
CrohnDB: A Web Database for Expression Profiling of Protein-Coding and Long Non-Coding RNA Genes in Crohn Disease




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. RNA-Seq Data Analysis and Visualization
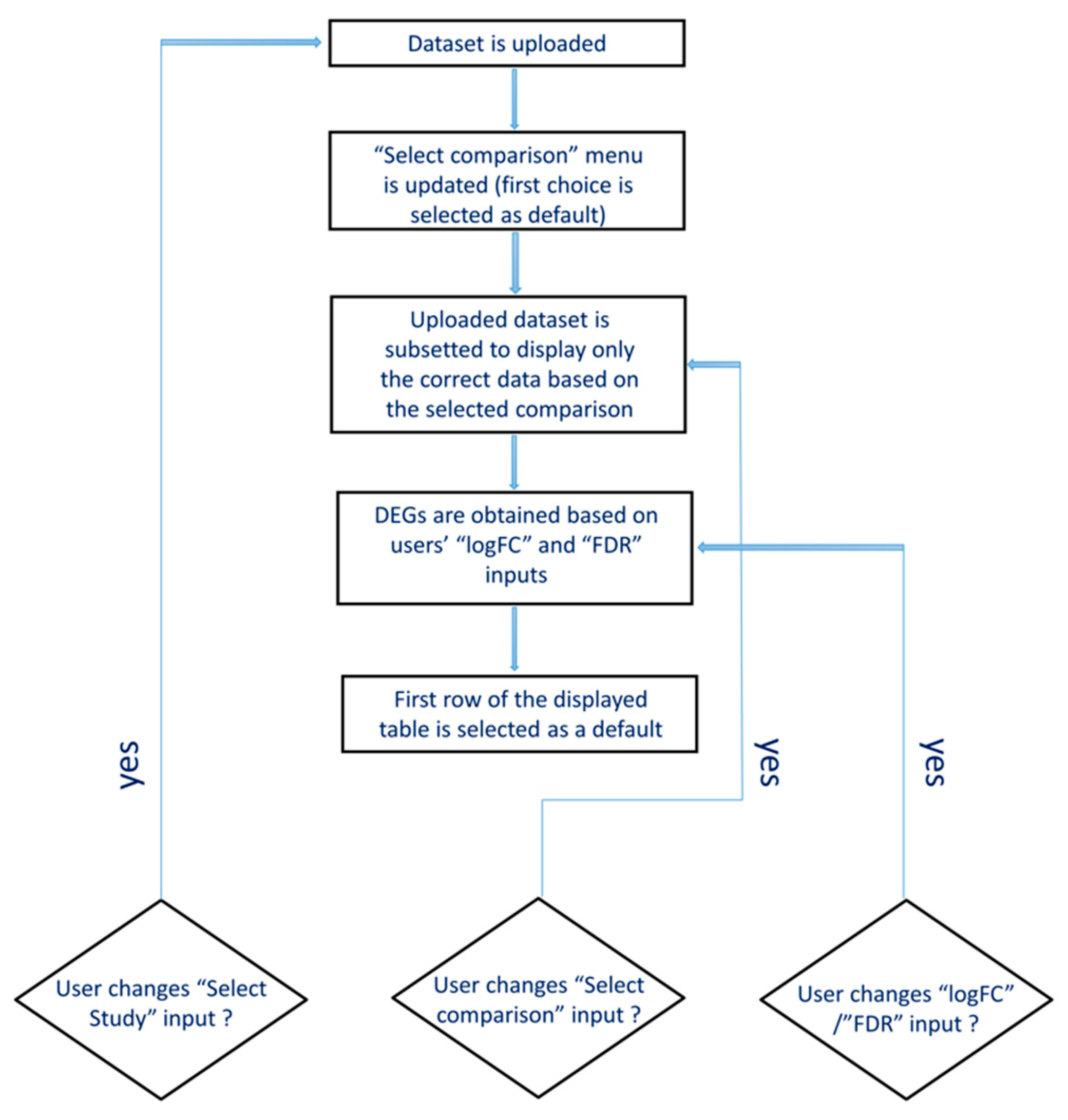
2.2. CrohnDB Web Database
3. Results
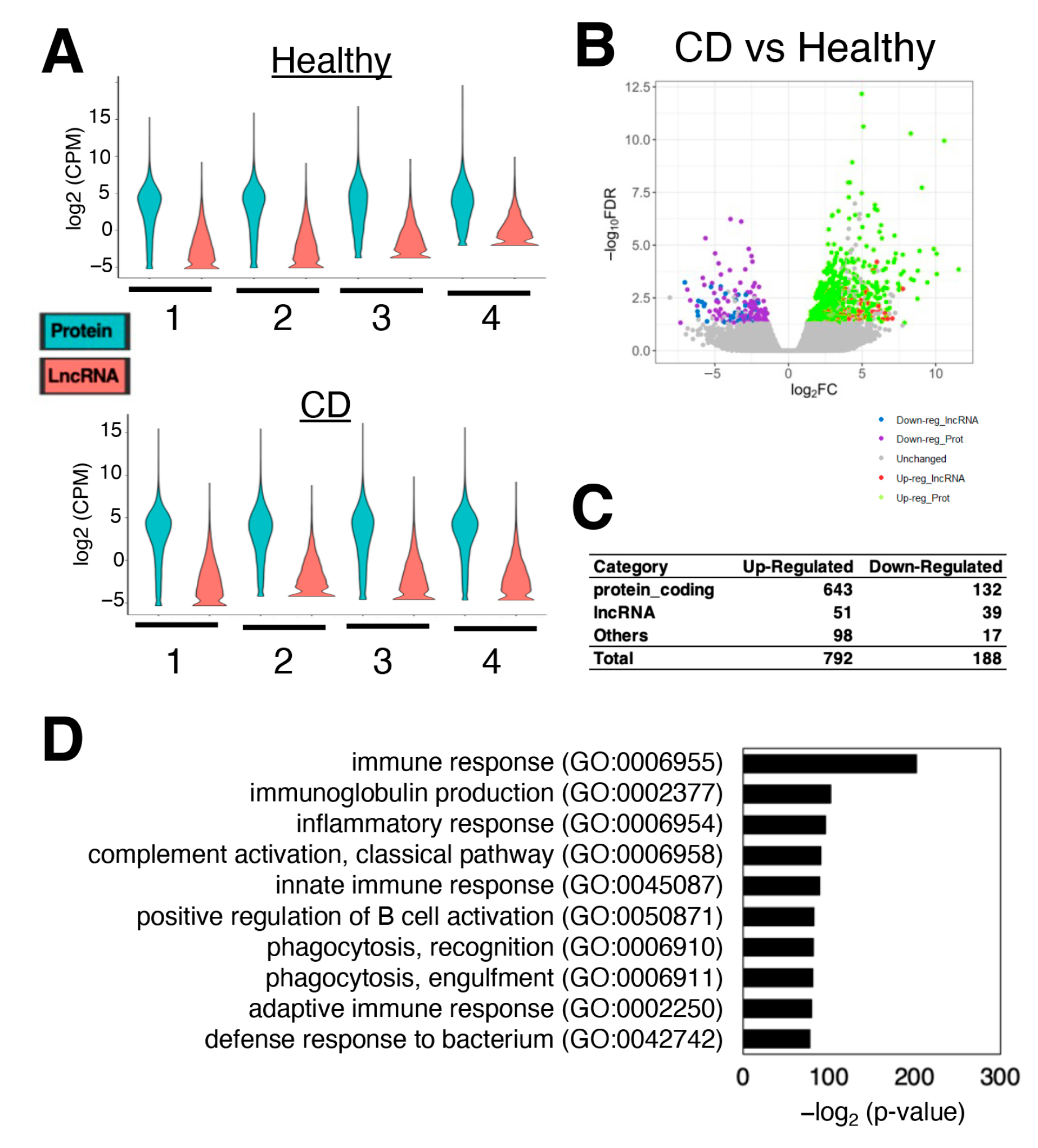
3.1. Hundreds of Genes Are Differentially Expressed in CD Patients Compared to Healthy Donors
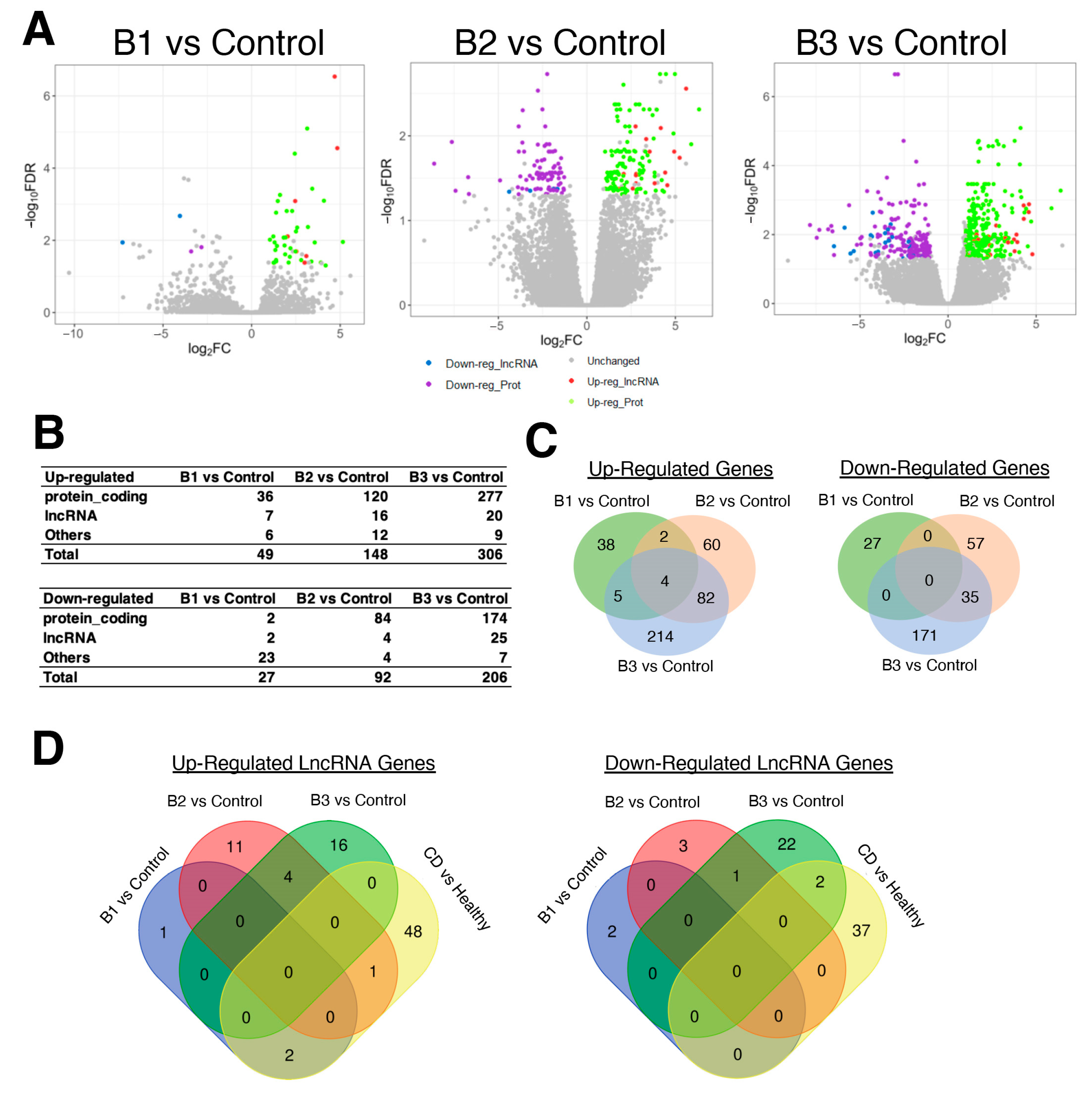
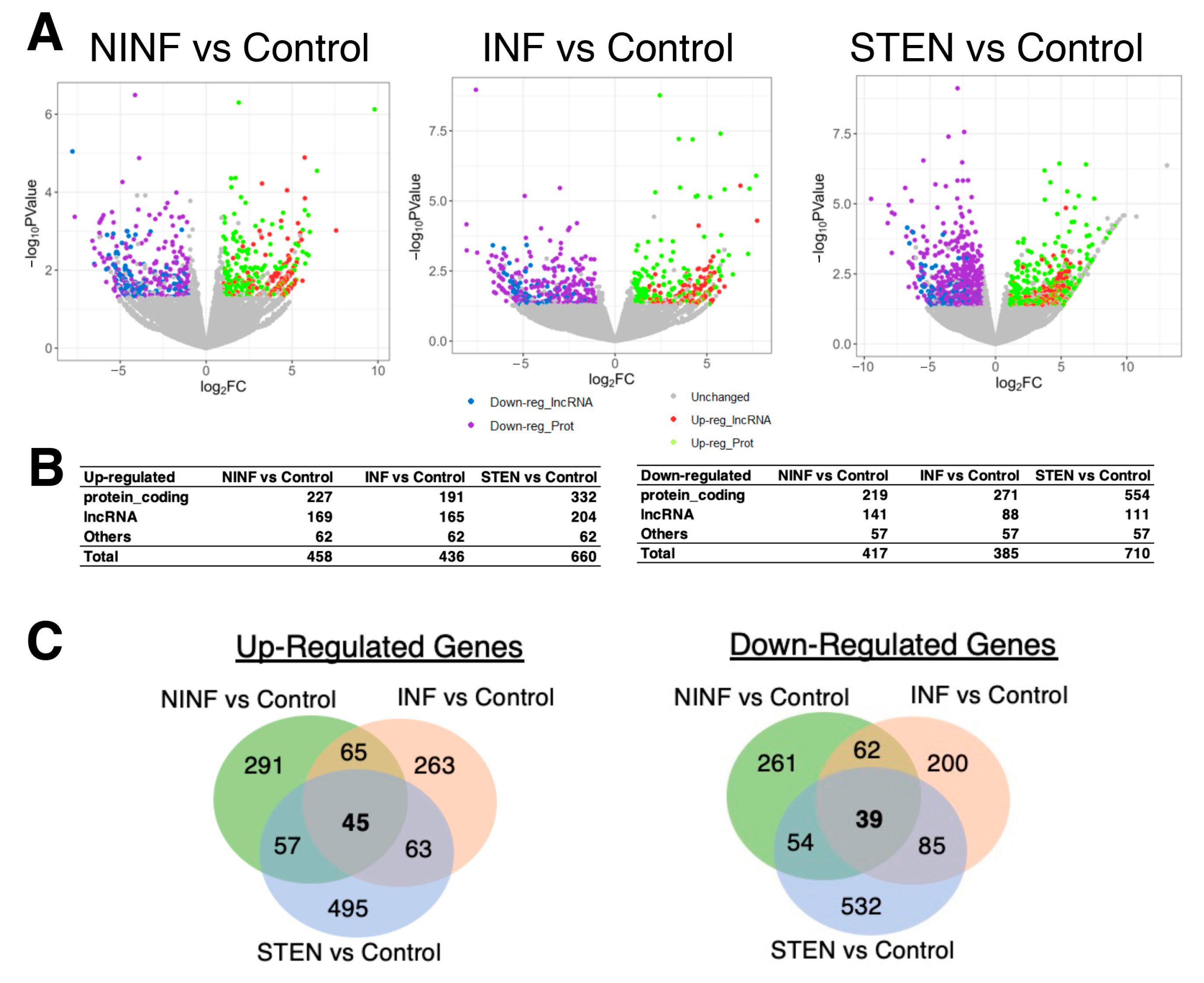
3.2. Severeal Differentially Expressed lncRNA Genes Are Shared in Fibroblasts Isolated from Different Etiologies of CD Patients Compared to Healthy Control Subjects
3.3. The Web Database, CrohnDB, for Screening of Protein-Coding and lncRNA Genes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Cushing, K.; Higgins, P.D.R. Management of Crohn Disease: A Review. JAMA 2021, 325, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Roda, G.; Chien Ng, S.; Kotze, P.G.; Argollo, M.; Panaccione, R.; Spinelli, A.; Kaser, A.; Peyrin-Biroulet, L.; Danese, S. Crohn’s disease. Nat. Rev. Dis. Primers 2020, 6, 22. [Google Scholar] [CrossRef] [PubMed]
- Gajendran, M.; Loganathan, P.; Catinella, A.P.; Hashash, J.G. A comprehensive review and update on Crohn’s disease. Dis. Mon. 2018, 64, 20–57. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.C.; Shi, H.Y.; Hamidi, N.; Underwood, F.E.; Tang, W.; Benchimol, E.I.; Panaccione, R.; Ghosh, S.; Wu, J.C.Y.; Chan, F.K.L.; et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: A systematic review of population-based studies. Lancet 2017, 390, 2769–2778. [Google Scholar] [CrossRef] [PubMed]
- Shaler, C.R.; Parco, A.A.; Elhenawy, W.; Dourka, J.; Jury, J.; Verdu, E.F.; Coombes, B.K. Psychological stress impairs IL22-driven protective gut mucosal immunity against colonising pathobionts. Nat. Commun. 2021, 12, 6664. [Google Scholar] [CrossRef]
- Sun, Y.; Li, L.; Xie, R.; Wang, B.; Jiang, K.; Cao, H. Stress Triggers Flare of Inflammatory Bowel Disease in Children and Adults. Front. Pediatr. 2019, 7, 432. [Google Scholar] [CrossRef]
- Lewis, J.D. The Role of Diet in Inflammatory Bowel Disease. Gastroenterol. Hepatol. 2016, 12, 51–53. [Google Scholar]
- Garza-Hernandez, D.; Sepulveda-Villegas, M.; Garcia-Pelaez, J.; Aguirre-Gamboa, R.; Lakatos, P.L.; Estrada, K.; Martinez-Vazquez, M.; Trevino, V. A systematic review and functional bioinformatics analysis of genes associated with Crohn’s disease identify more than 120 related genes. BMC Genom. 2022, 23, 302. [Google Scholar] [CrossRef]
- Elhenawy, W.; Hordienko, S.; Gould, S.; Oberc, A.M.; Tsai, C.N.; Hubbard, T.P.; Waldor, M.K.; Coombes, B.K. High-throughput fitness screening and transcriptomics identify a role for a type IV secretion system in the pathogenesis of Crohn’s disease-associated Escherichia coli. Nat. Commun. 2021, 12, 2032. [Google Scholar] [CrossRef]
- Jaeger, N.; Gamini, R.; Cella, M.; Schettini, J.L.; Bugatti, M.; Zhao, S.; Rosadini, C.V.; Esaulova, E.; Di Luccia, B.; Kinnett, B.; et al. Single-cell analyses of Crohn’s disease tissues reveal intestinal intraepithelial T cells heterogeneity and altered subset distributions. Nat. Commun. 2021, 12, 1921. [Google Scholar] [CrossRef]
- Di Narzo, A.F.; Brodmerkel, C.; Telesco, S.E.; Argmann, C.; Peters, L.A.; Li, K.; Kidd, B.; Dudley, J.; Cho, J.; Schadt, E.E.; et al. High-Throughput Identification of the Plasma Proteomic Signature of Inflammatory Bowel Disease. J. Crohns Colitis 2019, 13, 462–471. [Google Scholar] [CrossRef]
- Chan, S.N.; Low, E.N.D.; Raja Ali, R.A.; Mokhtar, N.M. Delineating inflammatory bowel disease through transcriptomic studies: Current review of progress and evidence. Intest. Res. 2018, 16, 374–383. [Google Scholar] [CrossRef]
- Ohno, S. So much “junk” DNA in our genome. Brookhaven Symp. Biol. 1972, 23, 366–370. [Google Scholar]
- Pertea, M. The human transcriptome: An unfinished story. Genes 2012, 3, 344–360. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Porta, M.; Frankish, A.; Rung, J.; Harrow, J.; Brazma, A. Transcriptome analysis of human tissues and cell lines reveals one dominant transcript per gene. Genome Biol. 2013, 14, R70. [Google Scholar] [CrossRef]
- Palazzo, A.F.; Lee, E.S. Non-coding RNA: What is functional and what is junk? Front. Genet. 2015, 6, 2. [Google Scholar] [CrossRef]
- Statello, L.; Guo, C.J.; Chen, L.L.; Huarte, M. Gene regulation by long non-coding RNAs and its biological functions. Nat. Rev. Mol. Cell. Biol. 2021, 22, 96–118. [Google Scholar] [CrossRef]
- Ruan, X.; Li, P.; Chen, Y.; Shi, Y.; Pirooznia, M.; Seifuddin, F.; Suemizu, H.; Ohnishi, Y.; Yoneda, N.; Nishiwaki, M.; et al. In vivo functional analysis of non-conserved human lncRNAs associated with cardiometabolic traits. Nat. Commun. 2020, 11, 45. [Google Scholar] [CrossRef]
- Sommerauer, C.; Kutter, C. Noncoding RNAs and RNA-binding proteins: Emerging governors of liver physiology and metabolic diseases. Am. J. Physiol. Cell. Physiol. 2022, 323, C1003–C1017. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, W.; Zhu, W.; Dong, J.; Cheng, Y.; Yin, Z.; Shen, F. Mechanisms and Functions of Long Non-Coding RNAs at Multiple Regulatory Levels. Int. J. Mol. Sci. 2019, 20, 5573. [Google Scholar] [CrossRef]
- Marchese, F.P.; Raimondi, I.; Huarte, M. The multidimensional mechanisms of long noncoding RNA function. Genome Biol. 2017, 18, 206. [Google Scholar] [CrossRef] [PubMed]
- Derrien, T.; Johnson, R.; Bussotti, G.; Tanzer, A.; Djebali, S.; Tilgner, H.; Guernec, G.; Martin, D.; Merkel, A.; Knowles, D.G.; et al. The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Res. 2012, 22, 1775–1789. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M. Non-coding RNAs in human disease. Nat. Rev. Genet. 2011, 12, 861–874. [Google Scholar] [CrossRef] [PubMed]
- DiStefano, J.K. The Emerging Role of Long Noncoding RNAs in Human Disease. Methods Mol. Biol. 2018, 1706, 91–110. [Google Scholar] [CrossRef]
- Yarani, R.; Mirza, A.H.; Kaur, S.; Pociot, F. The emerging role of lncRNAs in inflammatory bowel disease. Exp. Mol. Med. 2018, 50, 1–14. [Google Scholar] [CrossRef]
- Mirza, A.H.; Berthelsen, C.H.; Seemann, S.E.; Pan, X.; Frederiksen, K.S.; Vilien, M.; Gorodkin, J.; Pociot, F. Transcriptomic landscape of lncRNAs in inflammatory bowel disease. Genome Med. 2015, 7, 39. [Google Scholar] [CrossRef]
- Ilieva, M.; Dao, J.; Miller, H.E.; Madsen, J.H.; Bishop, A.J.R.; Kauppinen, S.; Uchida, S. Systematic Analysis of Long Non-Coding RNA Genes in Nonalcoholic Fatty Liver Disease. Noncoding RNA 2022, 8, 56. [Google Scholar] [CrossRef]
- Ilieva, M.; Miller, H.E.; Agarwal, A.; Paulus, G.K.; Madsen, J.H.; Bishop, A.J.R.; Kauppinen, S.; Uchida, S. FibroDB: Expression Analysis of Protein-Coding and Long Non-Coding RNA Genes in Fibrosis. Noncoding RNA 2022, 8, 13. [Google Scholar] [CrossRef]
- Leinonen, R.; Sugawara, H.; Shumway, M.; International Nucleotide Sequence Database, C. The sequence read archive. Nucleic Acids Res. 2011, 39, D19–D21. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis. In Use R! Springer International Publishing: Cham, Switzerland, 2016. [Google Scholar]
- da Huang, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- da Huang, W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef]
- Winston, C.; Joe, C.; JJ, A.; Carson, S.; Barret, S.; Yihui, X.; Jeff, A.; Jonathan, M.P.; Alan, D.; Barbara, B. Shiny: Web Application Framework for R. Available online: https://shiny.rstudio.com/ (accessed on 21 March 2023).
- Sievert, C. Interactive Web-Based Data Visualization with R, Plotly, and Shiny; Chapman and Hall/CRC. 2020. Available online: https://plotly-r.com (accessed on 22 May 2022).
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. g:Profiler: A web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019, 47, W191–W198. [Google Scholar] [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]
- Yu, G. Enrichplot: Visualization of Functional Enrichment Result. 2022. Available online: https://yulab-smu.top/biomedical-knowledge-mining-book/ (accessed on 21 March 2023).
- Larsson, J. eulerr: Area-Proportional Euler and Venn Diagrams with Ellipses, R package version 7.0.0; R Foundation for Statistical Computing: Vienna, Austria, 2022. [Google Scholar]
- Conway, J.R.; Lex, A.; Gehlenborg, N. UpSetR: An R package for the visualization of intersecting sets and their properties. Bioinformatics 2017, 33, 2938–2940. [Google Scholar] [CrossRef]
- Hedrick, T.L.; Friel, C.M. Colonic crohn disease. Clin. Colon. Rectal Surg. 2013, 26, 84–89. [Google Scholar] [CrossRef]
- Wang, F.; Liang, R.; Soibam, B.; Yang, J.; Liu, Y. Coregulatory long non-coding RNA and protein-coding genes in serum starved cells. Biochim. Biophys. Acta Gene Regul. Mech. 2019, 1862, 84–95. [Google Scholar] [CrossRef]
- Peck, B.C.; Weiser, M.; Lee, S.E.; Gipson, G.R.; Iyer, V.B.; Sartor, R.B.; Herfarth, H.H.; Long, M.D.; Hansen, J.J.; Isaacs, K.L.; et al. MicroRNAs Classify Different Disease Behavior Phenotypes of Crohn’s Disease and May Have Prognostic Utility. Inflamm. Bowel Dis. 2015, 21, 2178–2187. [Google Scholar] [CrossRef]
- Zhang, X.; Han, J.; Du, L.; Li, X.; Hao, J.; Wang, L.; Zheng, G.; Duan, W.; Xie, Y.; Zhao, Y.; et al. Unique metastasis-associated lncRNA signature optimizes prediction of tumor relapse in lung adenocarcinoma. Thorac. Cancer 2020, 11, 728–737. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Ramalingam, T.R. Mechanisms of fibrosis: Therapeutic translation for fibrotic disease. Nat. Med. 2012, 18, 1028–1040. [Google Scholar] [CrossRef] [PubMed]
- Ueha, S.; Shand, F.H.; Matsushima, K. Cellular and molecular mechanisms of chronic inflammation-associated organ fibrosis. Front. Immunol. 2012, 3, 71. [Google Scholar] [CrossRef] [PubMed]
- Mack, M. Inflammation and fibrosis. Matrix Biol. 2018, 68–69, 106–121. [Google Scholar] [CrossRef]
- Li Yim, A.Y.F.; de Bruyn, J.R.; Duijvis, N.W.; Sharp, C.; Ferrero, E.; de Jonge, W.J.; Wildenberg, M.E.; Mannens, M.; Buskens, C.J.; D’Haens, G.R.; et al. A distinct epigenetic profile distinguishes stenotic from non-inflamed fibroblasts in the ileal mucosa of Crohn’s disease patients. PLoS ONE 2018, 13, e0209656. [Google Scholar] [CrossRef]
- Chen, T.; Pei, J.; Wang, J.; Luo, R.; Liu, L.; Wang, L.; Jia, H. HBx-related long non-coding RNA 01152 promotes cell proliferation and survival by IL-23 in hepatocellular carcinoma. Biomed. Pharmacother. 2019, 115, 108877. [Google Scholar] [CrossRef]
- Wu, J.; Wang, N.; Yang, Y.; Jiang, G.; Mu, Q.; Zhan, H.; Li, F. Correction: LINC01152 upregulates MAML2 expression to modulate the progression of glioblastoma multiforme via Notch signaling pathway. Cell. Death Dis. 2021, 12, 862. [Google Scholar] [CrossRef]
- Zhou, Z.; Yang, Z.; Cui, Y.; Lu, S.; Huang, Y.; Che, X.; Yang, L.; Zhang, Y. Identification and Validation of a Ferroptosis-Related Long Non-Coding RNA (FRlncRNA) Signature to Predict Survival Outcomes and the Immune Microenvironment in Patients with Clear Cell Renal Cell Carcinoma. Front. Genet. 2022, 13, 787884. [Google Scholar] [CrossRef]
- Xuan, Y.; Chen, W.; Liu, K.; Gao, Y.; Zuo, S.; Wang, B.; Ma, X.; Zhang, X. A Risk Signature with Autophagy-Related Long Noncoding RNAs for Predicting the Prognosis of Clear Cell Renal Cell Carcinoma: Based on the TCGA Database and Bioinformatics. Dis. Markers 2021, 2021, 8849977. [Google Scholar] [CrossRef]
- Guo, J.; Ye, F.; Xie, W.; Zhang, X.; Zeng, R.; Sheng, W.; Mi, Y.; Sheng, X. The HOXC-AS2/miR-876-5p/HKDC1 axis regulates endometrial cancer progression in a high glucose-related tumor microenvironment. Cancer Sci. 2022, 113, 2297–2310. [Google Scholar] [CrossRef]
- Li, Y.; Zhao, Z.; Sun, D.; Li, Y. Novel long noncoding RNA LINC02323 promotes cell growth and migration of ovarian cancer via TGF-beta receptor 1 by miR-1343-3p. J. Clin. Lab. Anal. 2021, 35, e23651. [Google Scholar] [CrossRef]
- Zhang, X.; Du, L.; Han, J.; Li, X.; Wang, H.; Zheng, G.; Wang, Y.; Yang, Y.; Hu, Y.; Wang, C. Novel long non-coding RNA LINC02323 promotes epithelial-mesenchymal transition and metastasis via sponging miR-1343-3p in lung adenocarcinoma. Thorac. Cancer 2020, 11, 2506–2516. [Google Scholar] [CrossRef]
- Dong, N.; Guo, J.; Han, S.; Bao, L.; Diao, Y.; Lin, Z. Positive feedback loop of lncRNA HOXC-AS2/miR-876-5p/ZEB1 to regulate EMT in glioma. OncoTargets Ther. 2019, 12, 7601–7609. [Google Scholar] [CrossRef]
- Qiao, Y.Q.; Huang, M.L.; Xu, A.T.; Zhao, D.; Ran, Z.H.; Shen, J. LncRNA DQ786243 affects Treg related CREB and Foxp3 expression in Crohn’s disease. J. Biomed. Sci. 2013, 20, 87. [Google Scholar] [CrossRef]
- Barriocanal, M.; Prats-Mari, L.; Razquin, N.; Prior, C.; Unfried, J.P.; Fortes, P. ISR8/IRF1-AS1 Is Relevant for IFNalpha and NF-kappaB Responses. Front. Immunol. 2022, 13, 829335. [Google Scholar] [CrossRef]
- Chen, D.; Lu, M.M.; Wang, J.H.; Ren, Y.; Xu, L.L.; Cheng, W.X.; Wang, S.S.; Li, X.L.; Cheng, X.F.; Gao, J.G.; et al. High-fat diet aggravates colitis via mesenteric adipose tissue derived exosome metastasis-associated lung adenocarcinoma transcript 1. World J. Gastroenterol. 2022, 28, 3838–3853. [Google Scholar] [CrossRef]
- Fang, L.; Hu, M.; Xia, F.; Bai, W. LINC01272 activates epithelial-mesenchymal transition through miR-153-5p in Crohn’s disease. Am. J. Transl. Res. 2022, 14, 2331–2342. [Google Scholar]
- Zhang, Y.; Yang, L.; Chen, L.L. Life without A tail: New formats of long noncoding RNAs. Int. J. Biochem. Cell. Biol. 2014, 54, 338–349. [Google Scholar] [CrossRef]
- Lynch, M.D.; Watt, F.M. Fibroblast heterogeneity: Implications for human disease. J. Clin. Investig. 2018, 128, 26–35. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Distefano, R.; Ilieva, M.; Madsen, J.H.; Uchida, S. CrohnDB: A Web Database for Expression Profiling of Protein-Coding and Long Non-Coding RNA Genes in Crohn Disease. Computation 2023, 11, 105. https://doi.org/10.3390/computation11060105
Distefano R, Ilieva M, Madsen JH, Uchida S. CrohnDB: A Web Database for Expression Profiling of Protein-Coding and Long Non-Coding RNA Genes in Crohn Disease. Computation. 2023; 11(6):105. https://doi.org/10.3390/computation11060105
Chicago/Turabian StyleDistefano, Rebecca, Mirolyuba Ilieva, Jens Hedelund Madsen, and Shizuka Uchida. 2023. "CrohnDB: A Web Database for Expression Profiling of Protein-Coding and Long Non-Coding RNA Genes in Crohn Disease" Computation 11, no. 6: 105. https://doi.org/10.3390/computation11060105
APA StyleDistefano, R., Ilieva, M., Madsen, J. H., & Uchida, S. (2023). CrohnDB: A Web Database for Expression Profiling of Protein-Coding and Long Non-Coding RNA Genes in Crohn Disease. Computation, 11(6), 105. https://doi.org/10.3390/computation11060105