

CrystalShift: A Versatile Command-Line Tool for Crystallographic Structural Data Analysis, Modification, and Format Conversion Prior to Solid-State DFT Calculations of Organic Crystals


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
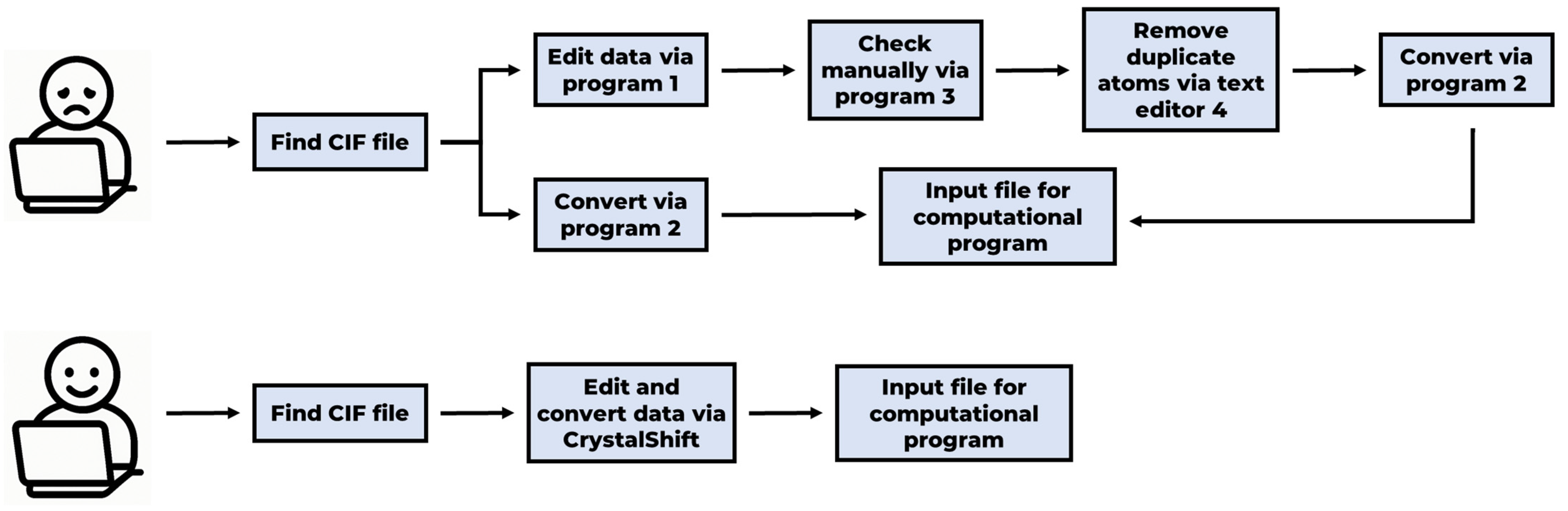
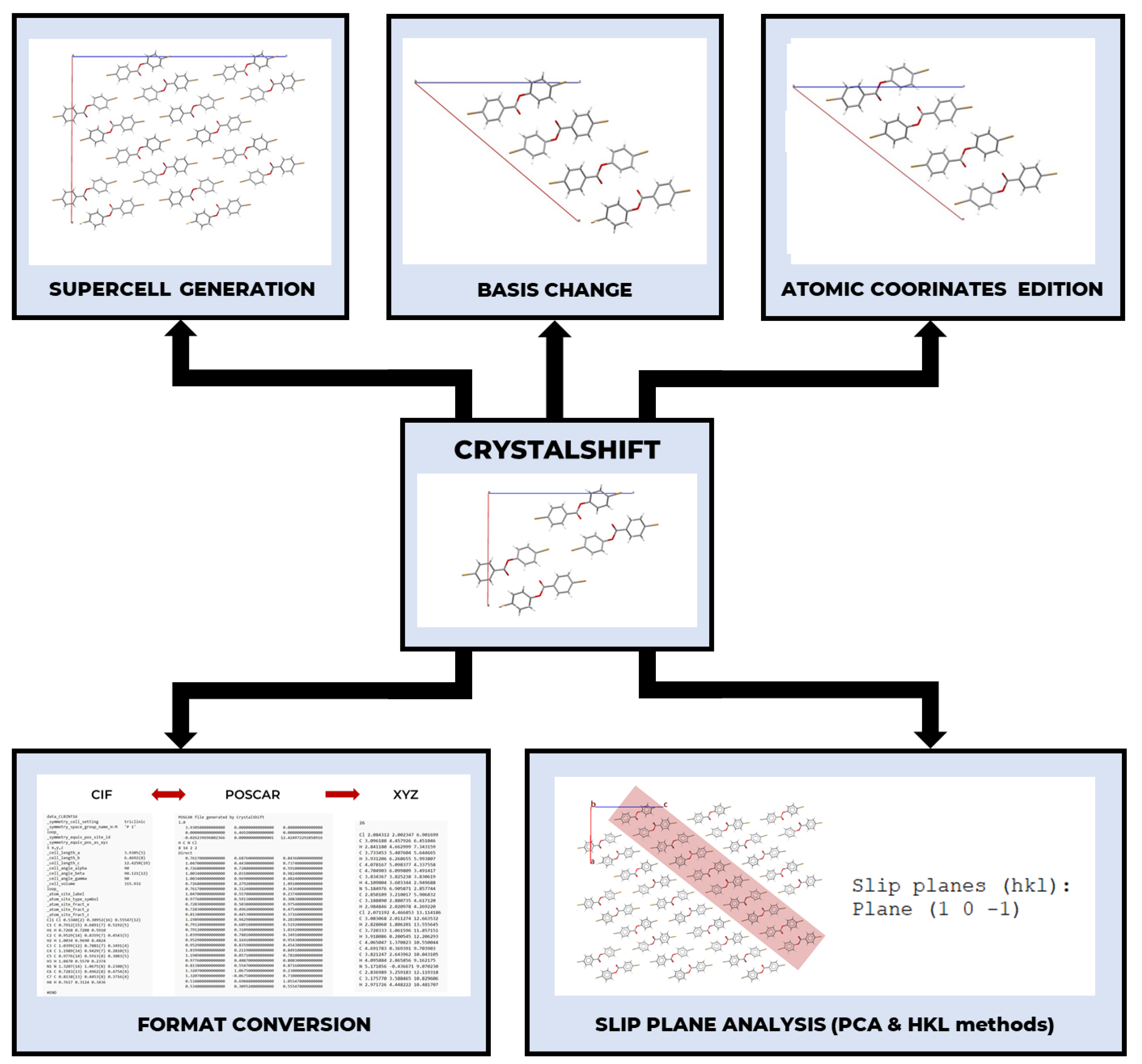
2. Software
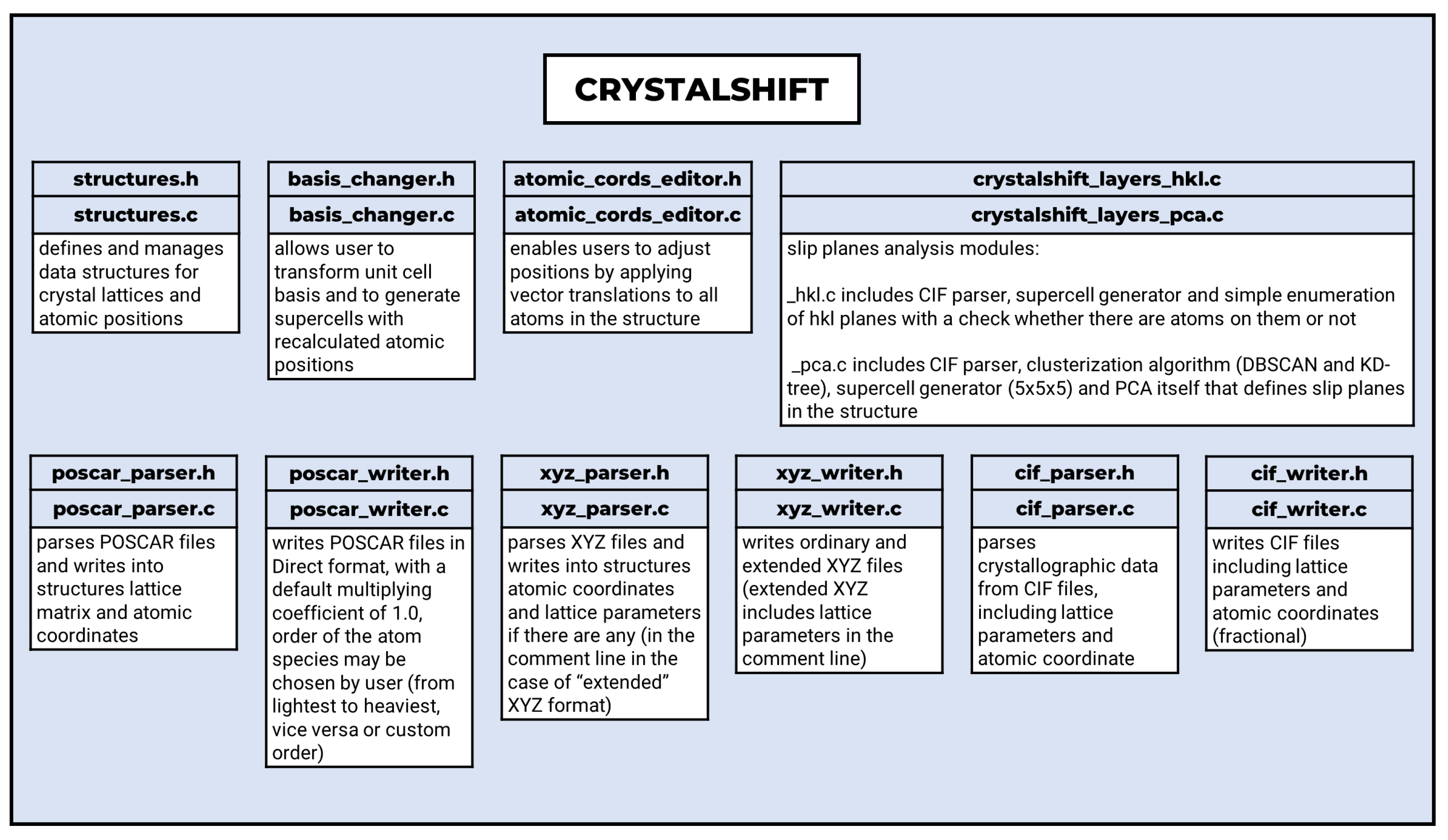
2.1. Software Design and Architecture
2.2. Input and Output Formats (Converter)
- CIF (Crystallographic Information File) is the most widely used format for crystallographic data. CIF files are parsed to extract lattice parameters, atomic coordinates, and additional data. Experimental error data and non-crystallographic information are stripped for clarity during editing. When writing CIF files, CrystalShift simplifies structures by assuming a triclinic lattice with space group P1, avoiding complications from symmetry operations.
- The POSCAR file format is specific to the Vienna Ab initio Simulation Package (VASP) [32,33,34,35], which is widely used for computational materials science. It represents lattice parameters in the form of a matrix and atomic coordinates. This format is extensively used in Density Functional Theory (DFT) calculations and other atomistic simulations. The output from CrystalShift POSCAR files is written in fractional (direct) coordinates, with support for reordering atomic species based on user-defined criteria (e.g., from lightest to heaviest element, otherwise, or user-defined order).
- The XYZ file format is a simple, human-readable format, used primarily for calculations and visualizations of single molecules. It lists the number of atoms and atomic coordinates only. In the extended version of XYZ, there are additionally added lattice parameters in the comment line. This format is very useful for further calculations using other computational software (ORCA [36], Gaussian [37], etc.).
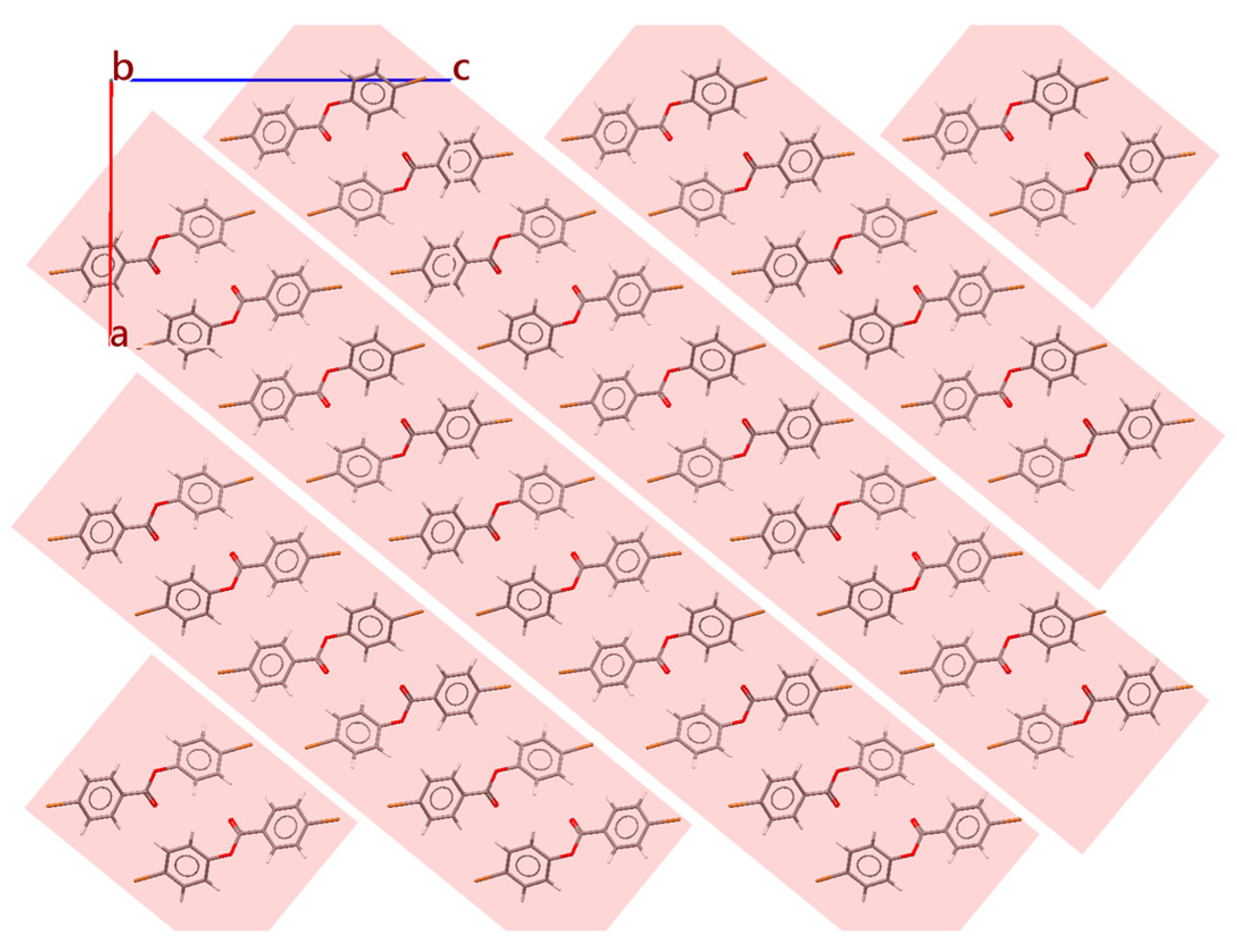
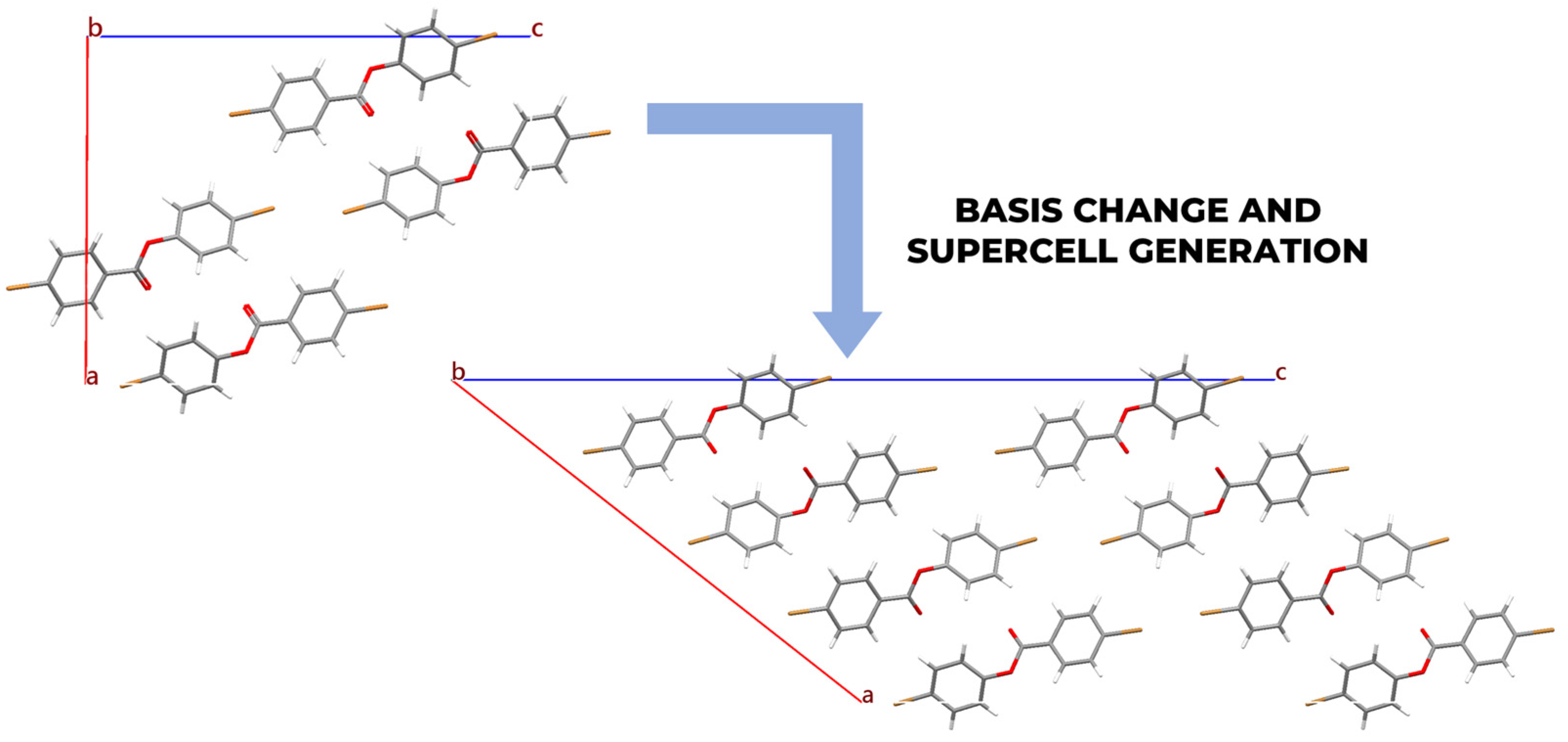
2.3. Basis Change and Supercell Generation
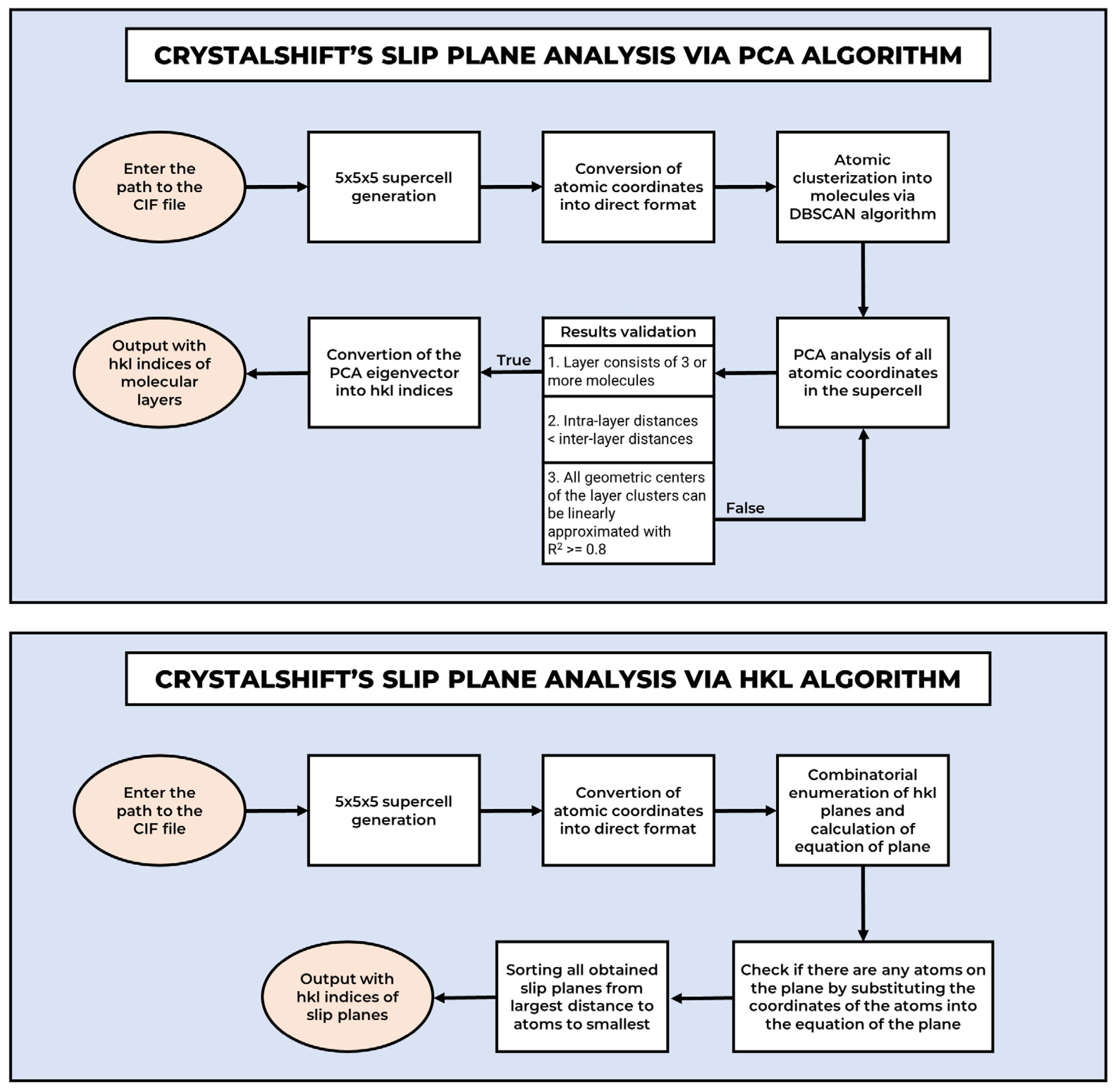
2.4. Molecular Layer Analysis
- Molecular layers should be “sufficiently” spaced apart.
- Errors may occur if the molecules are large and bulky. At best, the direction may be slightly incorrect, and at worst, an erroneous result may be obtained.
- PCA analyzes the spatial distribution of atoms rather than explicitly identifying unoccupied surfaces. Consequently, the obtained Miller indices (hkl) may be suboptimal for further analysis.
- To correctly check large Miller indices, it is necessary to construct supercells.
- The algorithm is unable to identify molecular layers if they correspond to multiple distinct sets of low Miller indices. For instance, if molecular layers are located on the (200) and (300) planes, the method cannot determine an appropriate index combination to define an intermediate plane between them.
2.5. Programming Requirements
3. Results
3.1. Feature Validation and Testing
- A semi-automated inspection of the correctness of the output file structure, as well as of the obtained results after editing the crystallographic data, starting with calculations in VASP (controlling possible errors during input file reading).
- A comparison with results from existing tools (e.g., Open Babel, cif2cell, pymatgen, ASE).
- The testing of converters was carried out automatically using Bash scripts. The conversion of CIF → POSCAR → CIF and CIF → XYZ → CIF was carried out. Due to the fact that atoms are recorded in groups classified by elements in POSCAR files, an additional program was written for sorting and obtaining statistics by comparing coordinates in the original CIF file with those in the CIF file obtained after conversion. In this way, 1000 structures, randomly selected from the CSD, were analyzed and showed a 100% success rate. Speed tests were conducted on 100 random structures from the CSD.
- The testing of the layer analysis module was carried out manually, by comparing visually observed molecular layers, slip planes calculated via CCDC Mercury, and results calculated by CrystalShift.
3.2. Error Handling
3.3. User Warnings
3.4. Limitations and Areas for Improvement
3.5. Worked Example
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bryant, M.J.; Maloney, A.G.P.; Sykes, R.A. Predicting Mechanical Properties of Crystalline Materials through Topological Analysis. CrystEngComm 2018, 20, 2698–2704. [Google Scholar] [CrossRef]
- Mondal, S.; Reddy, C.M.; Saha, S. Crystal Property Engineering Using Molecular-Supramolecular Equivalence: Mechanical Property Alteration in Hydrogen Bonded Systems. Chem. Sci. 2024, 15, 3578–3587. [Google Scholar] [CrossRef] [PubMed]
- Wahlberg, N.; Ciochoń, P.; Petriĉek, V.; Madsen, A.Ø. Polymorph Stability Prediction: On the Importance of Accurate Structures: A Case Study of Pyrazinamide. Cryst. Growth Des. 2014, 14, 381–388. [Google Scholar] [CrossRef]
- Gavezzotti, A. Crystal Formation and Stability: Physical Principles and Molecular Simulation. Cryst. Res. Technol. 2013, 48, 793–810. [Google Scholar] [CrossRef]
- Korabel’nikov, D.V.; Zhuravlev, Y.N. Semi-Empirical and Ab Initio Calculations for Crystals under Pressure at Fixed Temperatures: The Case of Guanidinium Perchlorate. RSC Adv. 2020, 10, 42204–42211. [Google Scholar] [CrossRef]
- Rychkov, D.A.; Hunter, S.; Kovalskii, V.Y.; Lomzov, A.A.; Pulham, C.R.; Boldyreva, E.V. Towards an Understanding of Crystallization from Solution. DFT Studies of Multi-Component Serotonin Crystals. Comput. Theor. Chem. 2016, 1088, 52–61. [Google Scholar] [CrossRef]
- Smirnova, V.Y.; Iurchenkova, A.A.; Rychkov, D.A. Computational Investigation of the Stability of Di-p-Tolyl Disulfide “Hidden” and “Conventional” Polymorphs at High Pressures. Crystals 2022, 12, 1157. [Google Scholar] [CrossRef]
- Mazurek, A.H.; Szeleszczuk, Ł.; Pisklak, D.M. Periodic DFT Calculations—Review of Applications in the Pharmaceutical Sciences. Pharmaceutics 2020, 12, 415. [Google Scholar] [CrossRef]
- Bučar, D.-K.; Lancaster, R.W.; Bernstein, J. Disappearing Polymorphs Revisited. Angew. Chemie Int. Ed. 2015, 54, 6972–6993. [Google Scholar] [CrossRef]
- Togo, A.; Tanaka, I. First Principles Phonon Calculations in Materials Science. Scr. Mater. 2015, 108, 1–5. [Google Scholar] [CrossRef]
- Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.P.; Taylor, R.; Towler, M.; van de Streek, J. Mercury: Visualization and Analysis of Crystal Structures. J. Appl. Crystallogr. 2006, 39, 453–457. [Google Scholar] [CrossRef]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From Visualization to Analysis, Design and Prediction. J. Appl. Crystallogr. 2020, 53, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Momma, K.; Izumi, F. VESTA: A Three-Dimensional Visualization System for Electronic and Structural Analysis. J. Appl. Crystallogr. 2008, 41, 653–658. [Google Scholar] [CrossRef]
- Available online: http://www.chemcraftprog.com (accessed on 29 April 2025).
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Blatov, V.A.; Shevchenko, A.P.; Proserpio, D.M. Applied Topological Analysis of Crystal Structures with the Program Package ToposPro. Cryst. Growth Des. 2014, 14, 3576–3586. [Google Scholar] [CrossRef]
- Spackman, P.R.; Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer: A Program for Hirshfeld Surface Analysis, Visualization and Quantitative Analysis of Molecular Crystals. J. Appl. Crystallogr. 2021, 54, 1006–1011. [Google Scholar] [CrossRef]
- OCC. Available online: https://github.com/peterspackman/occ (accessed on 29 April 2025).
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An Open Chemical Toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef]
- Björkman, T. CIF2Cell: Generating Geometries for Electronic Structure Programs. Comput. Phys. Commun. 2011, 182, 1183–1186. [Google Scholar] [CrossRef]
- Dubok, A.S.; Rychkov, D.A. Deformcell: A Python Script to Simplify and Fasten Mechanical Properties Calculations of Molecular Crystals in VASP Package for Research and Teaching Purposes. J. Struct. Chem. 2024, 65, 1784–1793. [Google Scholar] [CrossRef]
- Wang, V.; Xu, N.; Liu, J.-C.; Tang, G.; Geng, W.-T. VASPKIT: A User-Friendly Interface Facilitating High-Throughput Computing and Analysis Using VASP Code. Comput. Phys. Commun. 2021, 267, 108033. [Google Scholar] [CrossRef]
- Angel, R.J.; Alvaro, M.; Gonzalez-Platas, J. EosFit7c and a Fortran Module (Library) for Equation of State Calculations. Z. Für Krist.-Cryst. Mater. 2014, 229, 405–419. [Google Scholar] [CrossRef]
- Gonzalez-Platas, J.; Alvaro, M.; Nestola, F.; Angel, R. EosFit7-GUI: A New Graphical User Interface for Equation of State Calculations, Analyses and Teaching. J. Appl. Crystallogr. 2016, 49, 1377–1382. [Google Scholar] [CrossRef]
- Gaillac, R.; Pullumbi, P.; Coudert, F.-X. ELATE: An Open-Source Online Application for Analysis and Visualization of Elastic Tensors. J. Phys. Condens. Matter 2016, 28, 275201. [Google Scholar] [CrossRef] [PubMed]
- Vasp2cif Cif2vasp. Available online: https://www.nsc.liu.se/~pla/vasptools/ (accessed on 29 April 2025).
- Jain, A.; Hautier, G.; Moore, C.J.; Ping Ong, S.; Fischer, C.C.; Mueller, T.; Persson, K.A.; Ceder, G. A High-Throughput Infrastructure for Density Functional Theory Calculations. Comput. Mater. Sci. 2011, 50, 2295–2310. [Google Scholar] [CrossRef]
- Hjorth Larsen, A.; Jørgen Mortensen, J.; Blomqvist, J.; Castelli, I.E.; Christensen, R.; Dułak, M.; Friis, J.; Groves, M.N.; Hammer, B.; Hargus, C.; et al. The Atomic Simulation Environment—A Python Library for Working with Atoms. J. Phys. Condens. Matter 2017, 29, 273002. [Google Scholar] [CrossRef]
- Ester, M.; Kriegel, H.P.; Sander, J.; Xu, X. A Density-Based Algorithm for Discovering Clusters A Density-Based Algorithm for Discovering Clusters in Large Spatial Databases with Noise. In Proceedings of the 2nd International Conference on Knowledge Discovery and Data Mining, Portland, Oregon, 2–4 August 1996. [Google Scholar]
- Bentley, J.L. Multidimensional Binary Search Trees Used for Associative Searching. Commun. ACM 1975, 18, 509–517. [Google Scholar] [CrossRef]
- Krishna, G.R.; Devarapalli, R.; Lal, G.; Reddy, C.M. Mechanically Flexible Organic Crystals Achieved by Introducing Weak Interactions in Structure: Supramolecular Shape Synthons. J. Am. Chem. Soc. 2016, 138, 13561–13567. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab Initio Molecular Dynamics for Liquid Metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab Initio Molecular-Dynamics Simulation of the Liquid-Metal–Amorphous-Semiconductor Transition in Germanium. Phys. Rev. B 1994, 49, 14251–14269. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of Ab-Initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Neese, F. The ORCA Program System. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Pearson, K. LIII. On Lines and Planes of Closest Fit to Systems of Points in Space. Lond. Edinb. Dublin Philos. Mag. J. Sci. 1901, 2, 559–572. [Google Scholar] [CrossRef]
- Hotelling, H. Analysis of a Complex of Statistical Variables into Principal Components. J. Educ. Psychol. 1933, 24, 417–441. [Google Scholar] [CrossRef]
- Jolliffe, I.T.; Cadima, J. Principal Component Analysis: A Review and Recent Developments. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2016, 374, 20150202. [Google Scholar] [CrossRef]
- Saha, S.; Desiraju, G.R. Trimorphs of 4-Bromophenyl 4-Bromobenzoate. Elastic, Brittle, Plastic. Chem. Commun. 2018, 54, 6348–6351. [Google Scholar] [CrossRef] [PubMed]
- Masunov, A.E.; Wiratmo, M.; Dyakov, A.A.; Matveychuk, Y.V.; Bartashevich, E.V. Virtual Tensile Test for Brittle, Plastic, and Elastic Polymorphs of 4-Bromophenyl 4-Bromobenzoate. Cryst. Growth Des. 2020, 20, 6093–6100. [Google Scholar] [CrossRef]
- Masunov, A.E.; Wiratmo, M.; Dyakov, A.A.; Matveychuk, Y.V.; Bartashevich, E.V. Prediction of Crystal Structures and Mechanical Properties for Brittle, Plastic, and Elastic Polymorphs of 4-Bromophenyl 4-Bromobenzoate. Cryst. Growth Des. 2022, 22, 4546–4558. [Google Scholar] [CrossRef]
- Paikar, A.; Podder, D.; Chowdhury, S.R.; Sasmal, S.; Haldar, D. Bromine-Bromine Interactions Enhanced Plasticity for the Bending of a Single Crystal without Affecting Fluorescent Properties. CrystEngComm 2019, 21, 589–593. [Google Scholar] [CrossRef]
- Chen, K.; Wang, J.; Wu, W.; Shan, H.; Zhao, H.; Wang, N.; Wang, T.; Huang, X.; Hao, H. Multi-Flexible Organic Crystal Responding to Different Mechanical Forces for Flexible Optical Waveguides. Dye. Pigment. 2023, 219, 111536. [Google Scholar] [CrossRef]
- Wang, C.; Sun, C.C. Computational Techniques for Predicting Mechanical Properties of Organic Crystals: A Systematic Evaluation. Mol. Pharm. 2019, 16, 1732–1741. [Google Scholar] [CrossRef] [PubMed]
- Reddy, C.M.; Gundakaram, R.C.; Basavoju, S.; Kirchner, M.T.; Padmanabhan, K.A.; Desiraju, G.R. Structural Basis for Bending of Organic Crystals. Chem. Commun. 2005, 1, 3945. [Google Scholar] [CrossRef]
- Reddy, C.M.; Padmanabhan, K.A.; Desiraju, G.R. Structure−Property Correlations in Bending and Brittle Organic Crystals. Cryst. Growth Des. 2006, 6, 2720–2731. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector Augmented-Wave Method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Joubert, D. From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Monkhorst, H.J.; Pack, J.D. Special Points for Brillouin-Zone Integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Dubok, A.S.; Rychkov, D.A. What Is More Important When Calculating the Thermodynamic Properties of Organic Crystals, Density Functional, Supercell, or Energy Second-Order Derivative Method Choice? Crystals 2025, 15, 274. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Isupova, I.A.; Rychkov, D.A. CrystalShift: A Versatile Command-Line Tool for Crystallographic Structural Data Analysis, Modification, and Format Conversion Prior to Solid-State DFT Calculations of Organic Crystals. Computation 2025, 13, 138. https://doi.org/10.3390/computation13060138
Isupova IA, Rychkov DA. CrystalShift: A Versatile Command-Line Tool for Crystallographic Structural Data Analysis, Modification, and Format Conversion Prior to Solid-State DFT Calculations of Organic Crystals. Computation. 2025; 13(6):138. https://doi.org/10.3390/computation13060138
Chicago/Turabian StyleIsupova, Ilona A., and Denis A. Rychkov. 2025. "CrystalShift: A Versatile Command-Line Tool for Crystallographic Structural Data Analysis, Modification, and Format Conversion Prior to Solid-State DFT Calculations of Organic Crystals" Computation 13, no. 6: 138. https://doi.org/10.3390/computation13060138
APA StyleIsupova, I. A., & Rychkov, D. A. (2025). CrystalShift: A Versatile Command-Line Tool for Crystallographic Structural Data Analysis, Modification, and Format Conversion Prior to Solid-State DFT Calculations of Organic Crystals. Computation, 13(6), 138. https://doi.org/10.3390/computation13060138