Abstract
The enzyme ROCK1 plays a pivotal role in the disruption of the tight junction protein CLDN1, a downstream effector influencing various cellular functions such as cell migration, adhesion, and polarity. Elevated levels of ROCK1 pose challenges in HCV, where CLDN1 serves as a crucial entry factor for viral infections. This study integrates a drug screening protocol, employing a combination of quantitative structure–activity relationship machine learning (QSAR-ML) techniques; absorption, distribution, metabolism, and excretion (ADME) predictions; and molecular docking. This integrated approach allows for the effective screening of specific compounds, using their calculated features and properties as guidelines for selecting drug-like candidates targeting ROCK1 inhibition in HCV treatment. The QSAR-ML model, validated with scores of 0.54 (R2), 0.15 (RMSE), and 0.71 (CCC), demonstrates its predictive capabilities. The ADME-Docking study’s final results highlight notable compounds from ZINC15, specifically ZINC000071318464, ZINC000073170040, ZINC000058568630, ZINC000058591055, and ZINC000058574949. These compounds exhibit the best ranking Vina scores for protein–ligand binding with the crystal structure of ROCK1 at the C2 pocket site. The generated features and calculated pIC50 bioactivity of these compounds provide valuable insights, facilitating the identification of structurally similar candidates in the ongoing exploration of drugs for ROCK1 inhibition.
1. Introduction
Liver disease is regarded as a global health concern, contributing to a substantial amount of both mortality and morbidity worldwide. Within the Philippines, it has been implicated in 27.3 cases per 1000 deaths, underlining the gravity of the issue. Notably, hepatitis b, hepatitis c, and other viral infections targeting the liver inflict a substantial burden on low-income countries such as the Philippines. The nation faces challenges in this regard, including limited surveillance, epidemiological studies, and clinical practice guidelines, highlighting clinical and medical deficiencies in addressing liver diseases [1]. Analysis of over 144,000 blood screening results for hepatitis c virus (HCV) revealed prevalence rates of 0.3% among hospital-based blood donors and 0.9% among overseas Filipino worker (OFW) candidates [2]. These findings are in parallel with the global prevalence of viremic HCV infection ranging from 0.7% to 1.8% [3,4]. Additionally, risk factors such as unscreened blood transfusions, unsafe injections, and hemodialysis contribute to HCV infections [5]. The use of serological tests, primarily utilizing anti-HCV immunoassays among blood donors, serves as the primary screening method [1,6]. It is advised to utilize direct-acting antivirals (DAAs) as the primary approach for managing HCV. Combinations of the antiviral medicine sofosbuvir/daclatasvir or sofosbuvir/velpatasvir were the recommended antiviral drugs in the country [1,7,8]. Since HCV is a multistep process, it requires an entry factor. Various host-mediating factors play into the viral entry of HCV. The tight-junction protein (TJ) claudin-1 (CLDN1) plays a vital role in mediating HCV entrance. It is necessary for cell line infections in humans (hepatoma) and represents the first gateway protein that becomes vulnerable to HCV in cell line expressions (non-hepatocellular). It is also required in the late stage of the entry process [9,10]. Intestinal epithelial TJ proteins such as CLDN1 are responsible for these processes. Disruption of CLDN1 through pathogenic exposure relies on two main pathways; one of the pathways is rho-associated protein kinase 1 (ROCK1) as it regulates multiple cellular functions. Moderate activity of these kinases is linked closely to TJ protein formations and arrangement. Thus, excess activity of ROCK1/2 leads to defects in TJ protein pathogenesis such as inflammation and ethanol interruption [11,12,13]. The FDA-approved drug for HCV treatment dasabuvir has anti-tumor effects and suppresses esophageal squamous cell carcinoma (ESCC) proliferation. It was also revealed that the drug is an inhibitor of the enzyme ROCK1. The drug dasabuvir inhibits the kinase activity of ROCK1 by reducing the activation of ERK1/2 through phosphate addition and decreasing the expression of CDK4 and CD1. This demonstrates dasabuvir’s effectiveness in blocking the ROCK1 signaling pathway [14]. Understanding ROCK1’s structural information can lead to developing and adding potential inhibitors that can aid in therapeutic strategies, looking for specific molecule structures and relevant properties that may enhance and provide a more robust screening phase for selecting the compounds with accepted inhibitory properties against ROCK1. However, despite having DAAs, HCV infection still poses a major problem for liver diseases globally and domestically. Inhibition of these HCV entry factors can prevent chronic infection and prolonged liver damage. The continued search for inhibitor compounds and suppression of specific target pathways provides us with an approach to develop HCV antiviral strategies.
Older methods that manually isolate and characterize molecules of particular interest and evaluate their biological activity are tedious and consume considerable amounts of time, effort, and research expenses. Nevertheless, harnessing modern technology that is enabled by computers will help propel developments in drug discovery for HCV infections. Virtual screening is one of the most efficient and cost-effective techniques for finding such novel compounds. The identification of potential compounds from a chemical database is a characteristic of a structure-based drug development approach. These compounds are taken into consideration for their bioactivity and binding effects on the target protein structure. The number of compounds may be in the range of (n > 1000); thus, computer-based screening techniques are employed to filter out undesirable compounds and would significantly reduce the cost and time it takes to pick out only the top compounds that may elicit inhibitory effects on the target of interest for drug discovery. Technological advancements have enabled computers to predict bioactivity through quantitative structure–activity relationship (QSAR)-based machine learning (ML) of novel compounds [15,16,17]. In the context of the study, ROCK1’s half-maximal inhibitory concentration (IC50) bioactivity is used for QSAR implementation. The relationship between the generated QSAR features can be established to effectively train a dataset using inhibitors for pattern recognition by use of machine learning (ML) techniques, and the bioactivity of the compounds can be predicted as a function of the features using ML models [18]. These techniques demonstrated that developing a validated ML model has significance in drug discovery. Other studies have implemented QSAR for HCV, targeting inhibitors such as HCV NS5B polymerase, NS3, NS3/4A, and NS3 GT-3a protease inhibitors [18,19,20,21]. QSAR-ML is then combined with molecular docking as a way to further increase the study’s efficacy of effectively screening the potential compounds that were calculated from the predictive capabilities of the model from QSAR-ML.
In the case of ROCK1 inhibition, identification, and classification of these potential candidates for their desirable inhibitory properties to effectively screen the compounds in an abundant dataset, challenges such as emerging drug-resistant HCV variants are what fuel the search for continuous exploratory inhibitors of ROCK1. With these, we would enhance our current existing therapeutic capabilities, provide alternative methods of treatment, and lower the high costs of such treatments, especially in low-income countries such as the Philippines.
IC50 is a term used to describe the drug dosage required to suppress a particular biological or biochemical activity by half. It varies based on the specific conditions of the assay used to measure it. This measurement is crucial in drug discovery as it offers an indication of the drug’s potency against its target. Methods in determining the IC50 of a compound of pharmacological interest are based on the said assay conditions that use whole cell systems. IC50 is a valuable point of information for drug potency; however, it is still limited to the experimental cell lines used and may not inhibit specific compound interactions [22,23]. A limited number of studies have been performed on actual drugs that specifically target ROCK1 for HCV treatment since most DAAs for HCV are used in conjunction with other drugs; also, a lot of the DAAs used for HCV treatment are against other targets.
The primary aim of this study is to efficiently narrow down and evaluate ROCK1 inhibitor compounds based on their bioactivity (pIC50) values within the context of HCV treatment. The objective is to gain insights into the type, structures, molecular properties, drug-likeness candidacy, and potential ROCK1 binding sites for drug affinity selection. The study employs QSAR-based ML modeling as a robust tool to uncover these findings. Utilizing feature engineering and ML techniques, the research screens and predicts the top compounds that exhibit optimal inhibitory effects, emphasizing reliable and critical features as key criteria for drug discovery. The QSAR-ML approach is complemented with ADME prediction and molecular docking of the compounds. This comprehensive methodology aims to solidify the assessment of whether these compounds possess drug-like properties, feature structures relevant for comparative analysis with established DAAs, and, crucially, if they prove to be effective ROCK1 inhibitors for targeting HCV. This varied approach enhances the understanding and selection of promising compounds with potential therapeutic efficacy in the treatment of HCV through ROCK1 inhibition.
2. Materials and Methods
2.1. Outline and Computational Resources
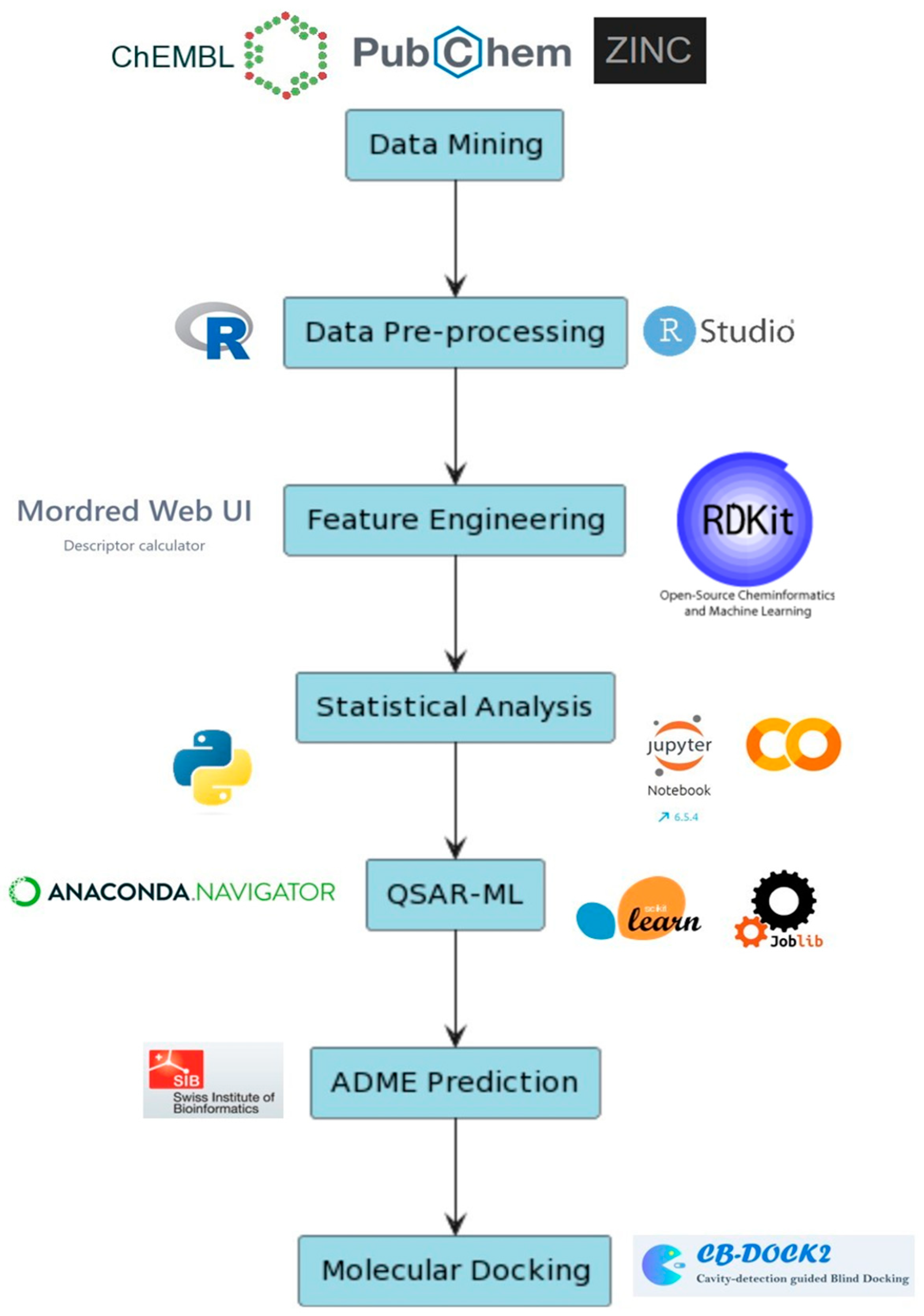
The study is organized into the following sections, as illustrated in Figure 1. However, certain limitations should be acknowledged. The classification of the compound bioactivity parameter relies on IC50 value ranges, and the handling of missing values, coupled with the dataset’s sample size, may introduce biases, rendering ML models susceptible to overfitting or underfitting. Another constraint arises in the construction and training of ML models, contingent upon the computational capacity and processing power of the hardware unit or allocated compute units, particularly when employing cloud services in free versions. This limitation becomes especially notable when fitting models with large numbers and fine parameter selections, significantly extending the runtime for computing processes (t > 6 h–days). It is essential to note that this study primarily concentrates on identifying a suitable criterion for screening drug-like compounds targeting ROCK1, with a focus on inhibitors’ pIC50 values and molecular features. The publicly available 166 MACCS keys are the only fingerprints generated in the following study.
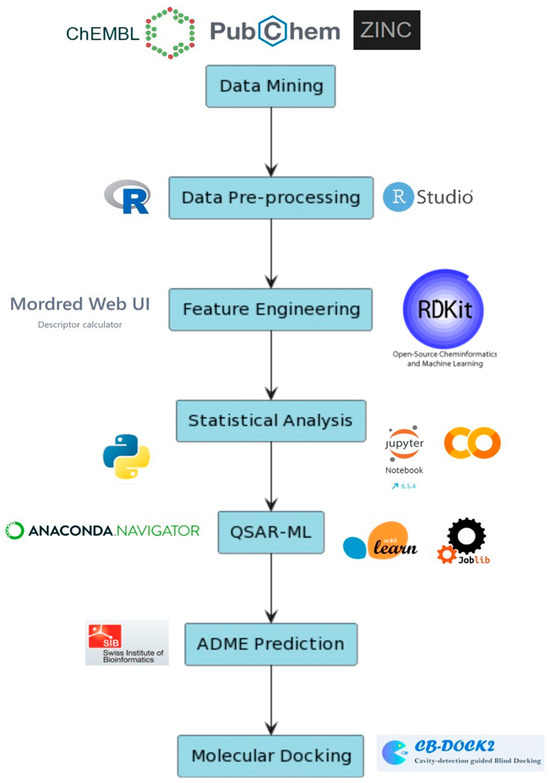
Figure 1.
Outline of the entire methods section as well as the programs and web servers used in the following study. Data mining (EBI-ChEMBL, PubChem and ZINC15 databases) and pre-processing (R language in RStudio IDE). Feature engineering, statistical analysis and QSAR-ML performed in all in Python scripting using Jupyter notebook and Google Colab alongside modules from Mordred, RDKit, scikit-learn and Joblib. ADME prediction for generating the bioavailability radar and BOILED-EGG representation of the compounds. Molecular docking carried out using CB-Dock2 for cavity detection and protein–ligand docking scoring calculation.
The computational methods employed in this study were executed on a computer with the following specifications: an Intel(R) Core(TM) i5-4200U CPU @ 1.60 GHz and 2.30 GHz processor equipped with 4.00 GB of RAM and operating on the Windows 10 Pro operating system. The majority of descriptor calculations, fingerprint generation, and ML scripts/codes are written on the Anaconda distribution version 2023.09-0 of Python 3 using Jupyter Notebook version 6.5.4 (https://www.anaconda.com/ (accessed on 26 November 2023)) [24,25]. The ML library used is the free library scikit-learn (sklearn) for Python (https://scikit-learn.org/stable/ (accessed on 26 November 2023)) [26]. The heavier process computations are leveraged using parallel computing from the module Joblib (https://joblib.readthedocs.io/en/stable/ (accessed on 26 November 2023)) as well as its arbitrary object persistence and caching-mechanism features that help in containing large data for reproducibility and working with long running jobs. In the study, all tasks within the QSAR-ML section were conducted using Google Colaboratory (Colab), a cloud computing service that hosts Jupyter Notebooks. This service utilizes a Python 3 Google Compute Engine backend and operates on Google’s servers (https://colab.research.google.com/ (accessed on 26 November 2023)), offering a powerful platform for running complex machine learning and data analysis jobs [27]. QSAR-ML in combination with an automatic blind docking study is the selected technique/protocol followed for effectively screening potential drug candidates for their major importance in ROCK1 inhibition [16]. All of the complete data procured and used in this study can be found in the Supplementary Materials. Documented R and Python scripts/codes and datasets can all be found in the Supplementary Material (Supplement S1: Code Repository and Dataset Repository).
2.2. Data Mining
Hepatitis C (HCV) is the chosen disease in the following study. Claudin-1 is searched in the chemical database ChEMBL for the enzyme candidate. The enzyme rho-associated protein kinase 1 (ROCK1) is the selected target enzyme for the study analysis and its inhibitors’ IC50 bioactivity [14,28,29]. The main dataset to be analyzed in the following study is named the “ROCK1 dataset (CHEMBL3231)”. The selection process must first be filtered using the following criteria: Eukaryotes, Mammalia, Primates, Homo Sapiens, Enzyme, and Activities (2490–345,557). The introduction of these filters showed the enzyme ROCK1 as one of the targets, gathering only the IC50 compounds with their ChEMBL details. An additive training dataset, referred to as the “HCV dataset”, was created using the following enzymes: NS5B polymerase (CHEMBL5375), NS3 (CHEMBL1293269), and NS3/4A (CHEMBL2095231) protease inhibitors. These enzymes, which have a specific number of compounds with IC50 values, played a role in HCV inhibition [18,19,20]. A combination of the “HCV-ROCK1 dataset” is used to train and test the regression models. A final dataset for external validation was also chosen containing only IC50 compounds, and cAMP-dependent protein kinase (PKA) enzyme [30,31]. The following dataset is named “PKA or External dataset”. All of the raw datasets are retrieved in a CSV format to be cleaned and pre-processed. Splitting of the datasets was performed in the following fashion: 80 (train)–10 (test)–10 (validation) ratio (HCV-ROCK1) and external validation (PKA dataset) [32]. Four inhibitor drugs were used for comparative analysis in the study: Dasabuvir (CHEMBL3137312), Sofosbuvir (CHEMBL1259059), Daclatasvir (CHEMBL2023898), and Velpatasvir (CHEMBL3545062). These drugs are the currently approved DAAs being used for HCV treatment [1,14]. Their pIC50 values are roughly estimated using the web app deployed (https://bioactivity.streamlit.app/ (accessed on 2 September 2023)), using their canonical smiles and ChEMBL IDs as an inputted text file [16].
2.3. Data Pre-Processing
The HCV-ROCK1 IC50 compounds were pre-processed using the R programming language, specifically version 4.1.0, within the RStudio environment [33,34]. Duplicate ChEMBL IDs are removed to reduce the number of compounds. IC50 values of the compounds are standardized, converting nanomolar (nM) units to molar (M) followed by the conversion to its logarithmic form in Equation (1).
The following are the only selected column variables to be shown for their degree of importance: Molecule.ChEMBL.ID (unique identifier), Smiles (canonical smiles), Standard.Relation (target-compound relationship type), Standard.Value (IC50 values), Document.ChEMBL.ID (source publication where bioactivity data were extracted) and the computed pIC50. The raw dataset provided a converted version of IC50 named pChEMBL.Value. It is ruled out as the selected variable since it has a large number of missing values in the observations; thus, the added standardized column variable named pIC50 is the one chosen for data analysis to avoid reducing the number of compounds further.
The compounds undergo bioactivity classification for indexing and classification purposes only to gauge/estimate the degree of bioactivity of the compounds based on their pIC50 values regarding the inhibitor drugs. Actual values of the drugs against specific assays are provided in the Supplementary Material (Table S1: Inhibitor Drugs). Missing values are converted to NA and are omitted if there are any in the dataset. Selecting the intermediate range for classification is performed in a percentile-based approach using the rough calculated estimate of velpatasvir’s pIC50 as the cutoff point for an “active compound” using a box and whisker plot for analyzing the dataset’s quartile values for both active and inactive compounds. It is then divided into the following three classes: “active” (pIC50 > 5.4), “intermediate” (5.00 < pIC50 > 5.39), and “inactive” (pIC50 < 4.99). The HCV-ROCK1 screened/pre-processed dataset is exported as a CSV file to be used for the generation of molecular descriptors and fingerprint keys.
2.4. Feature Engineering
Molecular descriptor (2D and 3D) calculations were performed using Mordred in Python scripting [35]. MACCS fingerprint keys were produced using RDKit version 2023.9.3, open-source cheminformatics software, along with Python scripting. This approach enabled the efficient generation of molecular fingerprints, facilitating the analysis and comparison of chemical structures within the study (https://www.rdkit.org (accessed on 26 November 2023)). The calculated descriptors and generated MACCS keys are exported as a CSV file for QSAR-ML analysis. The HCV-ROCK1 dataset is used for training, testing, and validating the model to be selected. All observations in the dataset were randomly shuffled with a fixed seed number (42) for reproducibility. Since there would inevitably be missing/NA/null values from the generated descriptors, all objects are coerced to be numeric/floating parameters, thus identifying those missing values. Simply dropping those null values would reduce the rows to unacceptable amounts; thus, they are then subjected to mean imputation using the SimpleImputer function from sklearn [36]. The dataset is separated as x and y for ML analysis in Equation (2).
where y is the response variable (pIC50) that the model is trying to predict, and x is the predictors (features)—the independent input variables, which include descriptors and keys. The model aims to learn patterns from these features to predict y in the regression analysis.
The complex dataset is simplified using feature selection, where highly correlated molecular descriptors are subjected to pairwise correlation with a threshold value of 0.8 and a variance threshold that removes low-variance descriptors of 0.1 [16]. The dataset is further pre-processed to center and scale all its features using the StandardScaler function from sklearn and log transformations on y. This step is crucial for many ML estimators/models as some may behave badly if the features are not standardized and/or normally distributed. Activity sampling is a sampling method employed to randomly select equal amounts of observations from bins (n = 5) based on the search space between the maximum and minimum values of y to avoid any discrepancies and biases when splitting the dataset [37]. It is then divided into the 80:10:10 ratio (train–test–validation) using the train_test_split function from sklearn [32,38]. All random states employed will have a seed number of 42 for reproducibility.
2.5. Statistical Analysis
Statistical analysis of y in the dataset is performed to assess its distribution and prevent overfitting. It is used to initially assess what kind of response the dataset has. Measurements of the mean, median, skewness, and kurtosis were calculated for the train, test, and validation sets of y [38,39]. The skewness of the dataset sample is computed using the Fischer–Pearson coefficient of skewness, in a biased sample skewness and an adjusted sample if the bias is false in Equations (3) and (4).
N represents the number of data points, m2 is the biased sample’s second central moment, and m3 is the biased sample’s third central moment (m4) by square of the variance, following Fischer’s definition. To align with a normal distribution, 3.0 is subtracted from the outcome, resulting in a ta value of 0.0 for a distribution that is perfectly normal in Equation (5).
2.6. QSAR-ML
The following study employed the QSAR technique to predict the bioactivity pIC50 of the compounds based on their generated chemical descriptors and fingerprints in a quantitative manner. This is achieved through ML to build the QSAR models that can forecast these properties, e.g., their features. The models learn from the datasets input with their known activities to then predict this in the model. The performances of the model are then used as metrics to gain insight into which models best fit the dataset and which features are the most important from the chosen model. Initial ML model analysis is performed using the module LazyRegressor from Lazy Predict to exhaust all of the possible regression models from sklearn that would be best suited for the dataset. The train and test sets were used to select the top-scoring model based on the following metrics: R2, root mean squared error (RMSE), and time taken [16,40,41,42]. The coefficient of determination scoring metric, represented as R2, measures the extent to which the variance of a feature is predictable from another feature. In the context of a performance metric, the best possible score for R2 is 1.0; this describes that the model explains the variability of the dependent response through its mean. R2 can also take on negative values. This scoring metric indicates the goodness of fit and, thus, how well external observations are likely to be forecasted using ML models, through the explained proportions of the variances [26]. RMSE, the square root of the mean squared error (MSE), computes as a risk metric that corresponds to the expected value of the quadratic (squared) error loss; an MSE close to zero is considered a good fit for the dataset. The selection of the best model was determined by the R2, RMSE, and time taken performances previously calculated using Lazy Predict paralleling the selection process carried out by Kumar et al. [16].
Based on the gathered results from Lazy Predict, NuSVR is the model best suited for the dataset. The following are the parameters/default settings used to build the NuSVR base model (nu = 0.5, C = 1.0, kernel = ’rbf’, degree = 3, gamma = ’scale’, coef0 = 0.0, shrinking = True, tol = 0.001, cache_size = 200, verbose = False, max_iter = −1). The study used multiprocessing from joblib’s parallel_backend to set the jobs to 200, which were then dumped using joblib. The test and validation performance of this model was analyzed, and the true vs. predicted values of each in the following plots were visualized [43,44,45,46,47,48,49,50]. The concordance correlation coefficient (CCC) is another metric used to measure the agreement between the true and predicted values for the test and validation sets using the model [51]. External validation of the model was conducted using the PKA dataset. Similar protocols were applied, including descriptor calculation, fingerprint generation, pre-processing, and feature selection, before being input into the selected model (NuSVR). Performance scores and CCC values were collected, along with visualized plots comparing the true and predicted values [31]. The features from the ROCK1 dataset that played the most significant role in the model were identified using the model inspection technique of permutation importance from sklearn [26]. This is approach useful as NuSVR is a non-linear model/estimator. In this approach, a single feature in the data is randomly permuted, which typically leads to a decrease in the model’s score. The scoring metric used is R2. The permutation process was repeated 5 times with a random state set to 42. Only the top 25 features were selected from the ROCK1 dataset, which were tested between the model and feature importance. Compounds with non-zero values in their MACCS keys from those 25 features were selected for further analysis of their ADME properties and molecular docking. These compounds are called “compounds of interest”. They are exported as a separate CSV file containing their respective data values and features.
2.7. ADME Prediction
The ADME properties of the compounds of interest were predicted using SwissADME, with their canonical smiles obtained from PubChem [52]. A comparison of the compounds to velpatasvir (+control) was carried out. Only the compounds that do not violate the screen in this section were used for molecular docking analysis.
2.8. Molecular Docking
The X-ray crystal structure of ROCK1 (PDB ID: 1S1C) at a 2.60 Å resolution from RCSB Protein Data Bank was retrieved [53]. The protein was prepared using the docking server CB-Dock2. By default, 55 water molecules and 66 other heteroatoms were deleted before docking by default [54]. The protein structure consists of four chains (A, B, X, and Y). Protein-surface-curvature-based cavity detection is employed to predict the binding modes without prior knowledge of the binding sites. This approach uses CurPocket detection from CB-Dock implementation; it automatically calculates the centers and sizes of the pockets with this approach [54,55]. Three-dimensional structures of the ligands of interests were retrieved from ZINC15 (https://zinc15.docking.org/ (accessed on 14 January 2024)) in SDF format, a database used to access commercially available compounds [56]. Some of the compounds of interest (ADME screened) not directly found in ZINC15 cross-referenced from the ChEMBL database are excluded from the retrieval for consistency purposes. Automatic blind docking is then performed using CB-Dock2; the web app uses Autodock Vina (version 1.1.2) as its backend [54,55,57,58].
3. Results
3.1. Data Mining and Pre-Processing
Listed are the datasets used; the following are the total number of observed ROCK1, HCV (NS5B polymerase, NS3, and NS3/4A proteases), and PKA (cAMP-dependent protein kinase) inhibitors gathered from ChEMBL (raw, screened): ROCK1 (1448, 1217), HCV (2901, 2246), and PKA (575, 575). HCV-ROCK1 was used as the train–test–validation set for ML models. The bioactivity classification of HCV and ROCK1 inhibitors is shown in Figure 2.
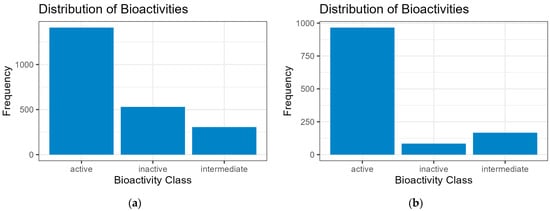
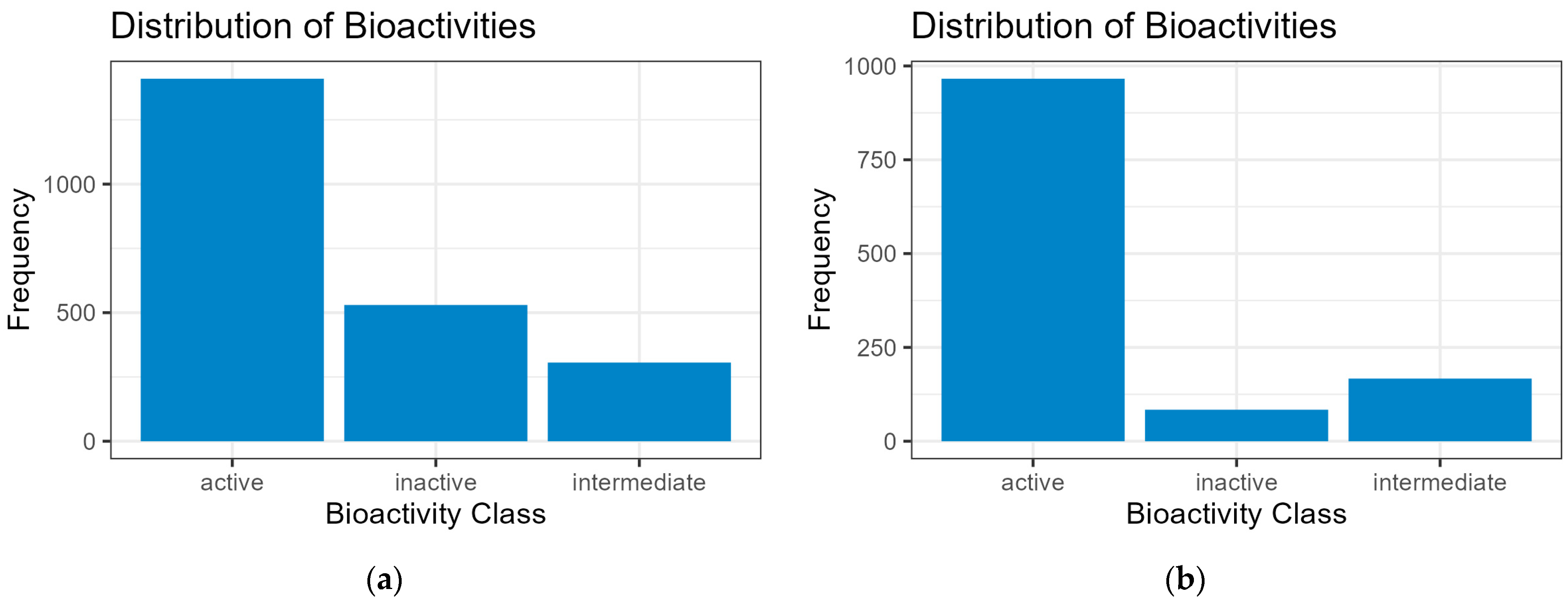
Figure 2.
Bioactivity distributions. (a) HCV (NS5B polymerase, NS3, and NS3/4A proteases) set; (b) ROCK1 set. The frequency distribution shows the number of each inhibitor compound considered in three classes as either “active” (pIC50 > 5.4), “intermediate” (5.00 < pIC50 > 5.39), or “inactive” (pIC50 < 4.99). The screening process for bioactivity distribution is detailed in the Supplementary Material (Supplement S1: Code Repository and Dataset Repository).
3.2. Mordred Descriptors and MACCS Keys
Mordred descriptor calculation through Python scripting was used to calculate the 1826 2D and 3D descriptors and generation of the publicly available 166 MACCS keys using RDKit for all of the dataset’s inhibitors; complete details are outlined in the Supplementary Material (Supplement S1: Code Repository and Dataset Repository).
3.3. Feature Selection and Extraction
Highly correlated descriptors are subjected to pairwise correlation, with a threshold of 0.8, and removal of low-variance features, with a threshold of 0.1 [16]. This reduces the 1826 initial descriptor count to 196. The final number of descriptors and 166 MACCS keys total 362. These are the features (x) selected for QSAR-ML analysis. The total shape of x in the HCV-ROCK1 set comes to 3463 rows × 362 columns, and the starting count point for indexing in Python is 0. After feature scaling and activity sampling, the set is split into train–test–validation. The final count to be used for QSAR-ML analysis is as follows: train set (2767, 362), test set (347, 362), and validation set (348, 362).
3.4. Statistical Measurements
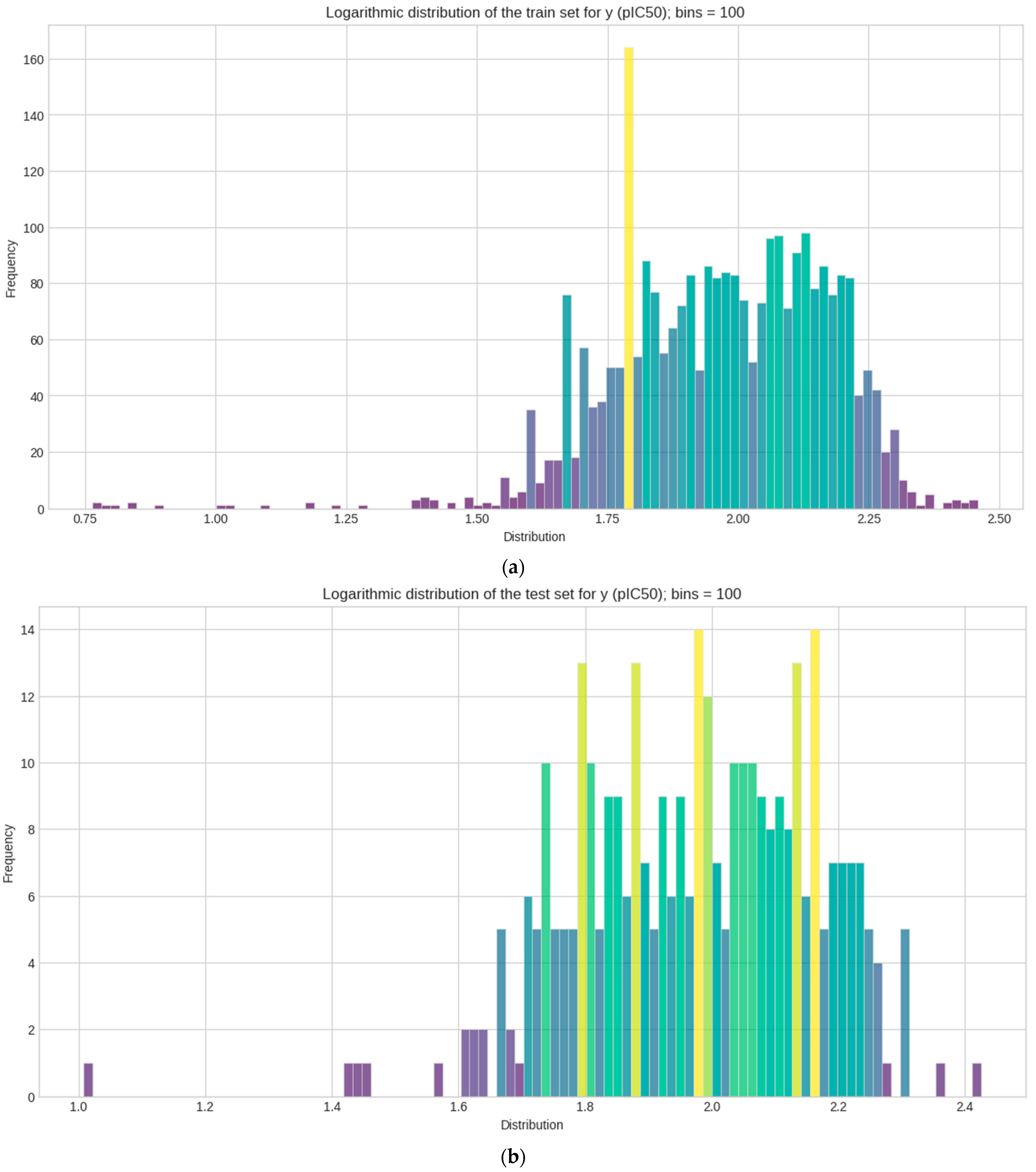
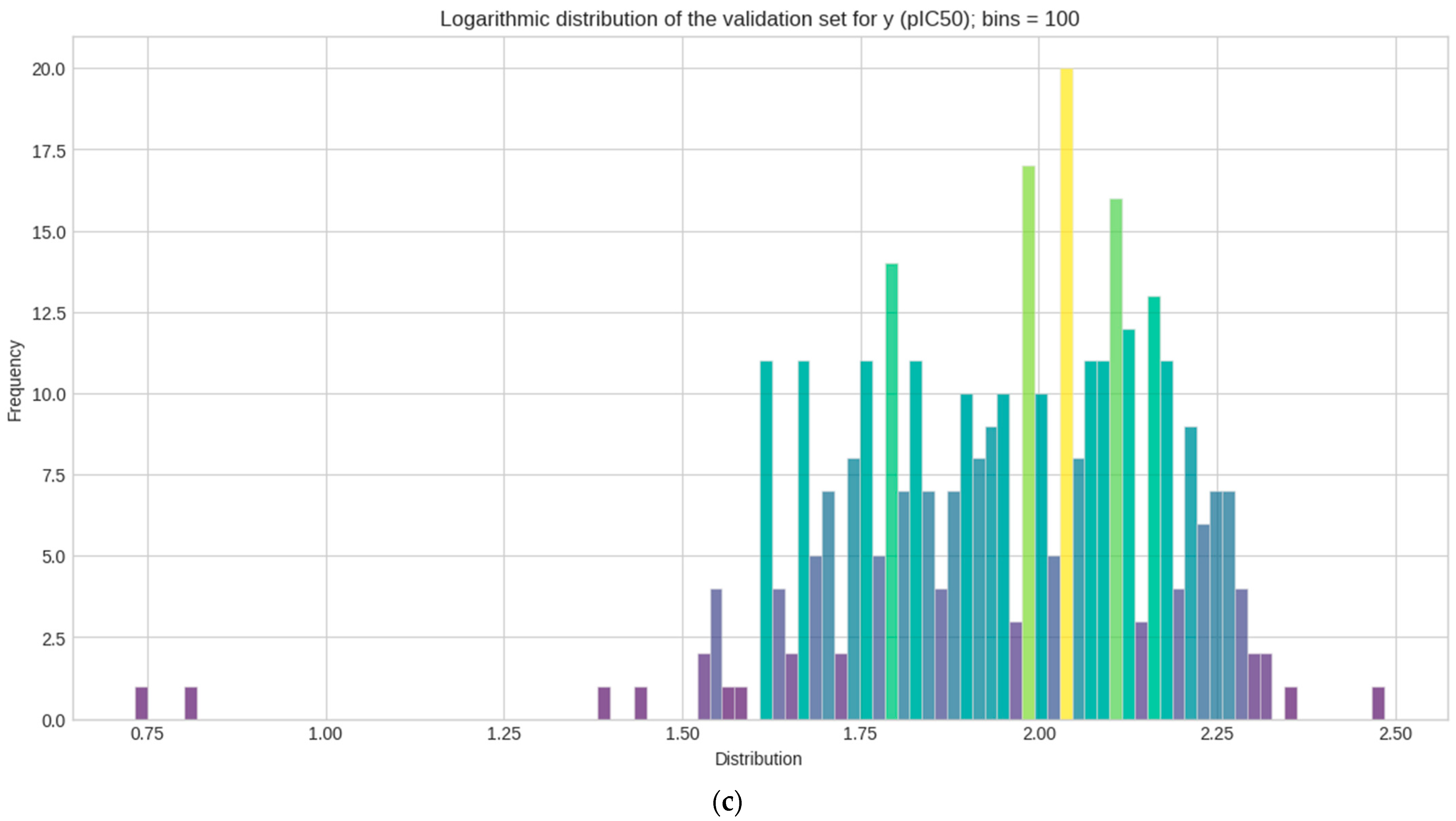
Logarithmic transformation is performed separately on y for each (train–test–validation) for the data to conform to normality [59]. The mean, median, skewness, and kurtosis are determined in Table 1. Figure 3 represents the distribution of each set in a histogram plot, bins (n = 100). Statistical measurements of each sets show how pIC50 (y) values are distributed across 100 bins in the histogram plot.

Table 1.
Statistical measurements of y (pIC50) in all sets.
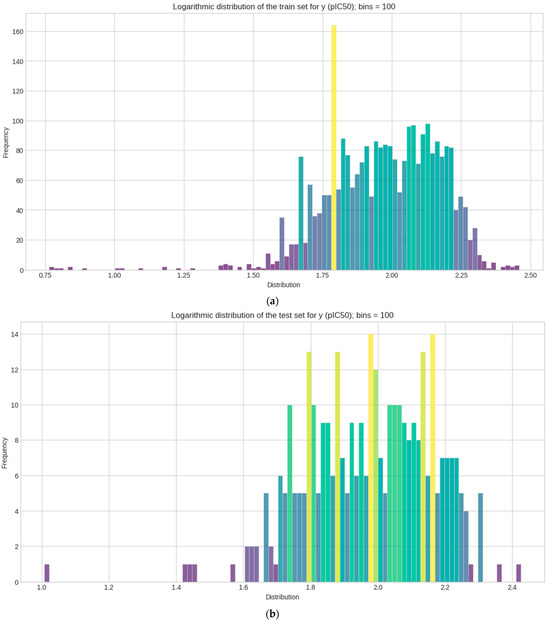
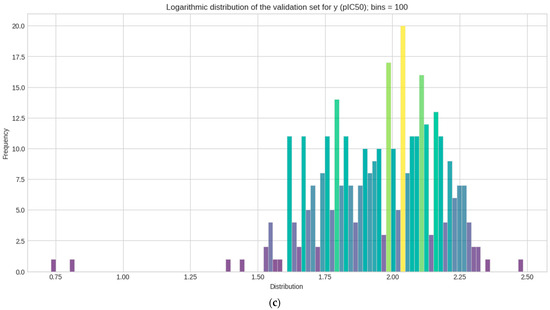
Figure 3.
Histogram plots of the logarithmic distribution of the y (pIC50) values visualized at 100 bins. Where frequency represents the count values and how each y datasets are logarithmically distributed after activity sampling is employed. (a) Train set; (b) test set; (c) validation set.
3.5. Regression Models
LazyRegressor computed 42 regression models from the sklearn library. Table 2 lists the top 10 models based on R2 scoring fitted using the train set and scored on the test set. NuSVR is the selected model suited for the HCV-ROCK1 set with the following scores: R2 = 0.67, RMSE = 0.11, and time taken = 15.80 s.
Table 2.
Top 10 best-suited ML models for HCV-ROCK1 dataset calculated using the LazyRegressor function from Lazy Predict.
3.6. NuSVR Performance
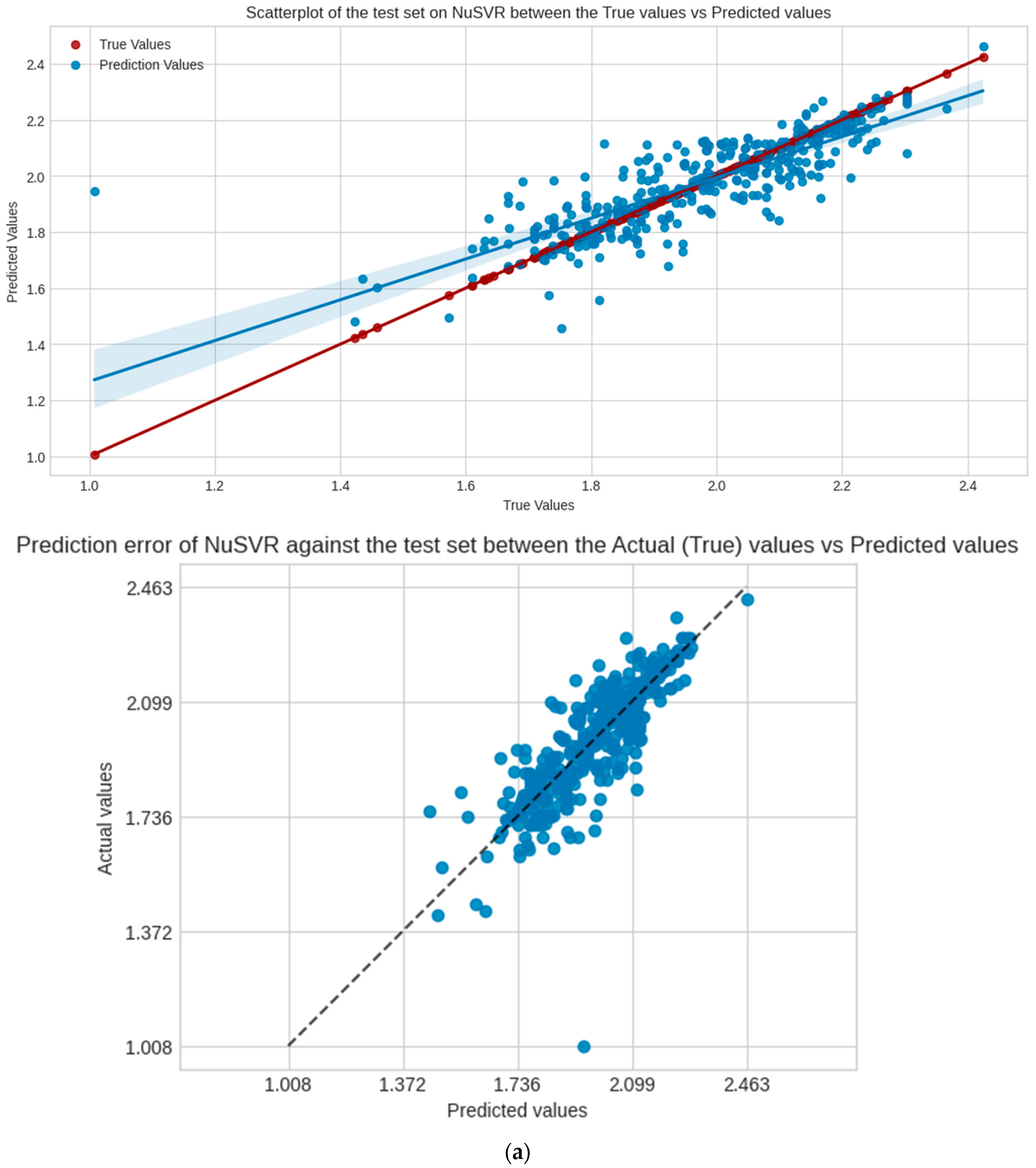
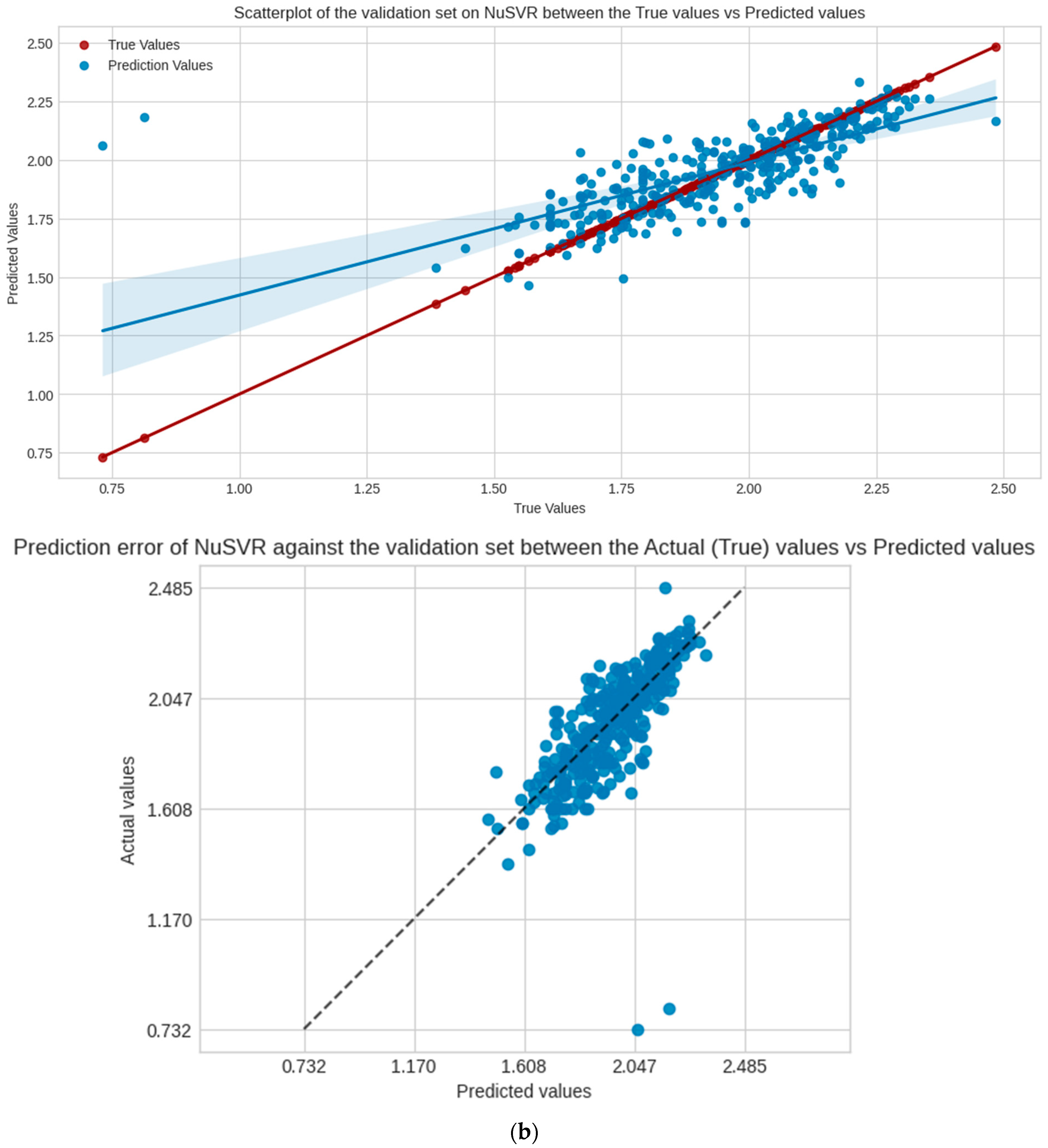
NuSVR is the selected model for evaluation, Figure 4 shows the regression scatter, prediction error plots for the test, and validation set of NuSVR, visualizing the true y values against the predicted y values. Table 3 shows the performance scores of the model in the following sets and scoring metrics. CCC measures the relational agreement between the true and predicted values using the model; these results indicate that NuSVR true and predicted y values are at 82%, 71%, and 27%, in agreement for the test, validation, and external validation sets, respectively.
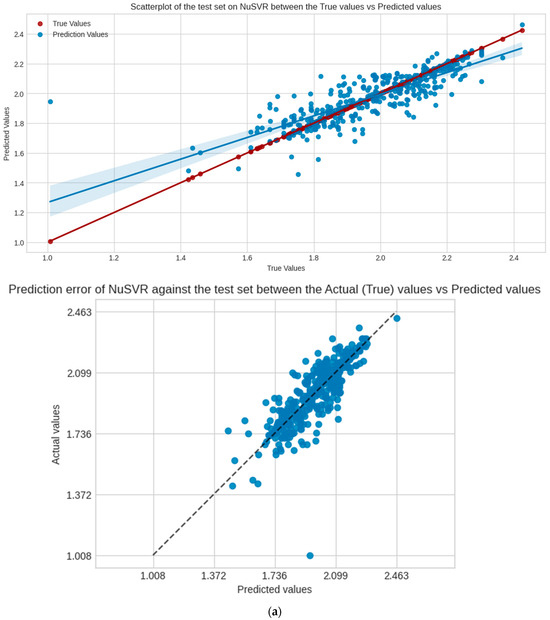
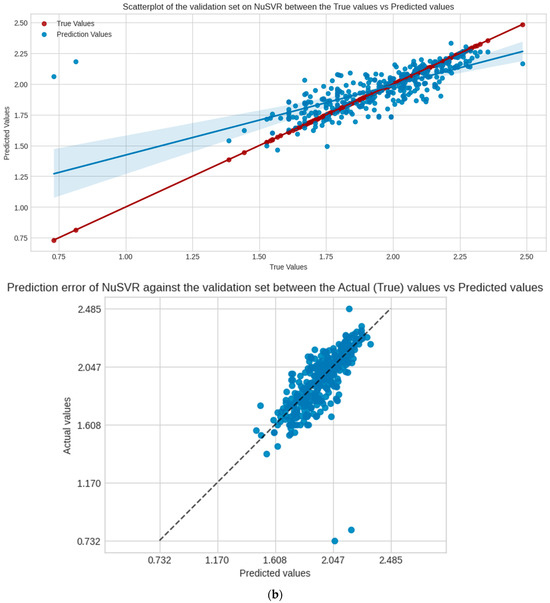
Figure 4.
Scatter and prediction error plots of NuSVR between the actual (true values of y) vs. predicted values of y in each dataset, not accounting for the training. (a) Test set; (b) validation set.
Table 3.
Performance scores of the model (NuSVR) against all of the datasets calculated using the models R2, RMSE, MSE and CCC.
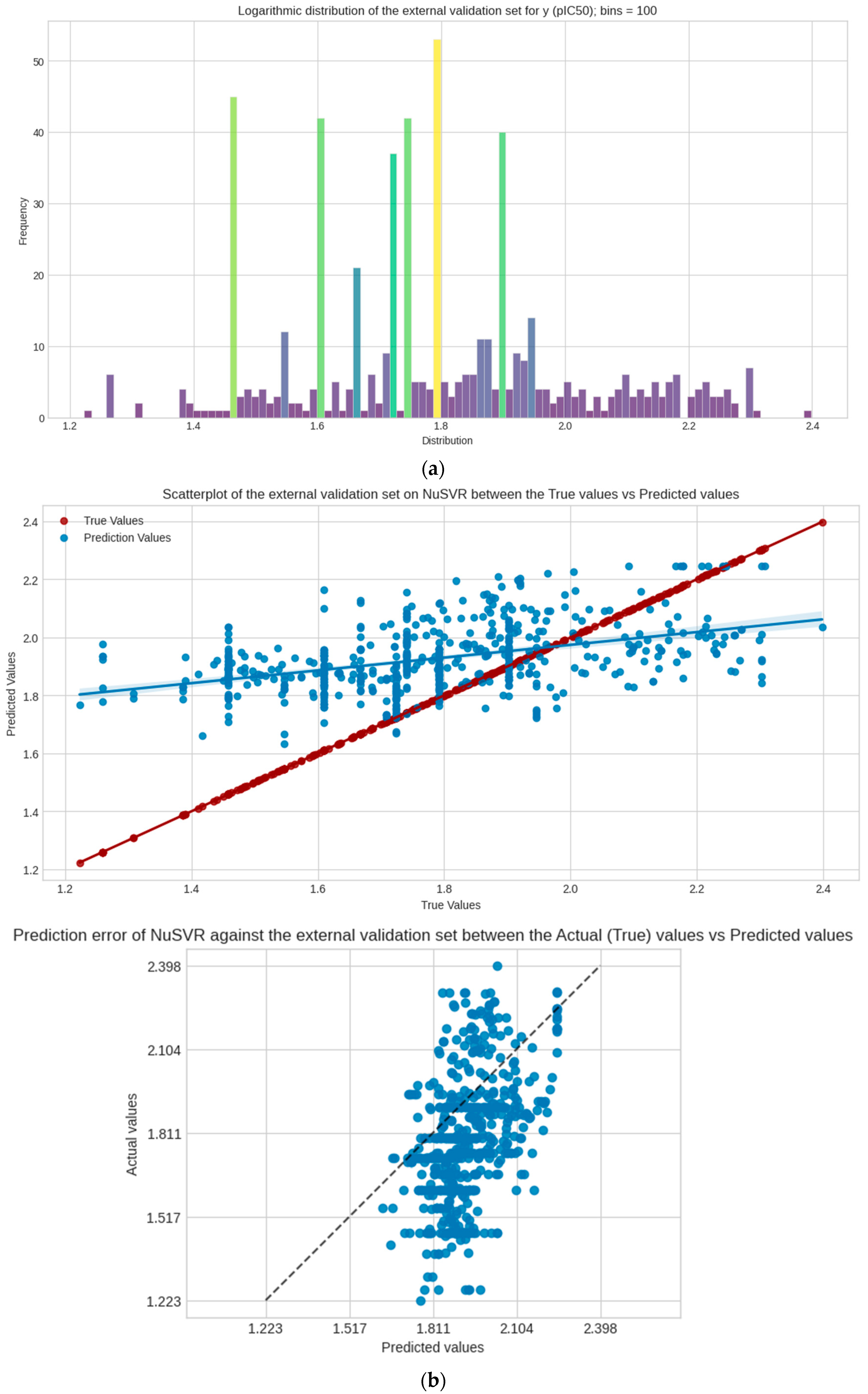
3.7. External Validation Metrics and Performance
The PKA dataset is used as a completely external dataset that also undergoes the same pre-processing and feature selection stage; it is worth noting that duplicate ChEMBL IDs are not removed here to account for the extreme loss of data since this set is significantly smaller than the test and validation sets combined. The same feature selection (imputation, scaling, and log transformation) is still followed. The scoring performance of the model is evaluated against this set, as shown in Table 3. Visualization of the regression and prediction error plots are represented in Figure 5.
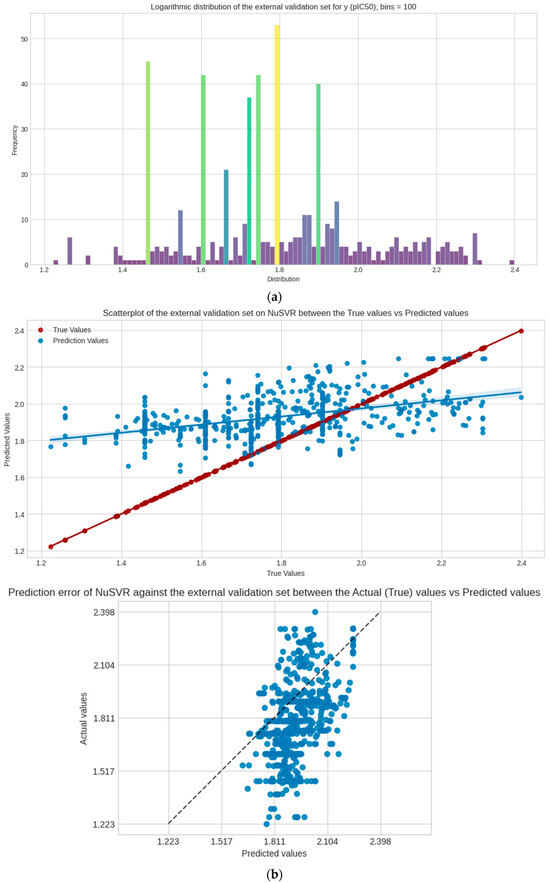
Figure 5.
The PKA dataset as an external validation and its metrics. (a) Logarithmic distribution of y; (b) regression and prediction error plots between true y and predicted y values in the set. The external validation dataset is computed separately but followed the same process as the other datasets. Supplementary Material (Supplement S1: Code and Dataset Repository).
3.8. Important Features and Compounds of Interest
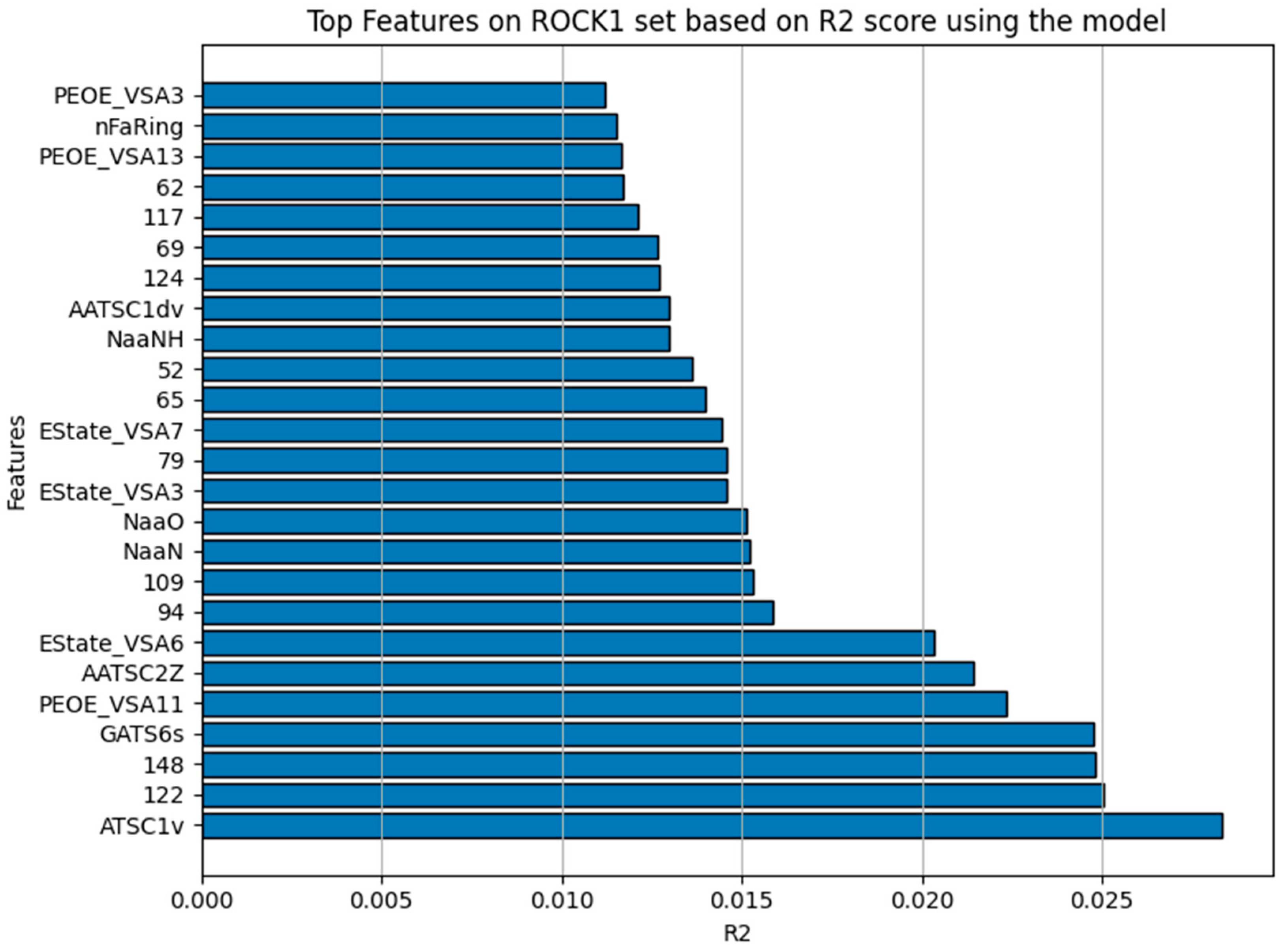
The model inspection technique of permutation importance calculates the feature importance of estimators for the dataset, in this case using only on the ROCK1 dataset. Five repeats are performed to randomly shuffle each feature and return a sample of its importance. It measures the contribution of each feature to the model’s statistical performance (R2). This involves randomly permuting/shuffling the values of each of the features and the degradation of the score is then observed. Figure 6 shows the 25 features the model majorly depends on for it to have a good R2 score. Table 4 summarizes these features, their details, and the score of impact it has on the model’s R2 [16,35]. The features described in the table are the criteria for compound selection to effectively screen ROCK1 inhibitors for their impact based on the bioactivity pIC50. The molecular fingerprint (MACCS keys) is an indication of the structural information of the compounds for drug screening/discovery selections.
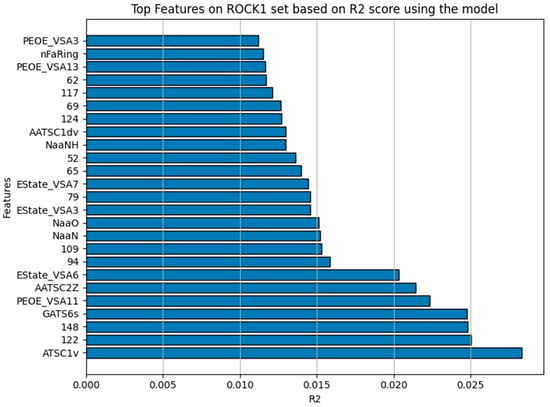
Figure 6.
The top 25 features in the y-axis that NuSVR mostly relies upon for it to have a good R2 represented in the x-axis after calculations were carried out using the permutation_importance function from scikit-learn.
Table 4.
Top 25 features that the model NuSVR depends upon to have a good R2. Permutation importance from scikit-learn is used to calculate the R2 performance of NuSVR based on the estimator/features only using the ROCK1 dataset at 5 repeats of random shuffling and return.
The last step for QSAR-ML is extracting these compounds of interest; only non-zero values from the MACCS keys that are part of the 25 features are selected for their ADME properties and molecular docking study. Performing QSAR-ML reduced the initial 1217 inhibitors from ROCK1 set to only 18; they are summarized in Table 5 with their relevant details in the Supplementary Material (Table S2: Compounds of Interest). Since velpatasvir is the cutoff point in determining the pIC50 values of the compounds, it is used as a positive control in the subsequent sections (ADME-DOCKING). Structural information about the compounds is represented in its fingerprint keys and the properties in its descriptors. The screened compounds that resulted from using QSAR-ML techniques are a starting point of reference for further testing of their ROCK1 inhibition and drug-like properties.
Table 5.
Summary of the compounds of interest gathered from QSAR-ML screening. Complete details of each compound after QSAR-ML screening are found in Supplementary Material Table S2.
3.9. Compound ADME Properties
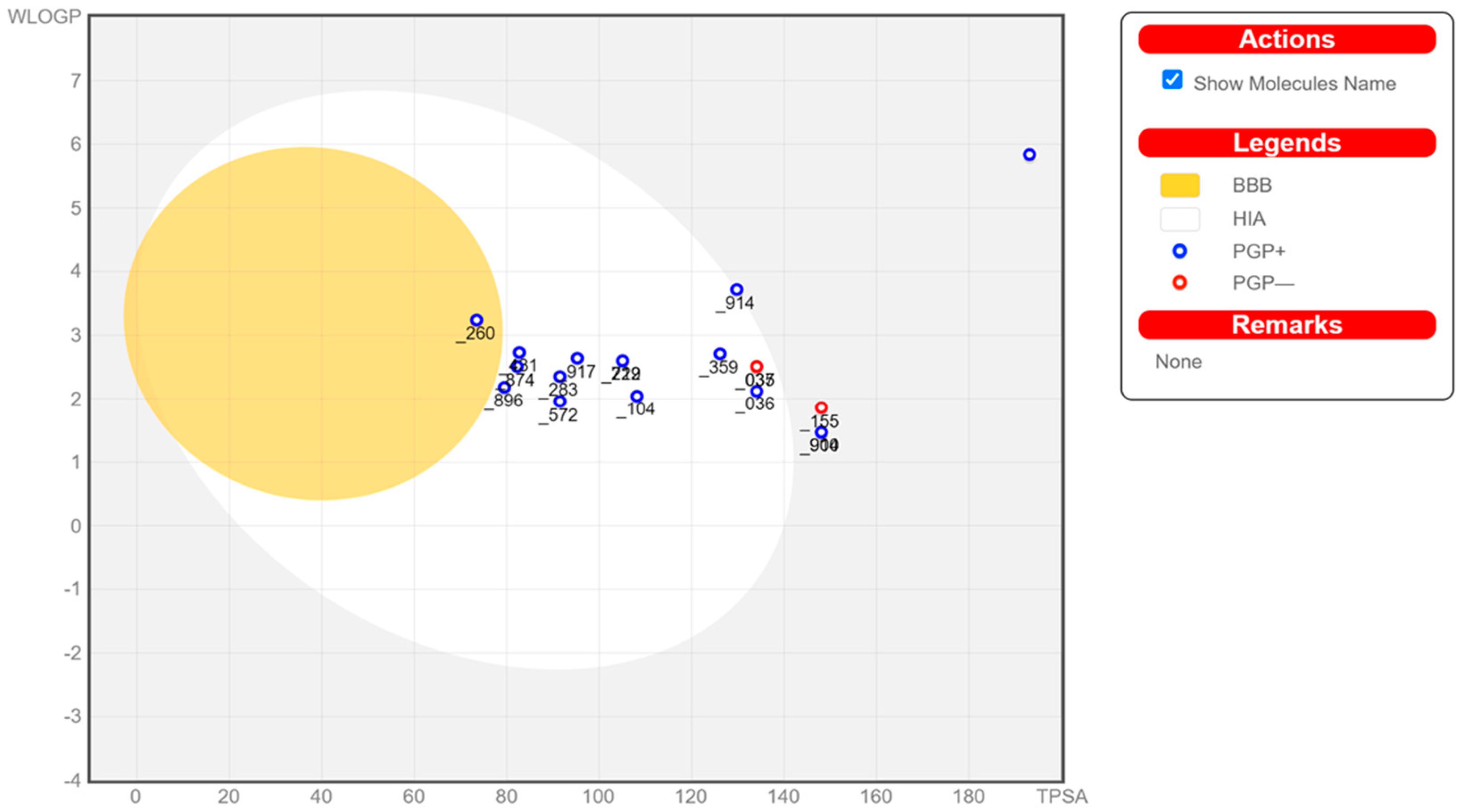
Prediction of the individual ADME (absorption, distribution, metabolism, and excretion) results of the compounds of interest is performed as an intermediary stage before the docking process. Calculation of these properties is performed in SwissADME and enables the visualization of a molecule’s drug likeliness [52]. Figure 7 reports each of the compounds that may or may not have passed the bioavailability radar, which is represented as the pink area for the optimal range in each property. The BOILED-Egg model in Figure 7 is a graphical representation for predictions for passive human gastrointestinal absorption (HIA) and blood–brain barrier (BBB) permeation. The egg whites (white-spaced region) in the model are the physicochemical space of molecules that has the highest probability of being absorbed in the GI tract, while the egg yolk (yellow-spaced region) is the space with the highest probability of molecules permeating the brain. Neither space is mutually exclusive [60]. A complete visual representation of the bioavailability radar for all of the compounds can be found in the Supplementary Material (Figure S1: Bioavailability Radar).
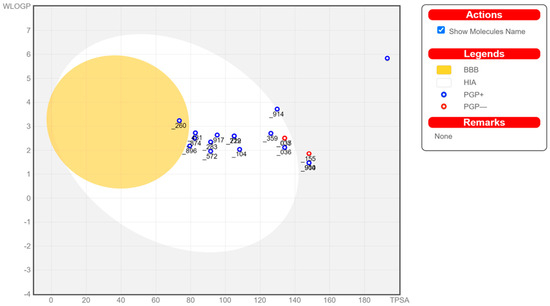
Figure 7.
BOILED-Egg representation in SwissADME of the screened compounds from QSAR-ML represented by their last three PubChem ID numbers for the evaluation of their gastrointestinal absorption and blood–brain barrier penetration.
3.10. Molecular Docking Results
Based on the bioavailability radar in the previous section, only 9 of the 18 compounds of interest are selected for docking analysis. Figure 8 shows the 3D crystal structure of ROCK1 (PDB: 1S1C). Detection of the pocket cavities using the CurPocket implementation from CB-Dock2 was performed showing the five calculated pocket sites—see Figure 8. Table 6 summarizes the following pockets with their cavity volume, centers, and sizes. The sequence chains in each query for the pockets are represented in different colors in the Supplementary Material (Figure S2: Chain Sequences). Based on the curvature-dependent surface area model from CurPocket, the protein’s convex, planar, and concave surfaces are calculated in hydrophobic free energy calculations. Calculating hydrophobic energy is too complex in nature as it plays a key role in biomolecular recognition. This is also estimated to provide approximately 75% of the free energy in most binding events. This implementation from CB-Dock2 is used to estimate the hydrophobic energy in protein–ligand binding. CB-Dock2 searches for concave surfaces for cavity detection; it generates a set of points for the representation of the solvent-accessible surface and calculates the curvature factors of each point using the curvature-dependent surface area model. The points are clustered by a density-peaked-based clustering algorithm [54,55,61].
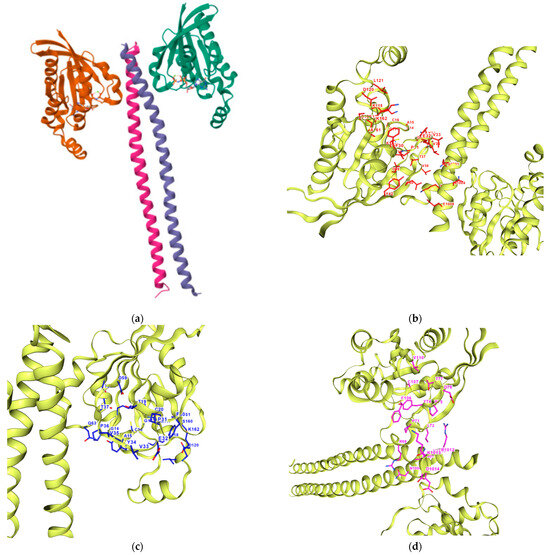
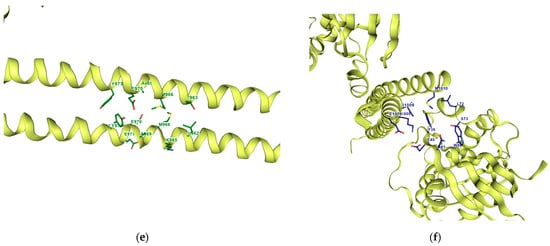
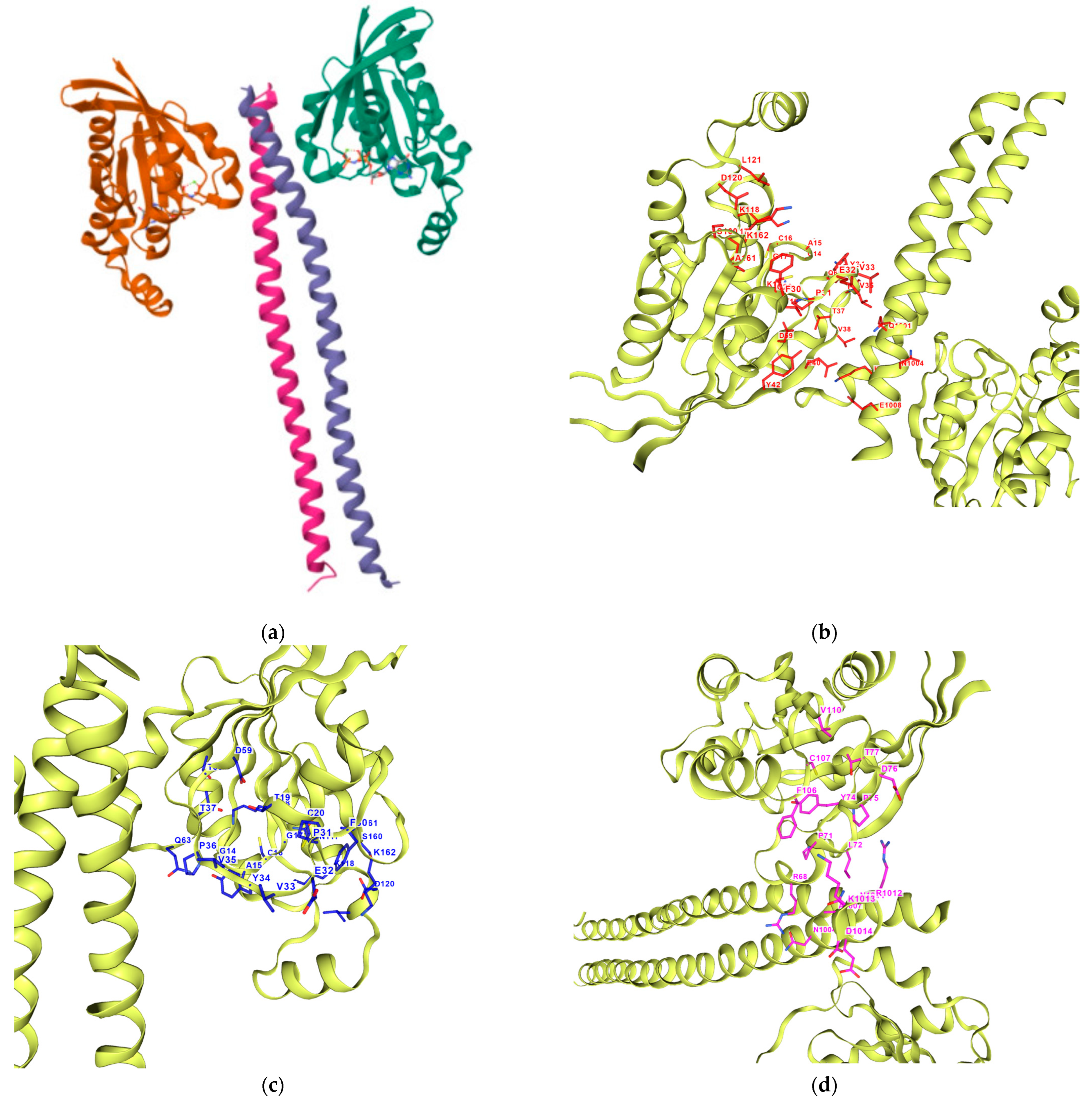
Figure 8.
Three-dimensional structure of ROCK1. (a) The retrieved X-ray crystal structure of ROCK1 (PDB ID: 1S1C) at 2.60 Å resolution from RCSB Protein Data Bank. Its cavity pockets were detected using CurPocket with their IDs from CB-Dock2: (b) C1; (c) C2; (d) C3; (e) C4; (f) C5.
Table 6.
The cavity pockets detected by CB-Dock2 for ROCK1 (PDB: 1S1C) calculated automatically using the CB-Dock2’s CurPocket detection algorithm.
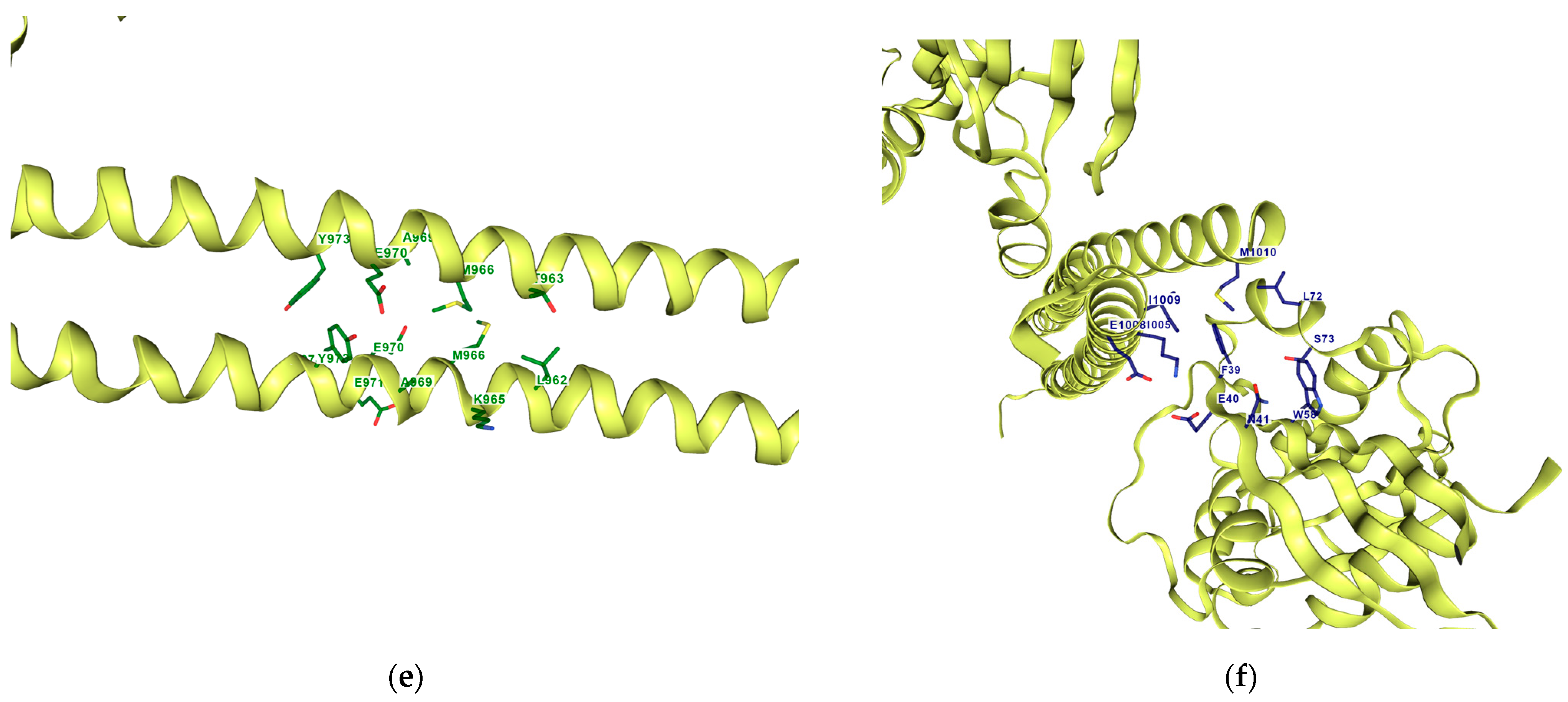
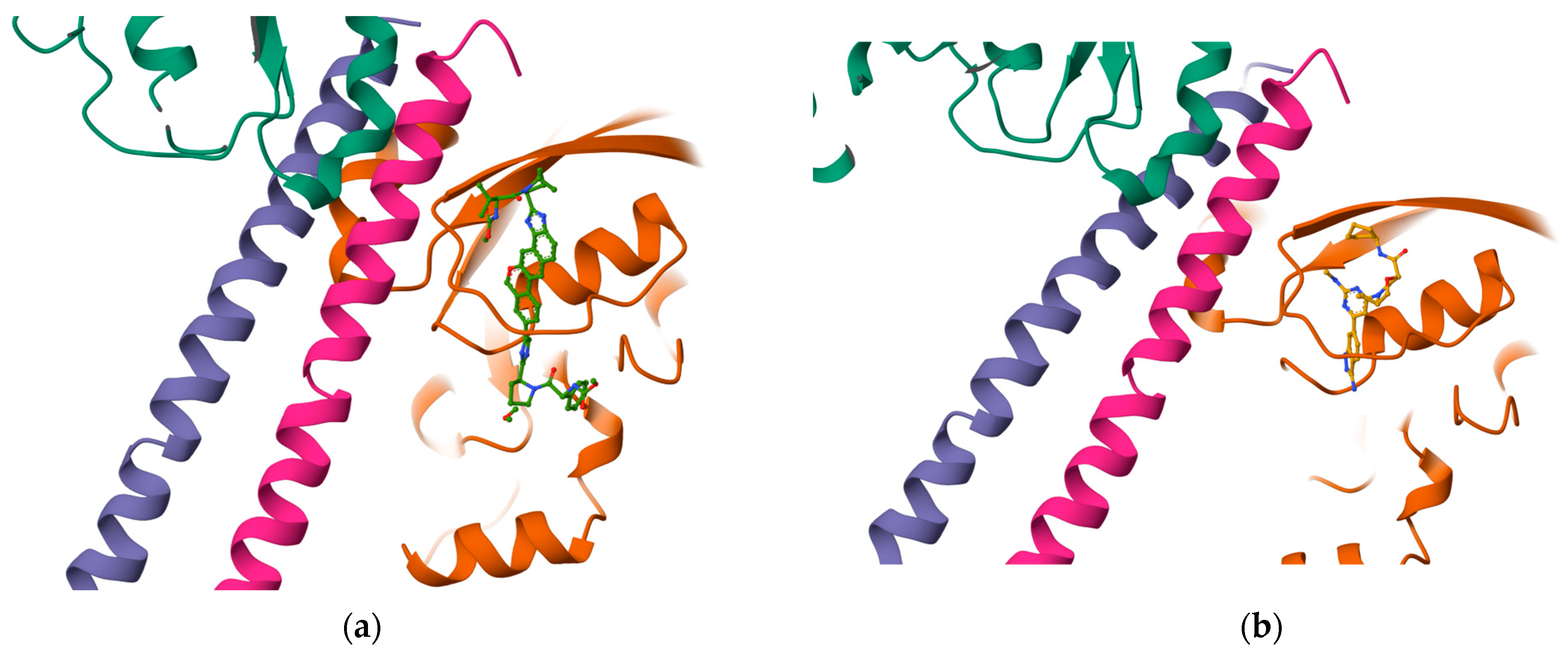
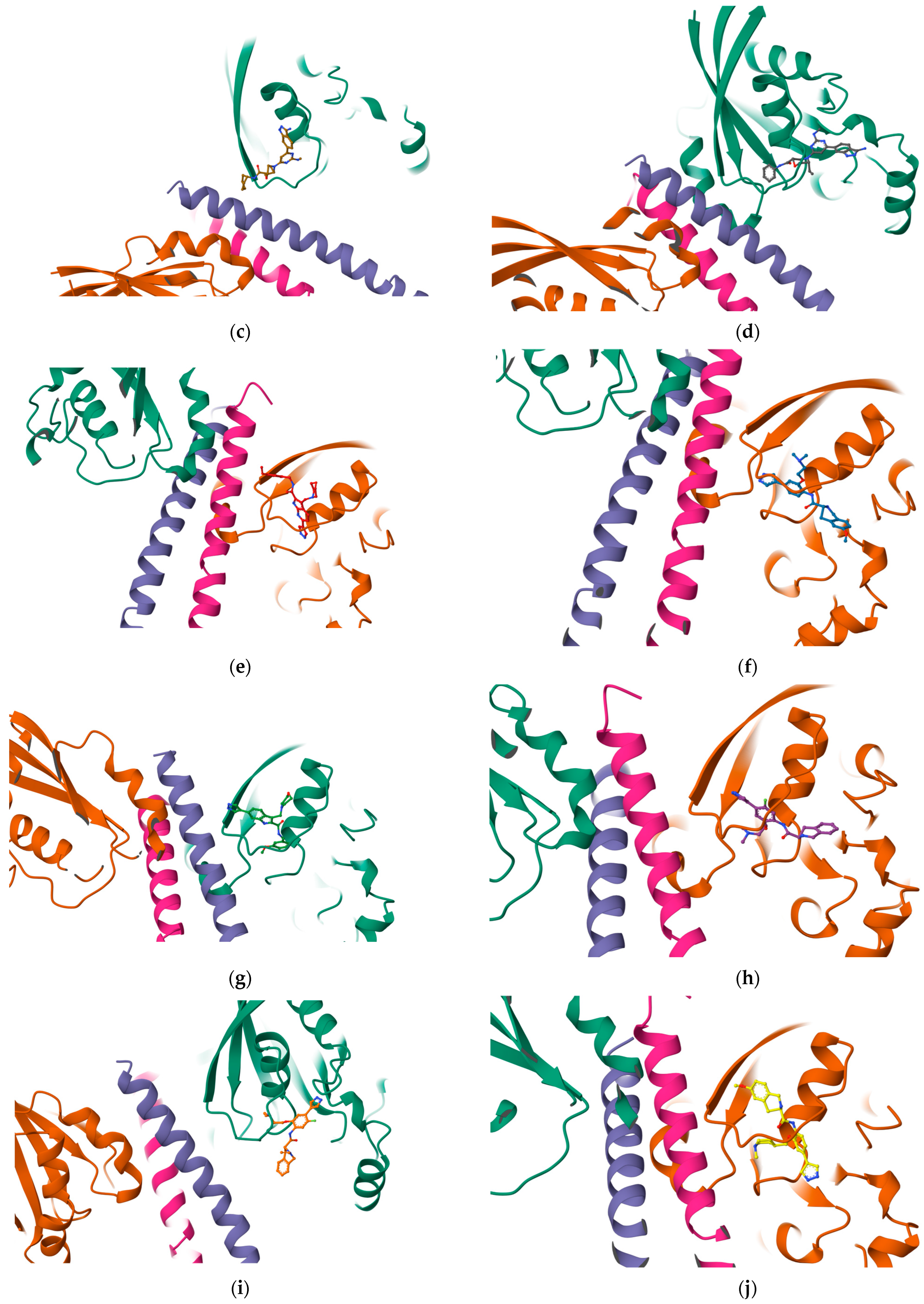
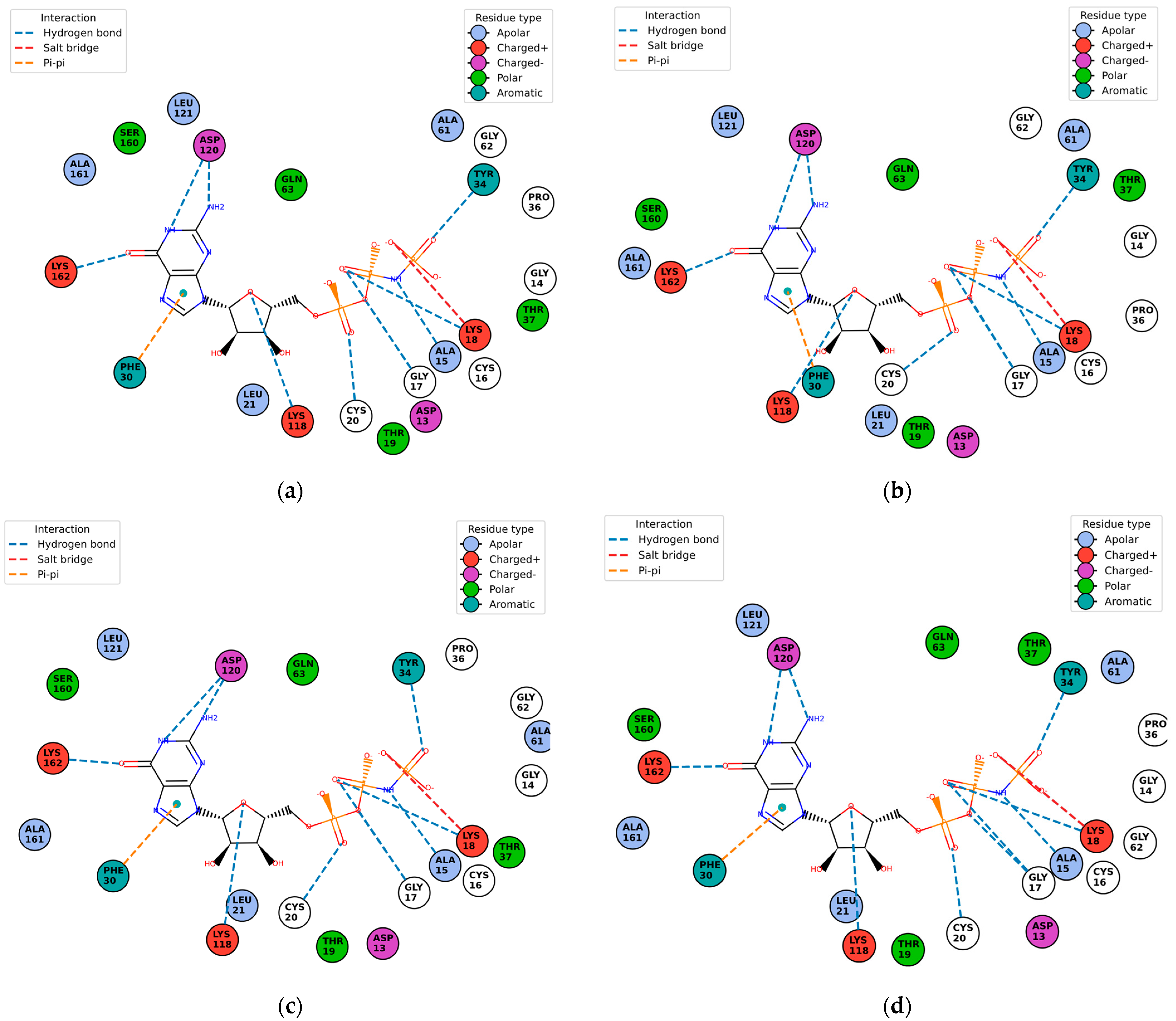
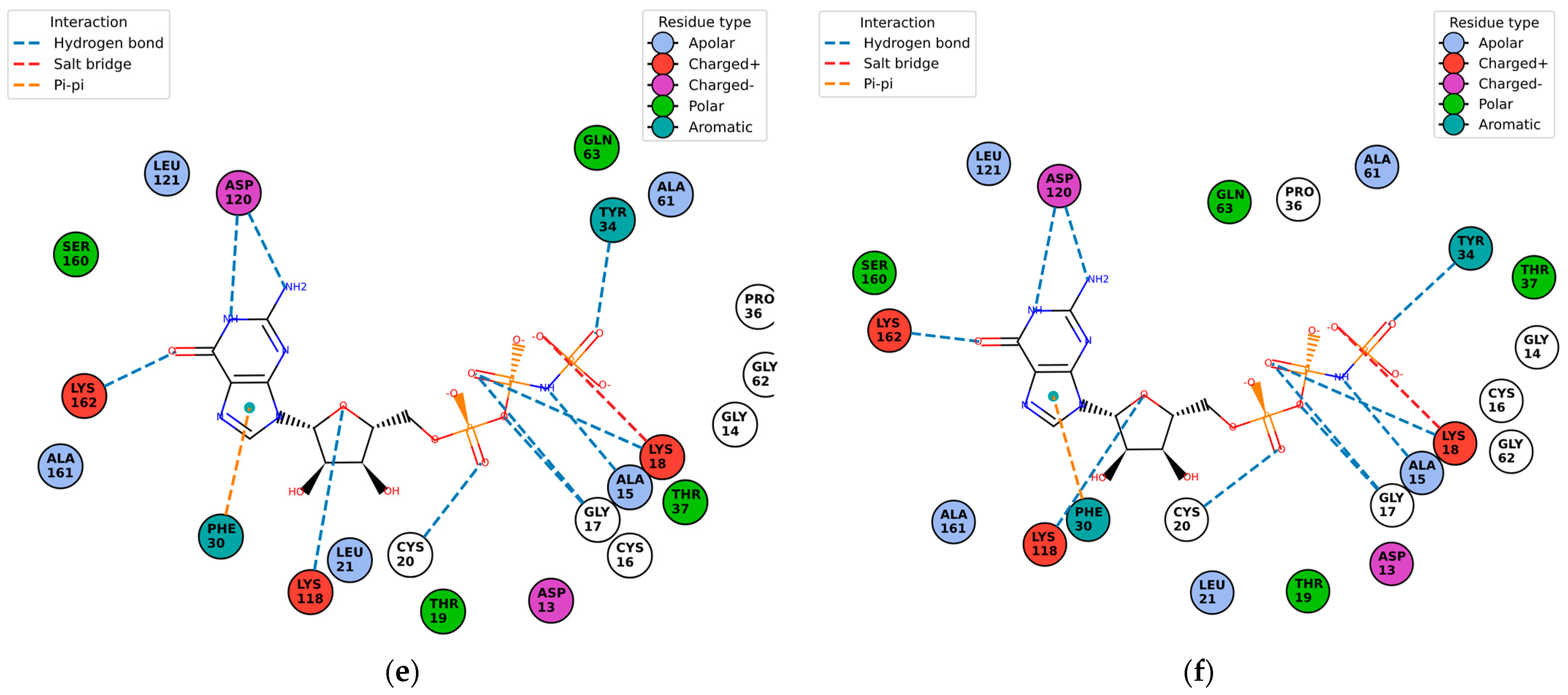
Once the detection of the pockets is complete, automatic blind docking using CB-Dock2 is run for the nine protein–ligand complexes. Figure 9 shows the best poses in each of the nine complex and velpatasvir, ranked by Autodock Vina scoring function in Table 7. Complete docking results with the ligand contact residues generated can be found in the Supplementary Material (Supplement S2 and Table S3). Contact residues and bonds of only the docked ligands at C2 pocket are represented in PlayMolecules Plexview application in Figure 10.
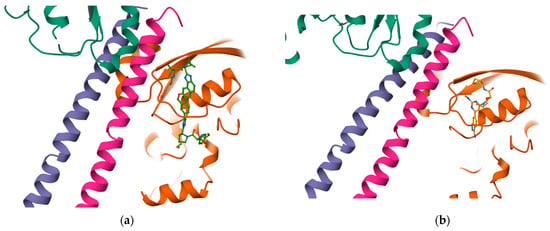
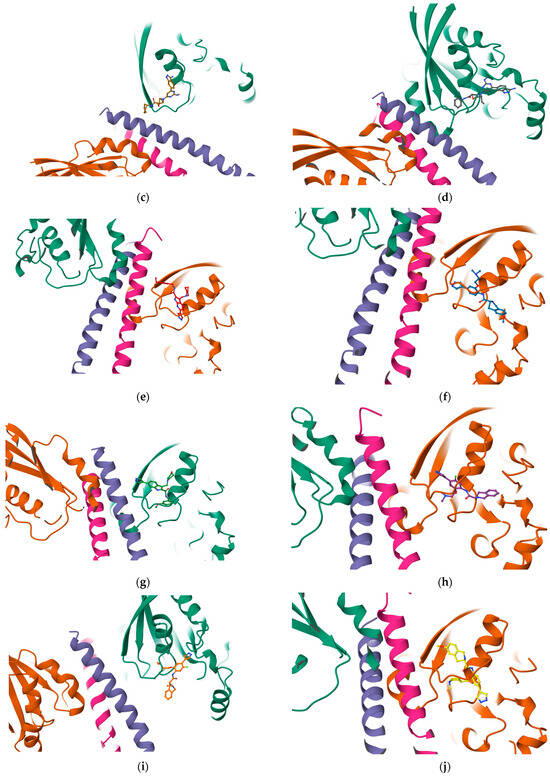
Figure 9.
Protein–ligand complexes are visualized in PlayMolecule. The top CurPockets ranked based on Autodock Vina’s scoring function in each of the protein–ligand complexes: (a) (Velpatasvir)1S1C-ZINC000203686879 at C2; (b) 1S1C-ZINC000071318464 at C2; (c) 1S1C-ZINC000071296700 at C1; (d) 1S1C-ZINC000071315829 at C1; (e) 1S1C-ZINC000073170040 at C2; (f) 1S1C-ZINC000058568630 at C2; (g) 1S1C-ZINC000073196364 at C1; (h) 1S1C-ZINC000058591055 at C2; (i) 1S1C-ZINC000058568675 at C1; (j) 1S1C-ZINC000058574949 at C2.
Table 7.
Summary of the best pockets in each of the complex using Vina scoring function highlighting that C2 is the best pocket site on the protein–ligand complexes docked on velpatasvir as the basis for their selection; full details of contact residues on specific sites and sequence chains are found in the Supplementary Material (Table S3).
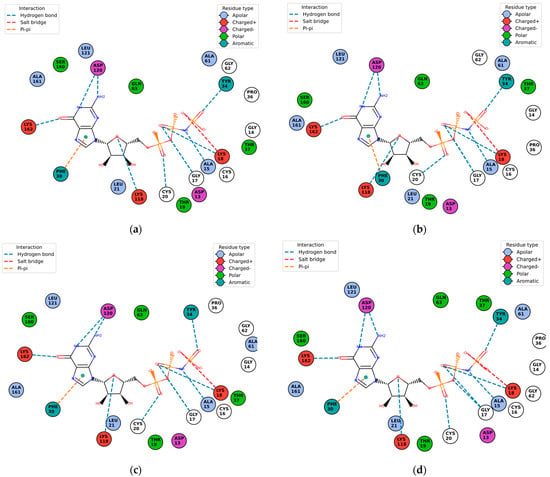
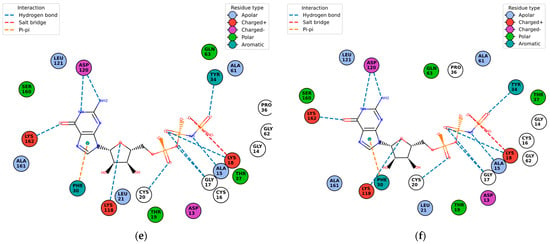
Figure 10.
Protein–ligand contact residues and bonds are in PlayMolecule’s Plexview. Only the docked complexes at the C2 site are represented, (a) (Velpatasvir)1S1C-ZINC000203686879; (b) 1S1C-ZINC000071318464; (c) 1S1C-ZINC000073170040; (d) 1S1C-ZINC000058568630; (e) 1S1C-ZINC000058591055; (f) 1S1C-ZINC000058574949.
4. Discussion
NS5B polymerase, a non-structural protein, functions as a viral polymerase for the replication process as a catalyst for synthesizing RNA based on an RNA template, generating new strands for viral replication. It converts the viral RNA into complementary RNA molecules within the replication process, thus allowing HCV to persist within the infected cells. Noviandy et al. used a QSAR-based stacked ensemble classifier to predict NS5B inhibitors using their IC50 [18]. NS3 protease, also a non-structural protein, alongside the viral cofactor NS4A peptide are also essential members for the HCV replication complexes. The NS3/4A protease has two domains: an N-terminal serine protease (AA 1-180) and an RNA helicase (AA 181-631) in the C-Terminal, which binds to the nucleic acid chains. The serine protease is considered one of the most promising HCV targets for drug development. Unfortunately, rapidly emerging resistant mutations within NS3/4A are reducing the sensitivity of drugs to protease inhibitors [19]. Ghiasi et al. developed robust and reliable QSAR models using NS3/4A protease inhibitors for IC50 prediction using the Monte Carlo technique [20]. cAMP-dependent protein kinase (PKA) phosphorylation is vital for the regulation of SR-BI expression in the liver. The inhibition of PKA can lead to the redistribution of CLDN1 from the plasma membrane and a reduction in viral entry, thus confirming its importance [30]. As described in the following study, inhibitors from NS5B polymerase (CHEMBL5375), NS3 (CHEMBL1293269), and NS3/4A (CHEMBL2095231) protease and the external validation dataset of cAMP-dependent protein kinase (PKA) enzyme are the basis for inclusion in building and externally validating the model.
In the context of HCV viral infection, a crucial aspect is the requirement for an entry factor. CLDN1 is highly present in the liver and is one of the key factors for entry, along with SRB1, CD81, and occludin. Evans et al. showed that expressing claudin-1 in non-hepatic cell lines makes them vulnerable to HCV infection. Claudin-1’s involvement is integral not only as an entry factor but also in the late-stage process following interaction with HCV and CD81 [9,10]. HCV particles specifically bind to the entry factors SR-BI and CD81 tetraspanin. The binding to CD81 triggers a diffusion of the virion-CD81 complex across the plasma membrane, heading towards sites for viral internalization. Consequently, claudin-1 plays a crucial role in the late-stage entry processes by interacting with CD81, especially in areas enriched with tetraspanin. CD81–claudin-1 complex actively participates in HCV endocytosis. These observations underscore the significant role of TJ proteins, particularly claudin-1, in the viral entry of HCV. However, much remains to be explored regarding the mechanisms of these proteins, and their regulation in the liver is yet to be fully understood. Limited information is available on claudin-1 trafficking and its interactions with other factors, such as CD81 and OCLN [62]. One of the main pathways for TJ disruption is ROCK1; this is also called the Rho/ROCK pathway. ROCK1 and ROCK2 are both downstream effectors that regulate multiple cellular functions, e.g., cell migration, adhesion, polarity, and proliferation. When ROCK1 activity levels are moderate, the formation and arrangement of TJ proteins occur. However, elevated levels lead to TJ defects. This pathway is key in molecular signal transduction. They are involved in TJ formation, intestinal permeability, and inflammation. ROCK1 is thus considered a key protein in the said pathway [29,63,64].
The negative skewed values of train, test, and validation sets in the statistical measurements indicate that the distribution of y in the sets have the characteristic of a longer tail on its left distribution; this is also representative in the histogram plot. Their distribution leans heavily on the right side of the curve. The distribution in the dataset can be considered leptokurtic, where a positive value (kurtosis > 0) shows that distribution has heavier tails and more peaked central regions. Outliers in the plot are also detected, as some values of y are clearly smaller or bigger than others. Initial analysis of y can help whether or not to check for these outliers and if the researcher chooses to remove them when inputting the data in ML models. In the following context, all values are kept for the prevention of the loss of data. These measurements of y are used for checking and comparative analysis with the external dataset (PKA).
NuSVR, as the chosen model, is a type of support vector machine for regression. This ML algorithm focuses on controlling the margin of errors by using support vectors. These support vectors are data points that have the most impact in determining the most optimal regression line. NuSVR uses the parameter nu to control the number of these support vectors, where nu is an upper bound on the fraction of training errors and a lower bound on the fraction of support vectors [26,43,46,48,49]. When the external dataset is compared with the HCV-ROCK1 set, the distribution of y in the following dataset is more skewed to the right, as indicated in its positive skewness value, and a negative kurtosis value indicates that this external dataset expresses a lighter tail and a flatter distribution, which is considered a platykurtic distribution; see Table 1. The R2 performance for this dataset displayed a negative score of −0.21870. This is slightly expected as support vector machines like NuSVR are non-linear regression models; this outcome arises when the predictions made on this set, compared to the actual outcome, have not been derived from model fitting using these data. This negative case may still arise since NuSVR uses a non-linear function in its algorithm. The chosen model still poses a great challenge for generalizability in using external datasets, including the following: ensuring all parameters are accounted for, such as tuning to the most optimized hyperparameters for support vector machines; the size of the training data; the compatibility of the chosen model and its decision space algorithm with the external dataset; and possibility that the response variable (pIC50) is only loosely associated with the features (e.g., molecular descriptors and fingerprints). Since the study has limited computational resources in its capabilities, these challenges are met whenever any ML model requires good transferability to other datasets.
For the compound to be an effective drug, it must reach its target in sufficient amounts of concentration and stay in its bioactive form long enough to elicit the expected biological events to happen. The molecules must exhibit high biological activity and low toxicity in the body. SwissADME reports the bioavailability radar to provide an overview of the drug likeness of the compounds of interest. Each property represents the following: LIPO (lipophilicity between −0.7 and +5.0 for XLOGP3), SIZE (MW between 150 and 500 g/mol), POLAR (polarity between 20 and 130 Å2 for TPSA), INSOLU (solubility logS not higher than 6), INSATU (fraction of carbons sp3 hybridized not less than 0.25), and FLEX (rotatable bonds must not exceed 9) [52]. The compounds that did not pass in this section are excluded in molecular docking analysis in Table 5; complete details of ADME properties are provided in the Supplementary Material (Figure S1: Bioavailability Radar). Many drug discovery and development failures are due to pharmacokinetics and bioavailability; gastrointestinal (GI) and brain access are behaviors that must be taken into account for estimation in the stages of such discovery processes.
The general scoring function of AutoDock Vina is represented in Equation (6).
where the sum of all pairs of atoms (i) that can move relative to each other, normally excluding atoms separated by three covalent bonds. This is assigned as type (ti), with a symmetric set of interaction functions (ftitj) of the distance (interatomic) (rij) defined. The optimization algorithm used finds the global minimum of c and other low-scoring confirmations and is ranked; then, the predicted free energy binding is calculated from the intermolecular part of the lowest-scoring confirmation in Equation (7).
where g can be any arbitrary strictly increasing smooth (possibly linear) function. CB-Dock2 uses the updated version of Vina scoring from version 1.2.0 of AutoDock Vina. The search algorithm continues to be used, but not with new features from AutoDock4 (AD4), such as batch processing and simultaneous docking of ligands. The search algorithm of Vina uses a Monte Carlo (MC) iterative search combined with the Broyden–Fletcher–Goldfarb–Shanno (BFGS) method for local optimization as a gradient-based local optimization. This allows MC to explore a broader conformational space, which is refined by BFGS to converge to a more energetically favorable structure.
The results reported in Table 7 suggest that C2 is the best-scoring pocket for velpatasvir. For the protein–ligand complexes, the following pocket IDs were observed: 1S1C-ZINC000071318464 at C2, 1S1C-ZINC000071296700 at C1, 1S1C-ZINC000071315829 at C1, 1S1C-ZINC000073170040 at C2, 1S1C-ZINC000058568630 at C2, 1S1C-ZINC000073196364 at C1, 1S1C-ZINC000058591055 at C2, 1S1C-ZINC000058568675 at C1, 1S1C-ZINC000058574949 at C2.
The results suggest that C2 is the best-ranked pocket ID for the complexes. The ligands for C2 are as follows: ZINC000071318464, ZINC000073170040, ZINC000058568630, ZINC000058591055, and ZINC000058574949. The ligand ZINC000071318464 is the only one that is considered as an intermediate in its bioactivity class; the rest are active (see Table 5). The five ligands that have the best scoring Vina for C2 all have the following structural properties embedded in their MACCS keys gathered from feature importance on the selected model. Figure 10 shows the contact residues of the complexes at the docked C2 site, which shows the H-bond, π–π, and salt bridge interactions between the residues and ligands [65]. The following residues interacting for velpatasvir at chain sequence B revealed the H-bond interactions at −ASP120, +LYS162, +LYS118, CYS20, GLY17, non-polar ALA15, +LYS18, and aromatic TYR34. π–π interaction was observed on the aromatic ring for PHE30. Hydrophobic interactions for the residues ALA161, LEU121, ALA61, LEU21 GLY62, and PRO36 were also observed. H-bond interactions are key in binding specificity while hydrophobic interactions contribute to binding affinity. The mobility and functional orientation of the hydrogen bonding hydrogens are of great pharmacological interest for influencing ligand bindings [66]. For the rest of the five screened ligands that show favorable energy at C2, such as ZINC000071318464, consistent residues paralleling that of velpatasvir on chain sequences B and Y were observed. Immune cells that are activated are dependent on enough SER and differentiation of T cells. GLN is essential for cell proliferation and immune system cells as GLN is a precursor in amino sugars and nucleotides. CYS availability has a key role in T cell functions because T cells do not have enzymes converting MET to CYS [67].
It is worth noting that these structural properties (e.g., an aromatic nitrogen atom that is part of an aromatic ring system, the presence of a quaternary ammonium cation, the presence of a quaternary nitrogen atom (-N) that is bonded to three other atoms, etc.) can be used as a screening criterion for finding other compounds of interest that may have the same embedded molecular fingerprints from QSAR-ML and molecular docking findings in the study. Selecting a value of pIC50 > 5 can also be taken into consideration for this inhibitor property (see Table 5). Drug–drug interactions can be predicted using similarity tests such as the cosine method, used to calculate the feature similarity of drugs and compounds where a high dimensionality vector is constructed; the five compounds were subjected to the following method to further probe their structural similarities alongside the already approved DAA for HCV treatments; Supplementary Material (Supplement S1) [68].
Currently, DAA combinations of sofosbuvir/daclatasvir and sofosbuvir/velpatasvir are the recommended antiviral treatments in the Philippines [1,8]. Dasabuvir is also another drug used to treat HCV in combination with other antivirals such as ombitasvir, paritprevir, and ritonavir. However, unlike sofosbuvir, dasabuvir is not used in the country for treatment [7]. In the context of ROCK1 inhibition, using these drugs as our metric can be beneficial for finding other drug compounds for HCV treatment. Identifying potential ROCK1 inhibitors could expand HCV therapeutic strategies. The structural information of ROCK1 can be exploited to develop these inhibitors. Screening compounds from chemical libraries for their impact on the protein structure and their bioactivity may lead to promising candidates [15].
5. Conclusions
The main motivation behind this study lies in the ever-increasing prevalence of drug-resistant HCV variants, propelling explorations of novel molecules specifically targeting enzymes like ROCK1, for which minimal studies have been conducted. A consideration to take when performing QSAR-ML is the limited research on ROCK1 and its inhibitors. The usage of datasets that did not use ROCK1 as its bioactivity assay for determining pIC50 represents a challenge, as the correlation between the following parameter and features of the inhibitors may vary. Moreover, the sample size is a challenge, resulting in a less generalized ML model. The presented results show the criteria a compound must meet in drug screening for ROCK1. Docking results highlight C2 as the best-ranked pocket for ligand binding. The ligands that exhibited the best scores on C2, paralleling those in the control (velpatasvir), are as follows: ZINC000071318464, ZINC000073170040, ZINC000058568630, ZINC000058591055, and ZINC000058574949. They have satisfied all of the requirements for their ADME properties, and the qualities they have exhibited can be considered drug-like. The structural properties of the compounds presented by the QSAR-ML with molecular docking provide valuable findings for identifying other potential candidates that inhibit ROCK1. The results in the study offer substantial utility in discovering compounds for ROCK1 inhibition. This information can guide the identification of significant binding properties and can contribute to the still ongoing drug discovery efforts involving ROCK1 inhibitors.
To enhance the generalizability of the ML model, researchers in future studies may opt for improved hyperparametization in the model, a technique that allows for the exploration of improved search spaces and optimization of model parameters. Additionally, employing multiple iterative processes to determine feature importance can contribute to a more refined model. While the present study faces constraints in terms of computational resources, future research endeavors in the domain of QSAR-ML, molecular docking and molecular dynamics may leverage the techniques and protocol outlined herein and further refine them according to their unique requirements. A substantial amount of attention should be dedicated to meticulously preparing the dataset, as the challenges encountered in this study primarily stem from the pre-processing of raw data sourced from chemical databases. The actions taken by researchers during the pre-processing stages wield significant influence over the subsequent phases of their investigation. As such, meticulous dataset preparation is imperative for laying a robust foundation for the entirety of the study. Another validation method involves performing molecular dynamics simulation on the protein–ligand complexes. This approach can validate the energy stability using the QM/MM inputs during simulation runs, providing a more accurate evaluation.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/computation12090175/s1: Supplement S1: Code and Dataset Repository; Supplement S2: Molecular Docking Files; Supplement S3: Molecular Dynamics Files; Figure S1: Bioavailability Radar; Figure S2: Chain Sequences; Table S1: Inhibitor Drugs; Table S2: Compounds of Interest; Table S3: CB-Dock2 Results.
Author Contributions
Conceptualization, J.R.D.B. and H.S.C.; methodology, J.R.D.B.; software, J.R.D.B.; validation, J.R.D.B. and H.S.C.; formal analysis, J.R.D.B.; investigation, J.R.D.B.; resources, J.R.D.B.; data curation, J.R.D.B.; writing—original draft preparation, J.R.D.B.; writing—review and editing, J.R.D.B. and H.S.C.; visualization, J.R.D.B.; supervision, H.S.C.; project administration, H.S.C.; funding acquisition, H.S.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
All data are presented in the main text and in the Supplementary Files.
Acknowledgments
We would like to acknowledge the School of Chemical, Biological, and Materials Engineering and Sciences (CBMES) as well as the Department of Biology, School of Health Sciences at Mapúa University for giving us this great opportunity to submit the following article in this journal to be peer reviewed.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Ornos, E.D.; Murillo, K.J.; Ong, J.P. Liver Diseases: Perspective from the Philippines. Ann. Hepatol. 2023, 28, 101085. [Google Scholar] [CrossRef] [PubMed]
- Yanase, Y.; Ohida, T.; Kaneita, Y.; Agdamag, D.M.D.; Leaño, P.S.A.; Gill, C.J. The Prevalence of HIV, HBV and HCV among Filipino Blood Donors and Overseas Work Visa Applicants. Bull. World Health Organ. 2007, 85, 131–137. [Google Scholar] [CrossRef]
- The Polaris Observatory HCV Collaborators. Global Change in Hepatitis C Virus Prevalence and Cascade of Care between 2015 and 2020: A Modelling Study. Lancet Gastroenterol. Hepatol. 2022, 7, 396–415. [Google Scholar] [CrossRef] [PubMed]
- Salari, N.; Kazeminia, M.; Hemati, N.; Ammari-Allahyari, M.; Mohammadi, M.; Shohaimi, S. Global Prevalence of Hepatitis C in General Population: A Systematic Review and Meta-Analysis. Travel Med. Infect. Dis. 2022, 46, 102255. [Google Scholar] [CrossRef] [PubMed]
- Manns, M.P.; Buti, M.; Gane, E.; Pawlotsky, J.M.; Razavi, H.; Terrault, N.; Younossi, Z. Hepatitis C Virus Infection. Nat. Rev. Dis. Primers 2017, 3, 17006. [Google Scholar] [CrossRef]
- Tsoi, W.C.; Simpson, C.; Jarvis, L.; Smith, A.; Robbins, N.; Sepetiene, R.; Bhatnagar, S. Multicenter Evaluation of the New Alinity s Anti-HCV II Assay for Routine Hepatitis C Virus Blood Screening. J. Clin. Virol. Plus 2023, 3, 100136. [Google Scholar] [CrossRef]
- Lim, S.G.; Aghemo, A.; Chen, P.J.; Dan, Y.Y.; Gane, E.; Gani, R.; Gish, R.G.; Guan, R.; Jia, J.D.; Lim, K.; et al. Management of Hepatitis C Virus Infection in the Asia-Pacific Region: An Update. Lancet Gastroenterol. Hepatol. 2017, 2, 52–62. [Google Scholar] [CrossRef]
- Espinosa, W.Z.; Jamias, J.D.; Limquiaco, J.L.; Macatula, T.C.; Sofia, K.; Calixto-Mercado, M.; Ong, J.P.; Tripon, E.S. 2020 Update to the Consensus Statements on the Diagnosis and Treatment of Hepatitis B: Special Populations Hepatology Society of the Philippines (HSP) Hepatitis B Virus (HBV) Consensus Core Group; Hepatology Society of the Philippines: Quezon, Philippines, 2020. [Google Scholar]
- Carriquí-Madroñal, B.; Lasswitz, L.; von Hahn, T.; Gerold, G. Genetic and Pharmacological Perturbation of Hepatitis-C Virus Entry. Curr. Opin. Virol. 2023, 62, 101362. [Google Scholar] [CrossRef]
- Evans, M.J.; Von Hahn, T.; Tscherne, D.M.; Syder, A.J.; Panis, M.; Wölk, B.; Hatziioannou, T.; McKeating, J.A.; Bieniasz, P.D.; Rice, C.M. Claudin-1 Is a Hepatitis C Virus Co-Receptor Required for a Late Step in Entry. Nature 2007, 446, 801–805. [Google Scholar] [CrossRef]
- Jiang, Y.; Song, J.; Xu, Y.; Liu, C.; Qian, W.; Bai, T.; Hou, X. Piezo1 Regulates Intestinal Epithelial Function by Affecting the Tight Junction Protein Claudin-1 via the ROCK Pathway. Life Sci. 2021, 275, 119254. [Google Scholar] [CrossRef]
- Elamin, E.; Masclee, A.; Dekker, J.; Jonkers, D. Ethanol Disrupts Intestinal Epithelial Tight Junction Integrity through Intracellular Calcium-Mediated Rho/ROCK Activation. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 306, G677–G685. [Google Scholar] [CrossRef]
- Utech, M.; Ivanov, A.I.; Samarin, S.N.; Bruewer, M.; Turner, J.R.; Mrsny, R.J.; Parkos, C.A.; Nusrat, A. Mechanism of IFN-γ-Induced Endocytosis of Tight Junction Proteins: Myosin II-Dependent Vacuolarization of the Apical Plasma Membrane. Mol. Biol. Cell 2005, 16, 5040–5052. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Jiang, Y.; Zhou, H.; Zhao, X.; Li, M.; Bao, Z.; Wang, Z.; Zhang, C.; Xie, Z.; Zhao, J.; et al. Dasabuvir Suppresses Esophageal Squamous Cell Carcinoma Growth In Vitro and In Vivo through Targeting ROCK1. Cell Death Dis. 2023, 14, 118. [Google Scholar] [CrossRef] [PubMed]
- Alotaibi, B.S.; Joshi, J.; Hasan, M.R.; Khan, M.S.; Alharethi, S.H.; Mohammad, T.; Alhumaydhi, F.A.; Elasbali, A.M.; Hassan, M.I. Identifying Isoononin and Candidissiol as Rho-Associated Protein Kinase 1 (ROCK1) Inhibitors: A Combined Virtual Screening and MD Simulation Approach. J. Biomol. Struct. Dyn. 2023, 41, 6749–6758. [Google Scholar] [CrossRef]
- Kumar, N.; Kaur, K.; Bedi, P.M.S. Hybridization of Molecular Docking Studies with Machine Learning Based QSAR Model for Prediction of Xanthine Oxidase Activity. Comput. Theor. Chem. 2023, 1227, 114262. [Google Scholar] [CrossRef]
- Lage, O.M.; Ramos, M.C.; Calisto, R.; Almeida, E.; Vasconcelos, V.; Vicente, F. Current Screening Methodologies in Drug Discovery for Selected Human Diseases. Mar. Drugs 2018, 16, 279. [Google Scholar] [CrossRef]
- Noviandy, T.R.; Maulana, A.; Idroes, G.M.; Irvanizam, I.; Subianto, M.; Idroes, R. QSAR-Based Stacked Ensemble Classifier for Hepatitis C NS5B Inhibitor Prediction. In Proceedings of the 2023 2nd International Conference on Computer System, Information Technology, and Electrical Engineering (COSITE), Banda Aceh, Indonesia, 2–3 August 2023. [Google Scholar]
- Lafridi, H.; Almalki, F.A.; Ben Hadda, T.; Berredjem, M.; Kawsar, S.M.A.; Alqahtani, A.M.; Esharkawy, E.R.; Lakhrissi, B.; Zgou, H. In Silico Evaluation of Molecular Interactions between Macrocyclic Inhibitors with the HCV NS3 Protease. Docking and Identification of Antiviral Pharmacophore Site. J. Biomol. Struct. Dyn. 2023, 41, 2260–2273. [Google Scholar] [CrossRef]
- Ghiasi, T.; Ahmadi, S.; Ahmadi, E.; Talei Bavil Olyai, M.R.; Khodadadi, Z. The Index of Ideality of Correlation: QSAR Studies of Hepatitis C Virus NS3/4A Protease Inhibitors Using SMILES Descriptors. SAR QSAR Environ. Res. 2021, 32, 495–520. [Google Scholar] [CrossRef] [PubMed]
- Ikram, S.; Ahmad, J.; Rehman, I.U.; Durdagi, S. Potent Novel Inhibitors against Hepatitis C Virus NS3 (HCV NS3 GT-3a) Protease Domain. J. Mol. Graph. Model. 2020, 101, 107727. [Google Scholar] [CrossRef]
- Aykul, S.; Martinez-Hackert, E. Determination of Half-Maximal Inhibitory Concentration Using Biosensor-Based Protein Interaction Analysis. Anal. Biochem. 2016, 508, 97–103. [Google Scholar] [CrossRef]
- Swinney, D.C. Molecular Mechanism of Action (MMoA) in Drug Discovery. Annu. Rep. Med. Chem. 2011, 46, 301–317. [Google Scholar] [CrossRef]
- Rolon-Mérette, D.; Ross, M.; Rolon-Mérette, T.; Church, K. Introduction to Anaconda and Python: Installation and Setup. Quant. Methods Psychol. 2020, 16, S3–S11. [Google Scholar] [CrossRef]
- Pérez, F.; Granger, B.E. IPython: A System for Interactive Scientific Computing. Comput. Sci. Eng. 2007, 9, 21–29. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-Learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Google Colaboratory. Available online: https://colab.research.google.com/ (accessed on 26 November 2023).
- Qiao, R.; Tang, W.; Li, J.; Li, C.; Zhao, C.; Wang, X.; Li, M.; Cui, Y.; Chen, Y.; Cai, G.; et al. Structure-Based Virtual Screening of ROCK1 Inhibitors for the Discovery of Enterovirus-A71 Antivirals. Virology 2023, 585, 205–214. [Google Scholar] [CrossRef]
- Liu, Z.; Li, C.; Chen, S.; Lin, H.; Zhao, H.; Liu, M.; Weng, J.; Liu, T.; Li, X.; Lei, C.; et al. MicroRNA-21 Increases the Expression Level of Occludin through Regulating ROCK1 in Prevention of Intestinal Barrier Dysfunction. J. Cell. Biochem. 2019, 120, 4545–4554. [Google Scholar] [CrossRef]
- Farquhar, M.J.; Harris, H.J.; Diskar, M.; Jones, S.; Mee, C.J.; Nielsen, S.U.; Brimacombe, C.L.; Molina, S.; Toms, G.L.; Maurel, P.; et al. Protein Kinase A-Dependent Step(s) in Hepatitis C Virus Entry and Infectivity. J. Virol. 2008, 82, 8797–8811. [Google Scholar] [CrossRef]
- Ho, S.Y.; Phua, K.; Wong, L.; Bin Goh, W.W. Extensions of the External Validation for Checking Learned Model Interpretability and Generalizability. Patterns 2020, 1, 100129. [Google Scholar] [CrossRef]
- Rácz, A.; Bajusz, D.; Héberger, K. Effect of Dataset Size and Train/Test Split Ratios in Qsar/Qspr Multiclass Classification. Molecules 2021, 26, 1111. [Google Scholar] [CrossRef]
- James, G.; Witten, D.; Hastie, T.; Tibshirani, R. An Introduction to Statistical Learning—With Applications in R; Springer: New York, NY, USA, 2013. [Google Scholar]
- Team, R.C. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2021. [Google Scholar]
- Moriwaki, H.; Tian, Y.S.; Kawashita, N.; Takagi, T. Mordred: A Molecular Descriptor Calculator. J. Cheminformatics 2018, 10, 4. [Google Scholar] [CrossRef]
- Little, R.J.A.; Rubin, D.B. Statistical Analysis with Missing Data; John Wiley & Sons: Hoboken, NJ, USA, 2014. [Google Scholar]
- Soltani, S.; Abolhasani, H.; Zarghi, A.; Jouyban, A. QSAR Analysis of Diaryl COX-2 Inhibitors: Comparison of Feature Selection and Train-Test Data Selection Methods. Eur. J. Med. Chem. 2010, 45, 2753–2760. [Google Scholar] [CrossRef]
- Hastie, T.; Tibshirani, R.; James, G.; Witten, D. An Introduction to Statistical Learning, 2nd ed.; Springer: New York, NY, USA, 2021; Volume 102. [Google Scholar]
- Kokoska, S.; Zwillinger, D. CRC Standard Probability and Statistics Tables and Formulae, Student Edition; CRC Press: Boca Raton, FL, USA, 2000. [Google Scholar]
- Cameron, A.C.; Windmeijer, F.A.G. An R-Squared Measure of Goodness of Fit for Some Common Nonlinear Regression Models. J. Econom. 1997, 77, 329–342. [Google Scholar] [CrossRef]
- Heijmans, R.D.H.; Neudecker, H. The Coefficient of Determination Revisited. In The Practice of Econometrics; Springer: Dordrecht, The Netherlands, 1987. [Google Scholar]
- Hyndman, R.J.; Koehler, A.B. Another Look at Measures of Forecast Accuracy. Int. J. Forecast. 2006, 22, 679–688. [Google Scholar] [CrossRef]
- Platt, J. Probabilistic Outputs for Support Vector Machines and Comparisons to Regularized Likelihood Methods. Adv. Large Margin Classif. 1999, 10, 61–74. [Google Scholar]
- Wu, T.F.; Lin, C.J.; Weng, R.C. Probability Estimates for Multi-Class Classification by Pairwise Coupling. J. Mach. Learn. Res. 2004, 5, 975–1005. [Google Scholar]
- Fan, R.E.; Chang, K.W.; Hsieh, C.J.; Wang, X.R.; Lin, C.J. LIBLINEAR: A Library for Large Linear Classification. J. Mach. Learn. Res. 2008, 9, 1871–1874. [Google Scholar]
- Chang, C.C.; Lin, C.J. LIBSVM: A Library for Support Vector Machines. ACM Trans. Intell. Syst. Technol. 2011, 2, 1–27. [Google Scholar] [CrossRef]
- Nasrabadi, N.M. Pattern Recognition and Machine Learning. J. Electron. Imaging 2007, 16, 049901. [Google Scholar] [CrossRef]
- Smola, A.J.; Schölkopf, B. A Tutorial on Support Vector Regression. Stat. Comput. 2004, 14, 199–222. [Google Scholar] [CrossRef]
- Schölkopf, B.; Smola, A.J.; Williamson, R.C.; Bartlett, P.L. New Support Vector Algorithms. Neural Comput. 2000, 12, 1207–1245. [Google Scholar] [CrossRef]
- Crammer, K.; Singer, Y. On the Algorithmic Implementation of Multiclass Kernel-Based Vector Machines. J. Mach. Learn. Res. 2001, 2, 265–292. [Google Scholar]
- Lin, L.I.-K. A Concordance Correlation Coefficient to Evaluate Reproducibility. Biometrics 1989, 45, 255–268. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Dvorsky, R.; Blumenstein, L.; Vetter, I.R.; Ahmadian, M.R. Structural Insights into the Interaction of ROCKI with the Switch Regions of RhoA. J. Biol. Chem. 2004, 279, 7098–7104. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yang, X.; Gan, J.; Chen, S.; Xiao, Z.X.; Cao, Y. CB-Dock2: Improved Protein-Ligand Blind Docking by Integrating Cavity Detection, Docking and Homologous Template Fitting. Nucleic Acids Res. 2022, 50, W159–W164. [Google Scholar] [CrossRef]
- Liu, Y.; Grimm, M.; Dai, W.; Hou, M.; Xiao, Z.X.; Cao, Y. CB-Dock: A Web Server for Cavity Detection-Guided Protein–Ligand Blind Docking. Acta Pharmacol. Sin. 2020, 41, 138–144. [Google Scholar] [CrossRef]
- Sterling, T.; Irwin, J.J. ZINC 15—Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef]
- Feng, C.; Wang, H.; Lu, N.; Chen, T.; He, H.; Lu, Y.; Tu, X.M. Log-Transformation and Its Implications for Data Analysis. Shanghai Arch. Psychiatry 2014, 26, 105–109. [Google Scholar] [CrossRef]
- Daina, A.; Zoete, V. A BOILED-Egg To Predict Gastrointestinal Absorption and Brain Penetration of Small Molecules. ChemMedChem 2016, 11, 1117–1121. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Li, L. Improved Protein-Ligand Binding Affinity Prediction by Using a Curvature-Dependent Surface-Area Model. Bioinformatics 2014, 30, 1674–1680. [Google Scholar] [CrossRef] [PubMed]
- Riva, L.; Song, O.; Prentoe, J.; Helle, F.; L’homme, L.; Gattolliat, C.-H.; Vandeputte, A.; Fénéant, L.; Belouzard, S.; Baumert, T.F.; et al. Identification of Piperazinylbenzenesulfonamides as New Inhibitors of Claudin-1 Trafficking and Hepatitis C Virus Entry. J. Virol. 2018, 92, 10. [Google Scholar] [CrossRef] [PubMed]
- Marchiando, A.M.; Shen, L.; Graham, W.V.; Edelblum, K.L.; Duckworth, C.A.; Guan, Y.; Montrose, M.H.; Turner, J.R.; Watson, A.J.M. The epithelial barrier is maintained by in vivo tight junction expansion during pathologic intestinal epithelial shedding. Gastroenterology 2011, 140, 1208–1218. [Google Scholar] [CrossRef]
- Chen, Y.; Huang, W.; Jiang, W.; Wu, X.; Ye, B.; Zhou, X. HIV-1 Tat Regulates Occludin and Aβ Transfer Receptor Expression in Brain Endothelial Cells via Rho/ROCK Signaling Pathway. Oxid. Med. Cell. Longev. 2016, 2016, 4196572. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Rosell, G.; Giorgino, T.; De Fabritiis, G. PlayMolecule ProteinPrepare: A Web Application for Protein Preparation for Molecular Dynamics Simulations. J. Chem. Inf. Model. 2017, 57, 1511–1516. [Google Scholar] [CrossRef]
- Wade, R.C.; Goodford, P.J. The role of hydrogen bonds in drug binding. Prog. Clin. Biol. Res. 1989, 289, 433–444. [Google Scholar]
- Tomé, D. Amino Acid Metabolism and Signalling Pathways: Potential Targets in the Control of Infection and Immunity. Nutr. Diabetes 2021, 11, 20. [Google Scholar] [CrossRef]
- Yan, C.; Duan, G.; Zhang, Y.; Wu, F.X.; Pan, Y.; Wang, J. Predicting Drug-Drug Interactions Based on Integrated Similarity and Semi-Supervised Learning. IEEE/ACM Trans. Comput. Biol. Bioinform. 2020, 19, 168–179. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).