
A Two-Step Machine Learning Method for Predicting the Formation Energy of Ternary Compounds

 , , ,
, , ,  , and
, and 
Abstract
1. Introduction
2. Methodology
2.1. Centered Adam
| Algorithm 1 Centered Adam algorithm |
Require: ▷ hyperparameters Require: ▷ loss function L Require: ▷ parameter of loss function All vector operations are element-wise while t ≤ maximum iterations or sufficient convergence of do ▷ calculate gradient ▷ calculate first moment ▷ calculate centered variance ▷ bias correction for first moment ▷ bias correction for centered variance ▷ update parameters end while |
Bias Correction for Centered Adam
3. Computational Details
4. Results and Discussion
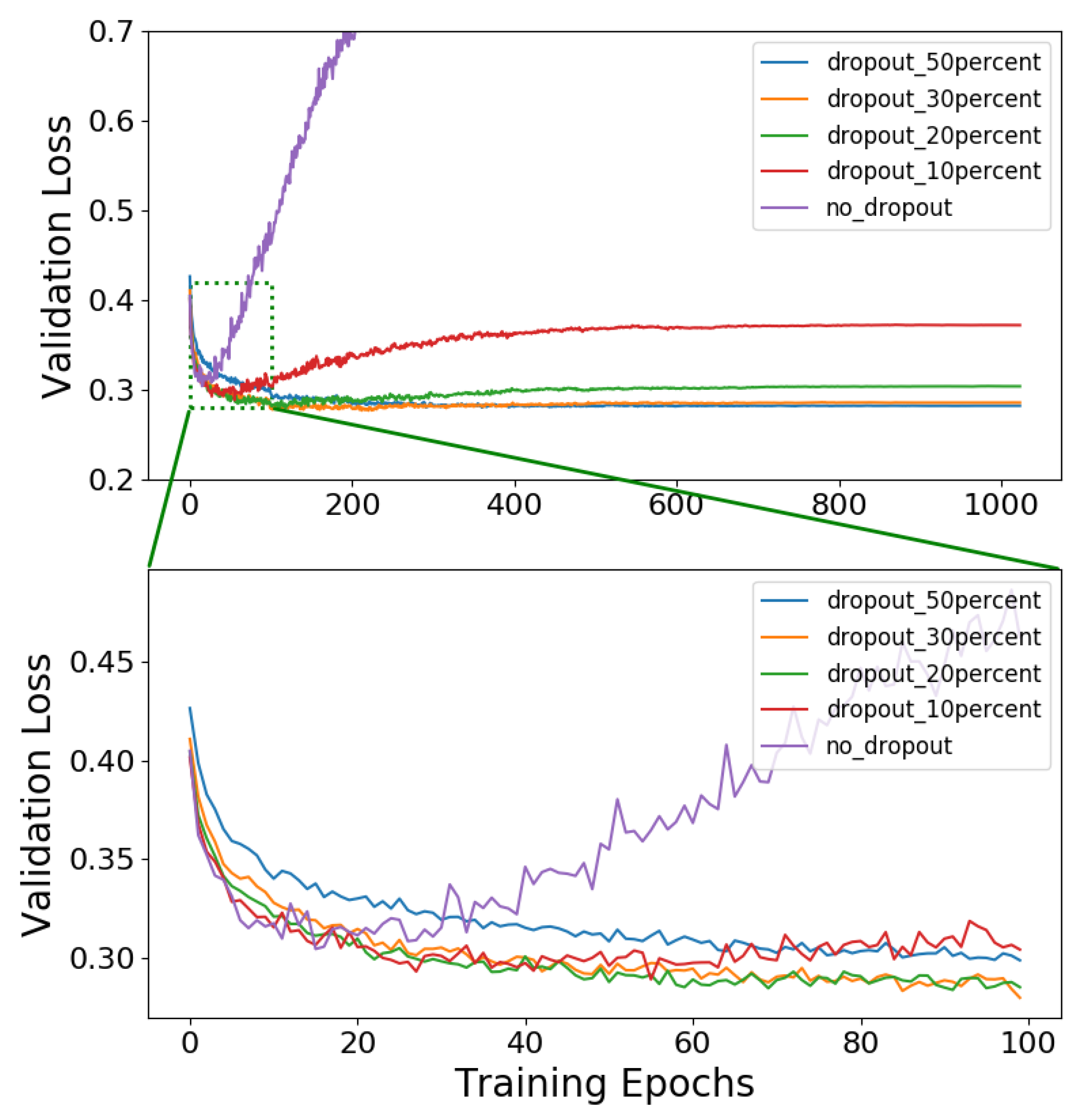
4.1. Classification
4.2. Regression
4.3. MNIST Classifier
5. Summary
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A. Descriptors
Appendix A.1. Classification
- Heuristic formation energy: heuristically computed formation energy of the ternary compound (eV/atom);
- Average Pauling electronegativity: average of the Pauling electronegativity values of the constituent elements of the ternary compound;
- Average group on the periodic table: average of the group numbers of the elements;
- Average row on the periodic table: average of the row numbers of the elements;
- Average atomic mass: average of the atomic mass values of the constituent elements of the ternary compound (u);
- Average ionic radius: average of the ionic radius values of the constituent elements of the ternary compound (Å);
- Average electron affinity: average of the electron affinity values of the constituent elements of the ternary compound (eV);
- Average first ionization energy: average of the first ionization energy values of the constituent elements of the ternary compound (eV);
- Average van der Waals radius: average of the van der Waals radius values of the constituent elements of the ternary compound (Å);
- Ratio of the electronegativity of each element in the compound to the average value of electronegativity of the ternary compound;
- Ratio of the group number of each constituent element to the average for the ternary compound;
- Ratio of the row number of each constituent element to the average for the ternary compound;
- Ratio of the atomic mass of each constituent element to the average for the ternary compound;
- Ratio of the ionic radius of each constituent element to the average for the ternary compound;
- Ratio of the electron affinity of each constituent element to the average for the ternary compound;
- Ratio of the first ionization energy of each constituent element to the average for the ternary compound;
- Ratio of the van der Waals radius of each constituent element to the average for the ternary compound;
- Orbital fraction of valence electrons s: ratio of the number of s electrons to the sum of the average of all electrons of the ternary compound;
- Orbital fraction of valence electrons p: ratio of the number of p electrons to the sum of the average of all electrons of the ternary compound;
- Orbital fraction of valence electrons d: ratio of the number of d electrons to the sum of the average of all electrons of the ternary compound;
- Orbital fraction of valence electrons f: ratio of the number of f electrons to the sum of the average of all electrons of the ternary compound.
- Weight: the percentage of a binary compound’s formation energy that contributes to the formation energy of the ternary compound;
- Formation energy: the formation energy of the binary constituent of the ternary compound;
- Ratio of anions to cations: ratio of the number of anions to the number of cations in a binary compound;
- Ratio of the cations in a constituent binary compound to its number in the ternary compound;
- Ratio of the anions in a constituent binary compound to its number in the ternary compound;
- Average Pauling electronegativity: average of the Pauling electronegativities of the constituent elements of the binary compound;
- Average group on the periodic table: average of the group number of constituent elements;
- Average row on the periodic table: average of the row number of constituent elements;
- Average atomic mass: average of the atomic masses of the constituent elements of the binary compound;
- Average ionic radius: average of the ionic radii of the constituent elements of the binary compound;
- Average electron affinity: average of the electron affinity values of the constituent elements of the binary compound;
- Average first ionization energy: average of the first ionization energy values of the constituent elements;
- Average van der Waals radius: average of the van der Waals radius values of the constituent elements;
- Absolute difference in the Pauling electronegativity of the constituent elements;
- Absolute difference in the group number of the constituent elements;
- Absolute difference in the row number of the constituent elements;
- Absolute difference in the atomic mass of the constituent elements;
- Absolute difference in the ionic radius of the constituent elements;
- Absolute difference in the electron affinity value of the constituent elements;
- Absolute difference in the first ionization energy of the constituent elements;
- Absolute difference in the van der Waals radius of the constituent elements;
- Difference in the group number of the constituent elements;
- Difference in the row number of the constituent elements;
- Difference in the atomic mass of the constituent elements;
- Difference in the ionic radius of the constituent elements;
- Difference in the electron affinity value of the constituent elements;
- Difference in the first ionization energy of the constituent elements;
- Difference in the van der Waals radius of the constituent elements;
- Ratio of the Pauling electronegativity of the cations to that of the anions;
- Ratio of the group number of the cations to that of the anions;
- Ratio of the row number of the cations to that of the anions;
- Ratio of the atomic mass of the cations to that of the anions;
- Ratio of the ionic radius of the cations to that of the anions;
- Ratio of the electron affinity of the cations to that of the anions;
- Ratio of the first ionization energy of the cations to that of the anions;
- Ratio of the van der Waals radius of the cations to that of the anions;
- Orbital fraction of valence electrons s: ratio of the number of s electrons to the sum of the average of all electrons of the binary compound;
- Orbital fraction of valence electrons p: ratio of the number of p electrons to the sum of the average of all electrons of the binary compound;
- Orbital fraction of valence electrons d: ratio of the number of d electrons to the sum of the average of all electrons of the binary compound;
- Orbital fraction of valence electrons f: ratio of the number of f electrons to the sum of the average of all electrons of the binary compound.
Appendix A.2. Regression
- Average Pauling electronegativity: the average of the Pauling electronegativity values of the constituent elements of the ternary compound;
- Average group on the periodic table: average of the group numbers of the constituent elements;
- Average row on the periodic table: average of the row numbers of the constituent elements;
- Average atomic mass: average of the atomic masses of the constituent elements;
- Average ionic radius: average of the ionic radii of the constituent elements;
- Average electron affinity: average of the electron affinity values of the constituent elements;
- Average first ionization energy: average of the first ionization energies of the constituent elements;
- Average van der Waals radius: average of the van der Waals radii of the constituent elements;
- Ratio of the electronegativity of each element in the compound to the average value of the electronegativity of the ternary compound;
- Ratio of the group number of each element in the compound to the average value of the group number of the ternary compound;
- Ratio of the row number of each element in the compound to the average value of the row number of the ternary compound;
- Ratio of the atomic mass of each element in the compound to the average value of the atomic mass of the ternary compound;
- Ratio of the ionic radius of each element in the compound to the average value of the ionic radius of the ternary compound;
- Ratio of the electron affinity of each element in the compound to the average value of the electron affinity of the ternary compound;
- Ratio of the first ionization energy of each element in the compound to the average value of the first ionization energy of the ternary compound;
- Ratio of the van der Waals radius of each element in the compound to the average value of the van der Waals radius of the ternary compound;
- Orbital fraction of valence electrons s: the ratio of the number of s electrons to the sum of the average of all electrons of the ternary compound;
- Orbital fraction of valence electrons p: the ratio of the number of p electrons to the sum of the average of all electrons of the ternary compound;
- Orbital fraction of valence electrons d: the ratio of the number of d electrons to the sum of the average of all electrons of the ternary compound;
- Orbital fraction of valence electrons f: the ratio of the number of f electrons to the sum of the average of all electrons of the ternary compound.
Appendix B. Dataset and Preprocessing
MNIST
Appendix C. Computational Resources
References
- Morgan, D.; Ceder, G.; Curtarolo, S. High-throughput and data mining with ab initio methods. Meas. Sci. Technol. 2004, 16, 296. [Google Scholar] [CrossRef]
- Li, W.; Jacobs, R.; Morgan, D. Predicting the thermodynamic stability of perovskite oxides using machine learning models. Comput. Mater. Sci. 2018, 150, 454–463. [Google Scholar] [CrossRef]
- Schmidt, J.; Chen, L.; Botti, S.; Marques, M.A.L. Predicting the stability of ternary intermetallics with density functional theory and machine learning. J. Chem. Phys. 2018, 148, 241728. [Google Scholar] [CrossRef] [PubMed]
- Davies, D.; Butler, K.; Jackson, A.; Morris, A.; Frost, J.; Skelton, J.; Walsh, A. Computational Screening of All Stoichiometric Inorganic Materials. Chem 2016, 1, 617–627. [Google Scholar] [CrossRef] [PubMed]
- Meredig, B.; Agrawal, A.; Kirklin, S.; Saal, J.E.; Doak, J.W.; Thompson, A.; Zhang, K.; Choudhary, A.; Wolverton, C. Combinatorial screening for new materials in unconstrained composition space with machine learning. Phys. Rev. B 2014, 89, 094104. [Google Scholar] [CrossRef]
- Liu, R.; Ward, L.; Wolverton, C.; Agrawal, A.; Liao, W.; Choudhary, A. Deep learning for chemical compound stability prediction. In Proceedings of the ACM SIGKDD Workshop on Large-Scale Deep Learning for Data Mining (DL-KDD), San Francisco, CA, USA, 13–17 August 2016; pp. 1–7. [Google Scholar]
- Jha, D.; Ward, L.; Paul, A.; Liao, W.k.; Choudhary, A.; Wolverton, C.; Agrawal, A. ElemNet: Deep Learning the Chemistry of Materials From Only Elemental Composition. Sci. Rep. 2018, 8, 17593. [Google Scholar] [CrossRef] [PubMed]
- Xie, T.; Grossman, J.C. Crystal Graph Convolutional Neural Networks for an Accurate and Interpretable Prediction of Material Properties. Phys. Rev. Lett. 2018, 120, 145301. [Google Scholar] [CrossRef] [PubMed]
- Schütt, K.T.; Kessel, P.; Gastegger, M.; Nicoli, K.A.; Tkatchenko, A.; Müller, K.R. SchNetPack: A Deep Learning Toolbox For Atomistic Systems. J. Chem. Theory Comput. 2019, 15, 448–455. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, K.; DeCost, B. Atomistic Line Graph Neural Network for improved materials property predictions. npj Comput. Mater. 2021, 7, 185. [Google Scholar] [CrossRef]
- Wang, A.Y.T.; Murdock, R.J.; Kauwe, S.K.; Oliynyk, A.O.; Gurlo, A.; Brgoch, J.; Persson, K.A.; Sparks, T.D. Machine Learning for Materials Scientists: An Introductory Guide toward Best Practices. Chem. Mater. 2020, 32, 4954–4965. [Google Scholar] [CrossRef]
- Peterson, G.G.C.; Brgoch, J. Materials discovery through machine learning formation energy. J. Phys. Energy 2021, 3, 022002. [Google Scholar] [CrossRef]
- Kirklin, S.; Saal, J.E.; Meredig, B.; Thompson, A.; Doak, J.W.; Aykol, M.; Rühl, S.; Wolverton, C. The Open Quantum Materials Database (OQMD): Assessing the accuracy of DFT formation energies. npj Comput. Mater. 2015, 1, 15010. [Google Scholar] [CrossRef]
- Reiser, P.; Neubert, M.; Eberhard, A.; Torresi, L.; Zhou, C.; Shao, C.; Metni, H.; van Hoesel, C.; Schopmans, H.; Sommer, T.; et al. Graph neural networks for materials science and chemistry. Commun. Mater. 2022, 3, 93. [Google Scholar] [CrossRef] [PubMed]
- Yan, K.; Liu, Y.; Lin, Y.; Ji, S. Periodic Graph Transformers for Crystal Material Property Prediction. In Proceedings of the 36th Annual Conference on Neural Information Processing Systems, New Orleans, LA, USA, 28 November–9 December 2022. [Google Scholar]
- Jain, A.; Ong, S.P.; Hautier, G.; Chen, W.; Richards, W.D.; Dacek, S.; Cholia, S.; Gunter, D.; Skinner, D.; Ceder, G.; et al. The Materials Project: A materials genome approach to accelerating materials innovation. APL Mater. 2013, 1, 011002. [Google Scholar] [CrossRef]
- Chen, C.; Ye, W.; Zuo, Y.; Zheng, C.; Ong, S.P. Graph Networks as a Universal Machine Learning Framework for Molecules and Crystals. Chem. Mater. 2019, 31, 3564–3572. [Google Scholar] [CrossRef]
- Hillert, M. Phase Equilibria, Phase Diagrams and Phase Transformations: Their Thermodynamic Basis, 2nd ed.; Cambridge University Press: Cambridge, UK, 2007. [Google Scholar] [CrossRef]
- Muggianu, Y.M.; Gambino, M.; Bros, J. Enthalpies of formation of liquid alloys bismuth-gallium-tin at 723k-choice of an analytical representation of integral and partial thermodynamic functions of mixing for this ternary-system. J. Chim. Phys. Phys.-Chim. Biol. 1975, 72, 83–88. [Google Scholar] [CrossRef]
- Kingma, D.P.; Ba, J. Adam: A Method for Stochastic Optimization. In Proceedings of the 3rd International Conference on Learning Representations, ICLR 2015, San Diego, CA, USA, 7–9 May 2015. Conference Track Proceedings; 2015. [Google Scholar]
- Jost, S. Comparison of Architectures and Parameters for Artificial Neural Networks. 2021. Available online: https://github.com/simulatedScience/Bachelorthesis (accessed on 4 May 2023).
- Chollet, F. Keras. 2015. Available online: https://keras.io (accessed on 4 May 2023).
- Abadi, M.; Agarwal, A.; Barham, P.; Brevdo, E.; Chen, Z.; Citro, C.; Corrado, G.S.; Davis, A.; Dean, J.; Devin, M.; et al. TensorFlow: Large-Scale Machine Learning on Heterogeneous Systems. 2015. Available online: https://www.tensorflow.org/ (accessed on 4 May 2023).
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Van der Walt, S.; Colbert, S.C.; Varoquaux, G. The NumPy array: A structure for efficient numerical computation. Comput. Sci. Eng. 2011, 13, 22–30. [Google Scholar] [CrossRef]
- Jones, E.; Oliphant, T.; Peterson, P. SciPy: Open Source Scientific Tools for Python. 2001. Available online: http://www.scipy.org (accessed on 4 May 2023).
- McKinney, W. Data Structures for Statistical Computing in Python. In Proceedings of the 9th Python in Science Conference, Austin, TX, USA, 28 June–3 July 2010; pp. 51–56. [Google Scholar]
- Hunter, J.D. Matplotlib: A 2D graphics environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Rengaraj, V. A Two-Step Machine Learning for Predicting the Stability of Chemical Compositions. Master’s Thesis, Paderborn University, Paderborn, Germany, 2019. [Google Scholar]
- LeCun, Y.; Cortes, C. MNIST Handwritten Digit Database. 2010. Available online: http://yann.lecun.com/exdb/mnist/ (accessed on 4 May 2023).
- Li, C.; Farkhoor, H.; Liu, R.; Yosinski, J. Measuring the Intrinsic Dimension of Objective Landscapes. In Proceedings of the 6th International Conference on Learning Representations, ICLR 2018, Vancouver, BC, Canada, 30 April–3 May 2018. [Google Scholar]



{kind=link}
{kind=link}
| Model Parameters | |
|---|---|
| Activation function | ReLU |
| Activation function | Sigmoid |
| Configuration | 117-117-1 |
| Loss function | Binary cross-entropy |
| Training parameters | |
| Batch size | 128 |
| Training cycles | 512 |
| Validation split | 0.1 |
| Test split | 0.1 |
| Optimizer parameters | |
| Optimizer | Adam |
| Learning rate | 0.001 |
| 0.9 | |
| 0.999 | |
| Model Parameters | |
|---|---|
| Activation function | ReLU/Sigmoid |
| Activation function | Linear |
| configuration | / / |
| Loss function | Log cosh/Mean squared error |
| Training parameters | |
| Batch size | 100/1000/10,000 |
| Training cycles | 500 |
| Validation split | 0.2 |
| Optimizer parameters | |
| Optimizer | Adam/cAdam |
| Learning rate | 0.01/0.001 |
| 0.9 | |
| 0.999 | |
| Model Parameters | |
|---|---|
| Activation function | ReLU/Sigmoid |
| Activation function | Softmax/Sigmoid |
| configuration | / / |
| Loss function | Categorical cross-entropy/ Mean squared error |
| Training parameters | |
| Batch size | 100/1000/10,000 |
| Training cycles | 5/25/50 |
| Validation split | 0.2 |
| Optimizer parameters | |
| Optimizer | Adam/cAdam |
| Learning rate | 0.1/0.01/0.001 |
| 1/ | |
| 0.9 | |
| 0.999 | |
| Model | Accuracy | Precision | Recall | AUROC |
|---|---|---|---|---|
| LR | 0.79 | 0.73 | 0.57 | 0.85 |
| LDA | 0.79 | 0.72 | 0.56 | 0.85 |
| KNN | 0.82 | 0.72 | 0.68 | 0.86 |
| RF | 0.74 | 0.74 | 0.29 | 0.72 |
| AB | 0.81 | 0.74 | 0.63 | 0.87 |
| bnn-c | 0.86 | 0.78 | 0.77 | 0.93 |
| Layout | Accuracy (%) | Precision | Recall | AUROC |
|---|---|---|---|---|
| 117-117-1 | 86.92 | 0.79 | 0.78 | 0.93 |
| 117-300-1 | 87.14 | 0.78 | 0.80 | 0.93 |
| 117-55-1 | 86.25 | 0.77 | 0.79 | 0.93 |
| 117-117-117-1 | 87.42 | 0.81 | 0.78 | 0.93 |
| 117-117-117-117-1 | 87.06 | 0.80 | 0.77 | 0.93 |
| Dropout in % | Accuracy (%) | Precision | Recall | AUROC |
|---|---|---|---|---|
| 50 | 87.82 | 0.86 | 0.72 | 0.93 |
| 30 | 88.24 | 0.82 | 0.79 | 0.94 |
| 20 | 88.29 | 0.84 | 0.77 | 0.94 |
| 10 | 88.51 | 0.83 | 0.78 | 0.94 |
| Parameter | Best Values | ||||
|---|---|---|---|---|---|
| Final val loss (MAE) | 0.1116 | 0.11208 | 0.11416 | 0.11493 | 0.11534 |
| Test loss (MAE) | 0.11389 | 0.11298 | 0.11493 | 0.11445 | 0.11539 |
| Test loss | 0.012971 | 0.012764 | 0.0065903 | 0.0065354 | 0.0066426 |
| Training time (in s) | 44.379 | 566.85 | 577.92 | 80.447 | 44.17 |
| Neurons per layer | (50, 10) | (50, 10) | (50, 10) | (30, 30, 10) | (40, 20) |
| Activation functions | Sigmoid | Sigmoid | Sigmoid | Sigmoid | Sigmoid |
| Last activation function | Linear | Linear | Linear | Linear | Linear |
| Loss function | MSE | MSE | Log cosh | Log cosh | Log cosh |
| Number of epochs | 500 | 500 | 500 | 500 | 500 |
| Batch size | 1000 | 100 | 100 | 1000 | 1000 |
| Optimizer | Adam | cAdam | cAdam | cAdam | Adam |
| Learning rate | 0.01 | 0.001 | 0.001 | 0.01 | 0.01 |
| Parameter | Best Values | ||||
|---|---|---|---|---|---|
| Final validation accuracy | 0.97 | 0.9691 | 0.96905 | 0.96885 | 0.9688 |
| Test accuracy | 0.97006 | 0.96906 | 0.9681 | 0.97048 | 0.97084 |
| Final validation loss | 0.01339 | 0.0066957 | 0.01449 | 0.0070507 | 0.96845 |
| Training time (in s) | 46.666 | 43.602 | 86.105 | 86.874 | 23.122 |
| Neurons per layer | |||||
| Activation function | ReLU | ReLU | ReLU | ReLU | ReLU |
| Activation function | Sigmoid | Sigmoid | Sigmoid | Softmax | Softmax |
| Loss function | Cat-cross | Cat-cross | Cat-cross | Cat-cross | Cat-cross |
| Training cycles | 50 | 25 | 50 | 50 | 25 |
| Batch size | 100 | 100 | 100 | 100 | 100 |
| Optimizer | Adam | cAdam | cAdam | cAdam | Adam |
| Learning rate | 0.001 | 0.1 | 0.1 | 0.1 | 0.1 |
| 1.0 | 1.0 | 1.0 | 1.0 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rengaraj, V.; Jost, S.; Bethke, F.; Plessl, C.; Mirhosseini, H.; Walther, A.; Kühne, T.D. A Two-Step Machine Learning Method for Predicting the Formation Energy of Ternary Compounds. Computation 2023, 11, 95. https://doi.org/10.3390/computation11050095
Rengaraj V, Jost S, Bethke F, Plessl C, Mirhosseini H, Walther A, Kühne TD. A Two-Step Machine Learning Method for Predicting the Formation Energy of Ternary Compounds. Computation. 2023; 11(5):95. https://doi.org/10.3390/computation11050095
Chicago/Turabian StyleRengaraj, Varadarajan, Sebastian Jost, Franz Bethke, Christian Plessl, Hossein Mirhosseini, Andrea Walther, and Thomas D. Kühne. 2023. "A Two-Step Machine Learning Method for Predicting the Formation Energy of Ternary Compounds" Computation 11, no. 5: 95. https://doi.org/10.3390/computation11050095
APA StyleRengaraj, V., Jost, S., Bethke, F., Plessl, C., Mirhosseini, H., Walther, A., & Kühne, T. D. (2023). A Two-Step Machine Learning Method for Predicting the Formation Energy of Ternary Compounds. Computation, 11(5), 95. https://doi.org/10.3390/computation11050095