranged such that there is only partial support for the tree–that is some sequences group to support the tree but there are others that do not. Many of the SNPs that appear to be supporting the tree are simply SNPs that are unique to ECTV (group 2); interestingly, most of the other SNPs fall into group 3 (do not support tree). Furthermore, these data sets indicate that most of the blocks of sequences (different from ECTV), which do not match the tree, are very small (10–100 bp) and contain only a few SNPs; however, the limitation of defining these regions is the frequency of the SNPs in the sequences. In reviewing the blocks of SNPs in the 97 kb core alignment, we found that there was no consistency to the arrangements of the sequences; thus, the short sequences represented in the panels of Figure 3, would each support a different phylogenetic tree.
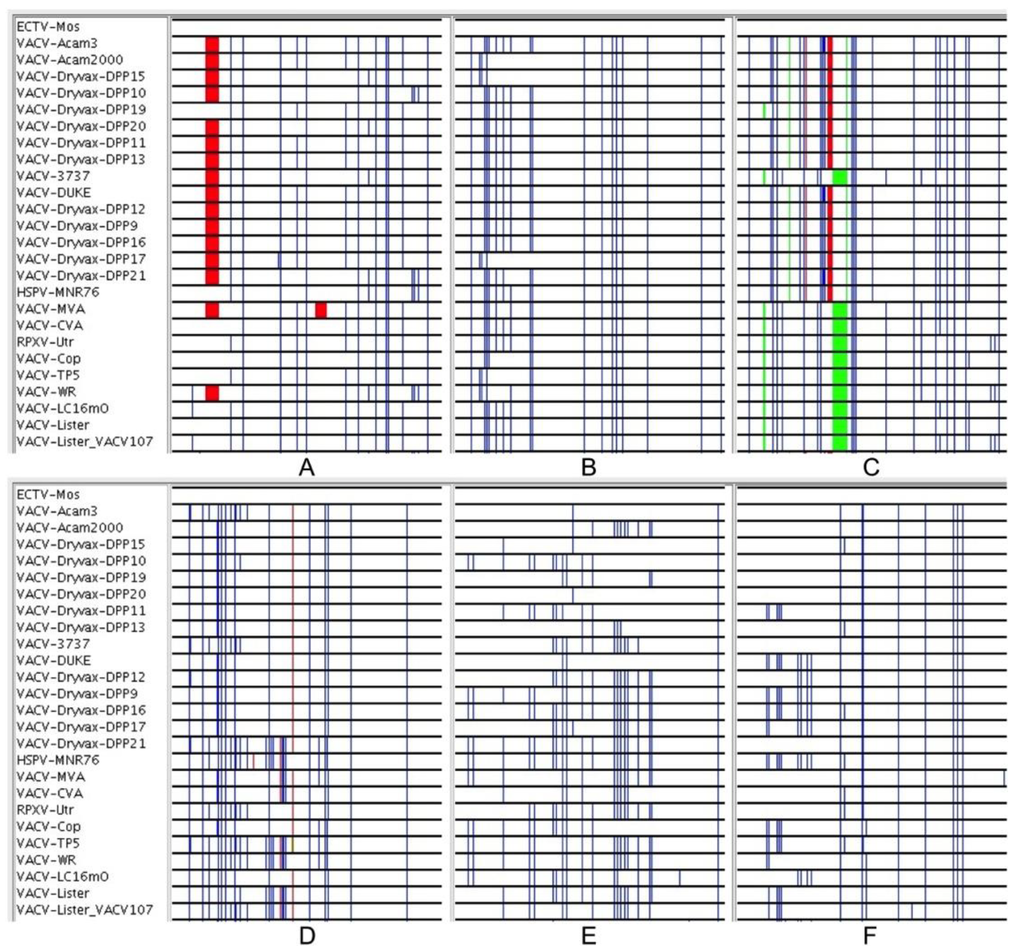
Figure 3.
(A–F): 350 nt segments of the core alignment (before gapped columns were removed) viewed with the Base-By-Base Visual Summary view. VACV sequences are all compared to ectromelia virus: Blue lines show SNPs; red and green show deletions and insertions, respectively.
Figure 3.
(A–F): 350 nt segments of the core alignment (before gapped columns were removed) viewed with the Base-By-Base Visual Summary view. VACV sequences are all compared to ectromelia virus: Blue lines show SNPs; red and green show deletions and insertions, respectively.
The Find Differences tool of Base-By-Base not only counts SNPs but also offers the opportunity to examine the arrangement of SNPs in MSAs, both along the physical length of sequences and their presence in individual and groups of sequences. These analyses also suggest extensive recombination has taken place between the VACVs. For example, Base-By-Base reports that there are 32 SNPs that are present in all VACVs and absent from all other orthopoxviruses in this alignment; however, 29/32 exist in the 3' half of the sequence, with 17/32 present in a single 10 kb region. Since it is reasonable to assume that the VACVs do not acquire mutations 10-fold more frequently in one half of the MSA than the other, the explanation must lie with either that: (1) the SNPs are not present in 100% of the VACV sequences; or (2) the SNPs are also shared by some of the non-VACV sequences in the alignment. To test this type of scenario, the Find Differences tool in Base-By-Base features a mechanism to permit any of the groups of sequences in a query to be less than a 100% match (i.e., Tolerance of mismatches). For example, Find SNPs in all sequences A, B, C and D (with tolerance 0%) and not in sequences E, F, G and H (with tolerance 25%) would hit nucleotides identical in sequences A–D and different in three or four of sequences E–H. However, it is also important to be aware of the limitations of this kind of analysis, which is affected by the number of sequences in the groups being compared as well as their relatedness; matching five closely related sequences from a single clade is very different to matching five sequences that are distributed among several clades that also contain non-matching sequences. Similarly, one needs to be aware which nucleotide is ancestral and which is the SNP when comparing large groups of sequences or allowing large tolerance values. For the above search requesting all VACVs to be the same and all others to be different, adding a Tolerance = 10 to the latter increases the number of SNPs matched to 290 although most do not match the maximum of 10 other sequences. Many of these SNPs are random mutations in the non-VACV sequences (terminal sequences or ancestors of particular clades). However, importantly, there are also several examples where the SNPs associated with a particular group of sequences are only found together in a small block of sequence, and importantly, these non-VACV sequences are not all associated with a single clade of viruses. Some of these patterns, which are the hallmark of recombination events, are shown in Table 2. Even the patterns composed of only two closely linked SNPs support a history of recombination because SNPs with the same particular set of “tolerated” sequences are not found anywhere else in the MSA.
Table 2.
Positions of SNPs where all VACVs are identical and all other sequences are different, with Tolerance of 10.
| Position | Tolerated Virus Set |
|---|---|
| 5685 | CPXV-AUS_1999, CPXV-HumLit08, CPXV-FIN_2000_MAN, CPXV-GRI, MPXV-ZAR |
| 5759 | CPXV-AUS_1999, CPXV-HumLit08, CPXV-FIN_2000_MAN, CPXV-GRI, MPXV-ZAR |
| 5789 | CPXV-AUS_1999, CPXV-HumLit08, CPXV-FIN_2000_MAN, CPXV-GRI, MPXV-ZAR |
| 5807 | CPXV-AUS_1999, CPXV-HumLit08, CPXV-FIN_2000_MAN, CPXV-GRI, MPXV-ZAR |
| 5820 | CPXV-AUS_1999, CPXV-HumLit08, CPXV-FIN_2000_MAN, CPXV-GRI, MPXV-ZAR |
| 5839 | CPXV-AUS_1999, CPXV-HumLit08, CPXV-FIN_2000_MAN, CPXV-GRI, MPXV-ZAR |
| 13452 | CPXV-HumLit08_1, VARV-GBR44_harv, CMLV-CMS, TATV-DAH68, CPXV-HumPad07_1, CPXV-GER_1980_EP4 |
| 13513 | CPXV-HumLit08_1, VARV-GBR44_harv, CMLV-CMS, TATV-DAH68, CPXV-HumPad07_1, CPXV-GER_1980_EP4 |
| 13514 | CPXV-HumLit08_1, VARV-GBR44_harv, CMLV-CMS, TATV-DAH68, CPXV-HumPad07_1, CPXV-GER_1980_EP4 |
| 13516 | CPXV-HumLit08_1, VARV-GBR44_harv, CMLV-CMS, TATV-DAH68, CPXV-HumPad07_1, CPXV-GER_1980_EP4 |
| 13554 | CPXV-HumLit08_1, VARV-GBR44_harv, CMLV-CMS, TATV-DAH68, CPXV-HumPad07_1, CPXV-GER_1980_EP4 |
| 19369 | CPXV-AUS_1999, CPXV-HumLit08_1, CPXV-FIN_2000_MAN, CPXV-GRI, MPXV-ZAR, CPXV-HumLan08_1 |
| 19378 | CPXV-AUS_1999, CPXV-HumLit08_1, CPXV-FIN_2000_MAN, CPXV-GRI, MPXV-ZAR, CPXV-HumLan08_1 |
| 19380 | CPXV-AUS_1999, CPXV-HumLit08_1, CPXV-FIN_2000_MAN, CPXV-GRI, MPXV-ZAR, CPXV-HumLan08_1 |
| 19387 | CPXV-AUS_1999, CPXV-HumLit08_1, CPXV-FIN_2000_MAN, CPXV-GRI, MPXV-ZAR, CPXV-HumLan08_1 |
| 59334 | CPXV-AUS_1999, CPXV-HumLit08_1, CPXV-FIN_2000_MAN, CPXV-GRI |
| 59343 | CPXV-AUS_1999, CPXV-HumLit08_1, CPXV-FIN_2000_MAN, CPXV-GRI |
| 59444 | CPXV-AUS_1999, CPXV-HumLit08_1, CPXV-FIN_2000_MAN, CPXV-GRI |
The three VACV-Lister sequences in this MSA form a well-defined clade and there are 22 SNPs that are common to these three sequences and absent from all the other sequences. When the tolerance level for the “different sequences” was relaxed to 10, we again saw several blocks of tightly linked additional SNPs (Table 3) that were shared by the Lister strains and other “more distantly related” viruses.
Table 3.
Positions of SNPs where the three VACV-Lister strains are identical and all other sequences are different, with Tolerance of 10.
| Position | Tolerated Virus Set |
|---|---|
| 61423 | VACV-Dryvax-DPP19, HSPV-MNR76, RPXV-Utr, VACV-Cop, VACV-TP5, VACV-WR, CPXV-AUS_1999 |
| 61426 | VACV-Dryvax-DPP19, HSPV-MNR76, RPXV-Utr, VACV-Cop, VACV-TP5, VACV-WR, CPXV-AUS_1999 |
| 61429 | VACV-Dryvax-DPP19, HSPV-MNR76, RPXV-Utr, VACV-Cop, VACV-TP5, VACV-WR, CPXV-AUS_1999 |
| 61435 | VACV-Dryvax-DPP19, HSPV-MNR76, RPXV-Utr, VACV-Cop, VACV-TP5, VACV-WR, CPXV-AUS_1999 |
| 61436 | VACV-Dryvax-DPP19, HSPV-MNR76, RPXV-Utr, VACV-Cop, VACV-TP5, VACV-WR, CPXV-AUS_1999 |
| 61438 | VACV-Dryvax-DPP19, HSPV-MNR76, RPXV-Utr, VACV-Cop, VACV-TP5, VACV-WR, CPXV-AUS_1999 |
| 87456 | VACV-Acam2000, -MVA, -CVA, -Cop, -WR, -TP5, CPXV-HumMag07_1, -HumGri07_1, -HumLan08_1, -FIN_2000_MAN |
| 87459 | VACV-Acam2000, -MVA, -CVA, -Cop, -WR, -TP5, CPXV-HumMag07_1, -HumGri07_1, -HumLan08_1, -FIN_2000_MAN |
| 87463 | VACV-Acam2000, -MVA, -CVA, -Cop, -WR, -TP5, CPXV-HumMag07_1, -HumGri07_1, -HumLan08_1, -FIN_2000_MAN |
| 87465 | VACV-Acam2000, -MVA, -CVA, -Cop, -WR, -TP5, CPXV-HumMag07_1, -HumGri07_1, -HumLan08_1, -FIN_2000_MAN |
| 66854 | VACV-Cop, CPXV-HumLit08_1, CPXV-HumPad07_1, CPXV-BR, CPXV-UK2000_K2984 |
| 66856 | VACV-Cop, CPXV-HumLit08_1, CPXV-HumPad07_1, CPXV-BR, CPXV-UK2000_K2984 |
3.4. Core Region SNPs Shared by the Variola Clade and More Distantly Related Viruses
It has previously noted that three (VARV, CMLV and TATV) of the viruses in the variola clade share some sets of SNPs with surprisingly distantly related viruses, suggesting ancestral recombination events [30]. More recent genome sequencing has added CPXV-like viruses to this clade (CPXV-HumGra07_1 in Figure 1); in the core MSA, there are 66 SNPs that are common to these 4 viruses but absent from all other sequences in the alignment. However, in the Base-By-Base search for such SNPs with a tolerance of 1 in the set of non-matching sequences, there were several distinct viruses that shared independent blocks of SNPs with the VARV clade (Table 4). For example: (1) the CPXV-GER91 sequence shares 10 otherwise unique SNPs with viruses in the VARV clade and nine of these are clustered within a 76 nt region; and (2) MPXV-ZAR shares nine SNPs with this group and seven are within a 52 nt region (resulting in a 2 aa change to a intracellular mature virus surface protein). Since this alignment contains a series of different orthopoxvirus clades, this search was rerun allowing a tolerance of 10, meaning that up to 10 viruses from the “different from the VARV clade” could in fact match these four sequences. Table 5 shows several blocks of SNPs that are shared by the VARV clade viruses and small groups (4–8) of other viruses. The key point is that in each of these examples, the groups are made up of different sets of viruses and that they do not constitute single or complete clades.
Table 4.
Positions where all four viruses of the variola virus (VARV) clade are the same and all others are different, except the single virus shown.
| CPXV-GER91 | MPXV-ZAR | CPXV-HumLit08_1 | CPXV-HumLue09_1 |
|---|---|---|---|
| 21074 | 52718 | 21693 | 95965 |
| 21092 | 52745 | 21702 | 96055 |
| 21093 | 52747 | 21705 | 96061 |
| 21116 | 52756 | 45848 | - |
| 21125 | 52760 | - | - |
| 21128 | 52766 | - | - |
| 21140 | 52769 | - | - |
| 21141 | 60513 | - | - |
| 21149 | 66305 | - | - |
| 23126 | - | - | - |
Table 5.
Positions where all four viruses of the VARV clade are the same and all others are different, except the set of viruses shown.
| Position | Tolerated Virus Set |
|---|---|
| 5006 | CPXV-HumGri07_1, CPXV-HumMag07_1, CPXV-GER_2002_MKY, CPXV-HumPad07_1, CPXV-GER_1980_EP4 |
| 5011 | CPXV-HumGri07_1, CPXV-HumMag07_1, CPXV-GER_2002_MKY, CPXV-HumPad07_1, CPXV-GER_1980_EP4 |
| 5035 | CPXV-HumGri07_1, CPXV-HumMag07_1, CPXV-GER_2002_MKY, CPXV-HumPad07_1, CPXV-GER_1980_EP4 |
| 5088 | CPXV-HumGri07_1, CPXV-HumMag07_1, CPXV-GER_2002_MKY, CPXV-HumPad07_1, CPXV-GER_1980_EP4 |
| 50919 | CPXV-HumGri07_1, CPXV-HumMag07_1, CPXV-GER_2002_MKY, CPXV-HumPad07_1, CPXV-GER_1980_EP4 |
| 21528 | CPXV-AUS_1999, CPXV-HumLit08_1, CPXV-HumPad07_1, CPXV-GER_1980_EP4, CPXV-MarLei07_1 |
| 21535 | CPXV-AUS_1999, CPXV-HumLit08_1, CPXV-HumPad07_1, CPXV-GER_1980_EP4, CPXV-MarLei07_1 |
| 21539 | CPXV-AUS_1999, CPXV-HumLit08_1, CPXV-HumPad07_1, CPXV-GER_1980_EP4, CPXV-MarLei07_1 |
| 21540 | CPXV-AUS_1999, CPXV-HumLit08_1, CPXV-HumPad07_1, CPXV-GER_1980_EP4, CPXV-MarLei07_1 |
| 21541 | CPXV-AUS_1999, CPXV-HumLit08_1, CPXV-HumPad07_1, CPXV-GER_1980_EP4, CPXV-MarLei07_1 |
| 21543 | CPXV-AUS_1999, CPXV-HumLit08_1, CPXV-HumPad07_1, CPXV-GER_1980_EP4, CPXV-MarLei07_1 |
| 21579 | CPXV-HumLit08_1, CPXV-HumPad07_1, CPXV-GER_1980_EP4, CPXV-MarLei07_1 |
| 21582 | CPXV-HumLit08_1, CPXV-HumPad07_1, CPXV-GER_1980_EP4, CPXV-MarLei07_1 |
| 21585 | CPXV-HumLit08_1, CPXV-HumPad07_1, CPXV-GER_1980_EP4, CPXV-MarLei07_1 |
| 21587 | CPXV-HumLit08_1, CPXV-HumPad07_1, CPXV-GER_1980_EP4, CPXV-MarLei07_1 |
| 21645 | CPXV-HumLit08_1, CPXV-HumPad07_1, CPXV-GER_1980_EP4, CPXV-MarLei07_1 |
| 21648 | CPXV-HumLit08_1, CPXV-HumPad07_1, CPXV-GER_1980_EP4, CPXV-MarLei07_1 |
| 98836 | HSPV-76, VACV-WR, CPXV-AUS_1999, CPXV-HumLit08_1, MPXV-ZAR, CPXV-FIN_2000_MAN, CPXV-GRI, CPXV-GER91 |
| 98839 | HSPV-76, VACV-WR, CPXV-AUS_1999, CPXV-HumLit08_1, MPXV-ZAR, CPXV-FIN_2000_MAN, CPXV-GRI, CPXV-GER91 |
| 98845 | HSPV-76, VACV-WR, CPXV-AUS_1999, CPXV-HumLit08_1, MPXV-ZAR, CPXV-FIN_2000_MAN, CPXV-GRI, CPXV-GER91 |
3.5. SNPs Associated with the Evolution of VACV-CVA and –MVA
One of the best established VACV lineages is the direct derivation of VACV-MVA from VACV-CVA by passaging the virus in CEF cells more than 570 times [31]; therefore we expected the patterns of SNPs in these sequences to display a simple evolutionary relationship. However, simple counting of unique SNPs revealed this to be untrue. Within the VACVs of the test MSA, Base-By-Base counted 15 SNPs common to VACV-CVA and -MVA, but different to all other sequences, and 36 SNPs unique to VACV-MVA. However, we were surprised to find 19 SNPs unique to VACV-CVA. These may have arisen during subsequent passaging of the VACV-CVA or reversion in VACV-MVA of SNPs common to VACV-CVA and -MVA; however, one would expect these events to be extremely rare considering only 36 SNPs unique to VACV-MVA were detected. Again, in a query to find SNPs unique to VACV-MVA, when the tolerance for the sequences to be different from VACV-MVA was relaxed (to 7), a large number of short blocks of different sets of sequences were observed (data not shown). The total number of SNPs counted was 350, this cannot be from random SNPs in VACV-MVA because 90% of the nucleotides in these sequences are 100% conserved and we only see 15 of the 350 SNPs in VACV-MVA hitting this 90% of the sequence. Interestingly, when this query was repeated, but it specified that VACV-CVA must be different from VACV-MVA and all other sequences, it still found 140 SNPs. Finally, when we looked at the positions of the SNPs unique to VACV-CVA, VACV-MVA, HSPV and all other VACV-Dryvax sequences, i.e., those SNPs that cause VACV-CVA and VACV-MVA to cluster with the Dryvax clade, only 10 were detected and these found in three very small regions (five in 122 nt, two in 5 nt and three in 72 nt) that were restricted to a <5 kb region towards the right end of the MSA.
4. Conclusions
Despite the fact that many phylogenetic trees have been drawn for the VACVs and other orthopoxviruses in the past, the detailed examination of the sequences presented here suggests that these genomes are, in fact, poorly suited for this type of analysis since it is assumed that the set of sequences evolved from a common ancestor without recombination events. In the same way that multiple alignment software will always give some kind of alignment, even if the sequences have translocations and inversions and are thus impossible to align in a meaningful way, phylogenetic analyses will always produce some kind of tree. The new software tools, described here and incorporated into Base-By-Base, have shown that the VACV sequences used here have considerably fewer positions that are unique to individual strains than predicted by their relationships to one another in the phylogenetic trees. Within the genomes, blocks of SNPs also confirm that relationships between the Dryvax isolates do not match the phylogenetic tree topology. These detailed comparisons of the VACV SNPs support previous suggestions of recombination and indicate that so many recombination events have taken place that the evolutionary signatures of SNP patterns from related sets of sequences are now very short. Given the long history of VACVs with their unnatural production in artificially infected animals and little if any attempts to purify stocks, it is easy to picture scenarios with high multiplicities of infection (MOI) that afford the opportunity for recombination. However, our comparison of the VACV sequences and the VARV clade sequences to other viruses, especially CPXVs, also revealed patterns of SNPs that indicate a history of ancient recombination events based on the sequences involved.
Understanding the origin and evolution of poxviruses is not only a matter of academic interest, it is relevant today because we need to understand the stability of virus-based therapeutics, which like VACV-Dryvax may also be grown at high MOIs. Similarly, genome comparisons that merely provide a measure of sequence identity or report the presence or absence of orthologs underestimate the complexity of the variation and similarities that exists between genomes. For example: (1) different regions of poxvirus genomes may be more or less conserved than others; and (2) different regions of a virus may be most closely related to different viruses (even a different species). When virulence studies compare genes and proteins, the relevance of small patterns may not be appreciated unless the origin of sequences is followed. Thus, differences such as the two amino acids shared between the IMV surface protein (VACV-Copenhagen H3L) from the VARV clade and MPXV might go unnoticed except that these changes result from the block of two nucleotides (within a span of 52 nt) unique to these two groups of viruses (Table 4). Because this region of unique identity between the MPX and VAR viruses is small, it does not outweigh the “normal” evolutionary signals in these genes and therefore these unexpected matches are not picked up in standard similarity searches by BLAST [32].
Finally, users of poxvirus phylogenetic trees should remember (1) that these represent a summary of the relationships between the various input sequences, which themselves may be small or large, and that due to past recombination events this phylogenetic summary may not accurately portray these relationships in many regions of the sequences; (2) that the fraction of the total SNPs contributing to a valid tree may be small; and (3) that the number of recombination events appears to be very large such that the resulting regions of micro-heterogeneity are not readily detected by eye or by recombination detection programs.
Acknowledgments
The authors thank the University of Victoria Computer Science Co-op students who have helped develop the Virology.ca bioinformatics tools, and especially Jeremy Ho for writing the python scripts, and Alex Purdy and Tim Van der Wekken for modifications to Base-By-Base. Funded by a Natural Sciences and Engineering Research Council Discovery Grant to Chris Upton.
Author Contributions
Chad Smithson, Samantha Kampman and Benjamin M. Hetman all contributed equally to the analyses and writing of the paper. Chris Upton conceived the project and helped write the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Upton, C.; Slack, S.; Hunter, A.L.; Ehlers, A.; Roper, R.L. Poxvirus orthologous clusters: Toward defining the minimum essential poxvirus genome. J. Virol. 2003, 77, 7590–7600. [Google Scholar] [CrossRef] [PubMed]
- Gubser, C.; Hué, S.; Kellam, P.; Smith, G.L. Poxvirus genomes: A phylogenetic analysis. J. Gen. Virol. 2004, 85, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Baroudy, B.M.; Venkatesan, S.; Moss, B. Incompletely base-paired flip-flop terminal loops link the two DNA strands of the vaccinia virus genome into one uninterrupted polynucleotide chain. Cell 1982, 28, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Parrino, J.; Graham, B.S. Smallpox vaccines: Past, present, and future. J. Allergy Clin. Immunol. 2006, 118, 1320–1326. [Google Scholar] [CrossRef] [PubMed]
- Verardi, P.H.; Titong, A.; Hagen, C.J. A vaccinia virus renaissance: New vaccine and immunotherapeutic uses after smallpox eradication. Hum. Vaccines Immunother. 2012, 8, 961–970. [Google Scholar] [CrossRef]
- Rosenthal, S.R.; Merchlinsky, M.; Kleppinger, C.; Goldenthal, K.L. Developing new smallpox vaccines. Emerg. Infect. Dis. 2001, 7, 920–926. [Google Scholar] [CrossRef] [PubMed]
- Carroll, D.S.; Emerson, G.L.; Li, Y.; Sammons, S.; Olson, V.; Frace, M.; Nakazawa, Y.; Czerny, C.P.; Tryland, M.; Kolodziejek, J.; et al. Chasing Jenner’s vaccine: Revisiting cowpox virus classification. PLoS One 2011, 6, e23086. [Google Scholar] [CrossRef] [PubMed]
- Dabrowski, P.W.; Radonić, A.; Kurth, A.; Nitsche, A. Genome-wide comparison of cowpox viruses reveals a new clade related to variola virus. PLoS One 2013, 8, e79953. [Google Scholar] [CrossRef] [PubMed]
- Hughes, A.L.; Irausquin, S.; Friedman, R. The evolutionary biology of poxviruses. Infect. Genet. Evol. 2010, 10, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Coulson, D.; Upton, C. Characterization of indels in poxvirus genomes. Virus Genes 2011, 42, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Evans, D.H. Genome scale patterns of recombination between co-infecting vaccinia viruses. J. Virol. 2014, 88, 5277–5286. [Google Scholar] [CrossRef] [PubMed]
- Bratke, K.A.; McLysaght, A. Identification of multiple independent horizontal gene transfers into poxviruses using a comparative genomics approach. BMC Evol. Biol. 2008, 8, 67. [Google Scholar] [CrossRef] [PubMed]
- Posada, D.; Crandall, K.A.; Holmes, E.C. Recombination in evolutionary genomics. Annu. Rev. Genet. 2003, 36, 75–97. [Google Scholar] [CrossRef]
- Xing, K.; Deng, R.; Wang, J.; Feng, J.; Huang, M.; Wang, X. Genome-based phylogeny of poxvirus. Intervirology 2006, 49, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Upton, C.; Hazes, B.; Evans, D.H. Genomic analysis of the vaccinia virus strain variants found in Dryvax vaccine. J. Virol. 2011, 85, 13049–13060. [Google Scholar] [CrossRef] [PubMed]
- Brodie, R.; Smith, A.J.; Roper, R.L.; Tcherepanov, V.; Upton, C. Base-By-Base: Single nucleotide-level analysis of whole viral genome alignments. BMC Bioinform. 2004, 5, 96. [Google Scholar] [CrossRef]
- Hillary, W.; Lin, S.-H.; Upton, C. Base-By-Base version 2: Single nucleotide-level analysis of whole viral genome alignments. Microb. Inform. Exp. 2011, 1, 2. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Asimenos, G.; Toh, H. Multiple alignment of DNA sequences with MAFFT. Methods Mol. Biol. 2009, 537, 39–64. [Google Scholar] [PubMed]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef] [PubMed]
- Strope, C.L.; Abel, K.; Scott, S.D.; Moriyama, E.N. Biological sequence simulation for testing complex evolutionary hypotheses: Indel-Seq-Gen version 2.0. Mol. Biol. Evol. 2009, 26, 2581–2593. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, M.; Kishino, H.; Yano, T. Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J. Mol. Evol. 1985, 22, 160–174. [Google Scholar] [CrossRef] [PubMed]
- Blair, C.; Murphy, R.W. Recent trends in molecular phylogenetic analysis: Where to next? J. Hered. 2011, 102, 130–138. [Google Scholar] [CrossRef]
- Paran, N.; Sutter, G. Smallpox vaccines: New formulations and revised strategies for vaccination. Hum. Vaccin. 2009, 5, 824–831. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, B.L.; Langland, J.O.; Kibler, K.V.; Denzler, K.L.; White, S.D.; Holechek, S.A.; Wong, S.; Huynh, T.; Baskin, C.R. Vaccinia virus vaccines: Past, present and future. Antivir. Res. 2009, 84, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Chen, N.; Roper, R.L.; Feng, Z.; Hunter, A.; Danila, M.I.; Lefkowitz, E.J.; Buller, R.M.L.; Upton, C. Complete coding sequences of the rabbitpox virus genome. J. Gen. Virol. 2005, 86, 2969–2977. [Google Scholar] [CrossRef] [PubMed]
- Tulman, E.R.; Delhon, G.; Afonso, C.L.; Lu, Z.; Zsak, L.; Sandybaev, N.T.; Kerembekova, U.Z.; Zaitsev, V.L.; Kutish, G.F.; Rock, D.L. Genome of horsepox virus. J. Virol. 2006, 80, 9244–9258. [Google Scholar] [CrossRef] [PubMed]
- Trindade, G.S.; Emerson, G.L.; Carroll, D.S.; Kroon, E.G.; Damon, I.K. Brazilian vaccinia viruses and their origins. Emerg. Infect. Dis. 2007, 13, 965–972. [Google Scholar] [CrossRef] [PubMed]
- Da Fonseca, F.G.; Trindade, G.S.; Silva, R.L.A.; Bonjardim, C.A.; Ferreira, P.C.P.; Kroon, E.G. Characterization of a vaccinia-like virus isolated in a Brazilian forest. J. Gen. Virol. 2002, 83, 223–228. [Google Scholar] [PubMed]
- Katoh, K.; Toh, H. Recent developments in the MAFFT multiple sequence alignment program. Brief. Bioinform. 2008, 9, 286–298. [Google Scholar] [CrossRef] [PubMed]
- Smithson, C.; Purdy, A.; Verster, A.J.; Upton, C. Prediction of Steps in the Evolution of Variola Virus Host Range. PLoS One 2014, 9, e91520. [Google Scholar] [CrossRef] [PubMed]
- Antoine, G.; Scheiflinger, F.; Dorner, F.; Falkner, F.G. The complete genomic sequence of the modified vaccinia Ankara strain: Comparison with other orthopoxviruses. Biochemistry 1998, 244, 365–396. [Google Scholar]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).