
Adsorption of SO2 Molecule on Pristine, N, Ga-Doped and -Ga-N- co-Doped Graphene: A DFT Study

Abstract
:1. Introduction
2. Computational Method
3. Results and Discussion
3.1. Properties of the Structure
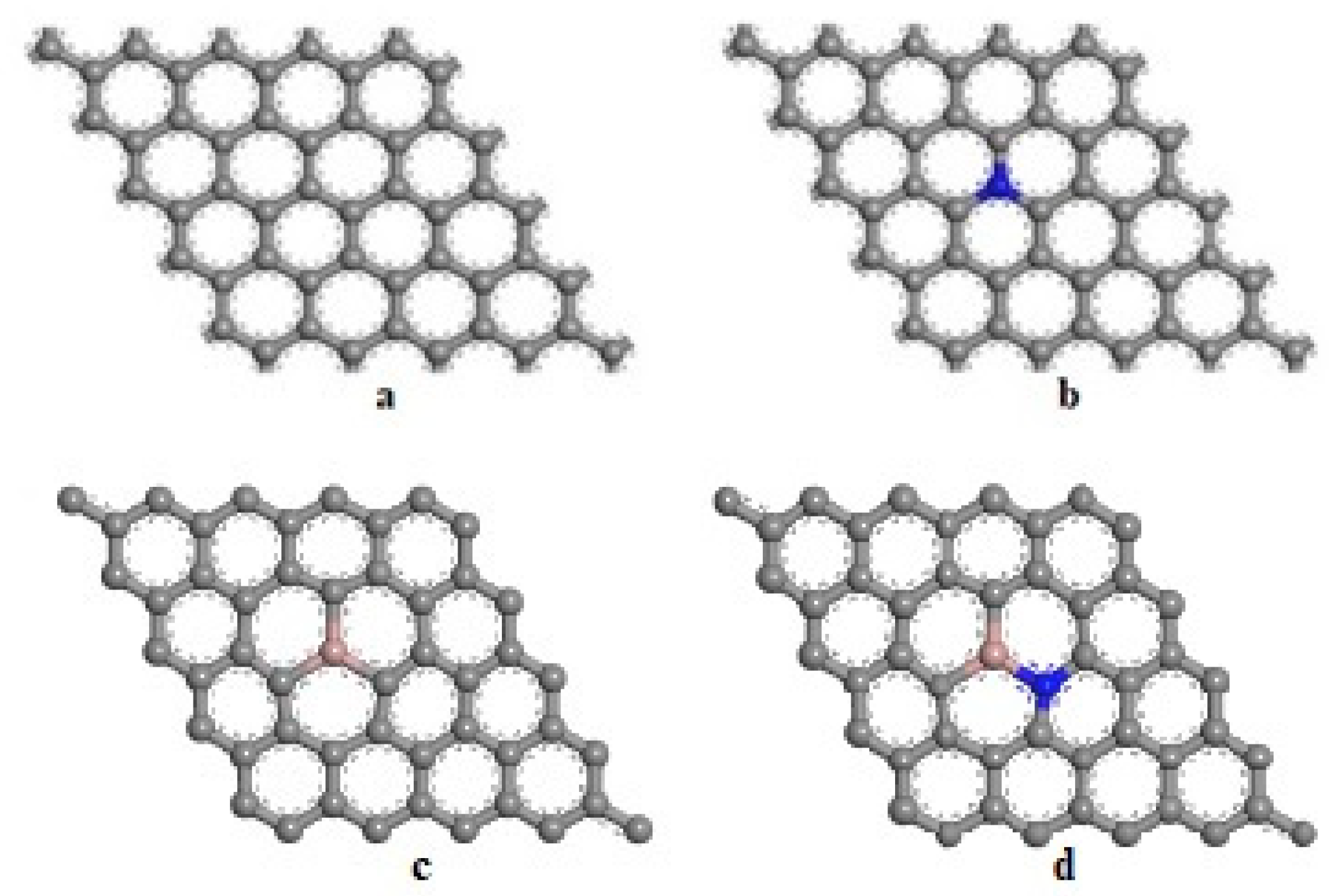
3.2. Geometry of Pristine Graphene and Doped Graphene
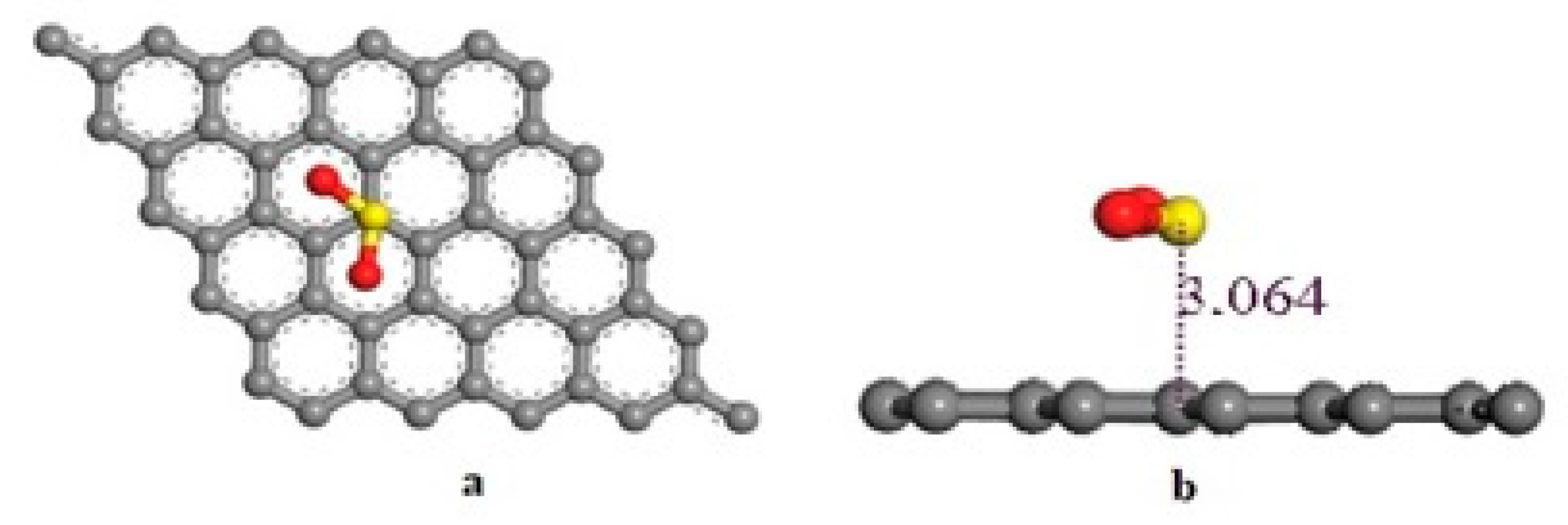
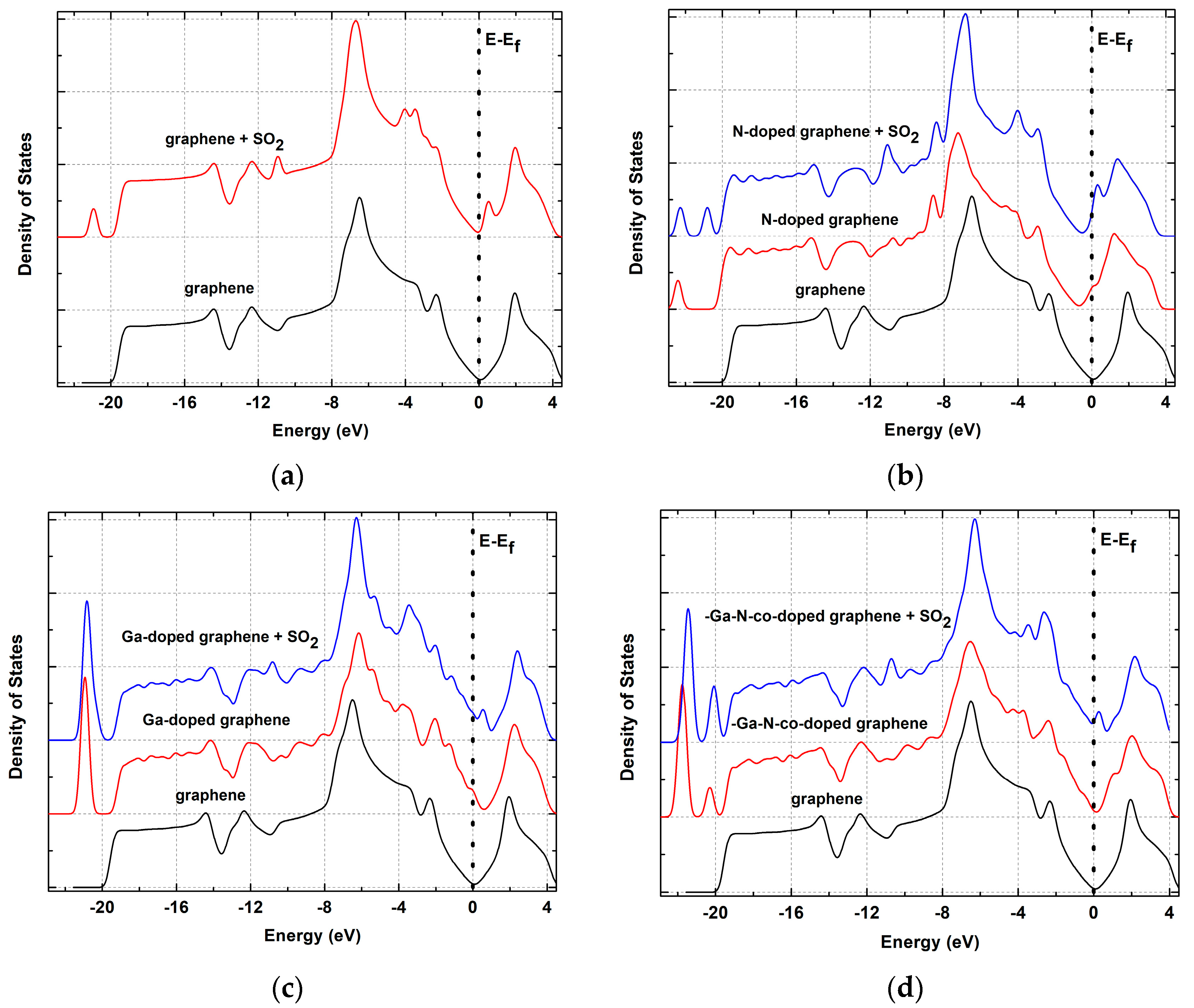
3.3. Adsorption of SO2 on Pristine Graphene
3.4. Adsorption of SO2 on N-Doped Graphene
3.5. Adsorption of SO2 on Ga-Doped Graphene
3.6. Adsorption of SO2 on -Ga-N- co-Doped Graphene
4. Summary and Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tsai, H.-S.; Wang, Y.; Liu, C.; Wang, T.; Huo, M. The Elemental 2D Materials beyond Graphene Potentially Used as Hazardous Gas Sensors for Environmental Protection. J. Hazard. Mater. 2022, 423, 127148. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Yang, D.; Zhang, G.; Chen, L.; Liu, D.; Cai, M.; Fan, X. The Effects of Graphene Stacking on the Performance of Methane Sensor: A First-Principles Study on the Adsorption, Band Gap and Doping of Graphene. Sensors 2018, 18, 422. [Google Scholar] [CrossRef] [PubMed]
- Jaaniso, R. (Ed.) Semiconductor Gas Sensors; Matthew Deans: Duxford, UK, 2020; ISBN 9780081025598. [Google Scholar]
- Chai, H.; Zheng, Z.; Liu, K.; Xu, J.; Wu, K.; Luo, Y.; Liao, H.; Debliquy, M.; Zhang, C. Stability of Metal Oxide Semiconductor Gas Sensors: A Review. IEEE Sens. J. 2022, 22, 5470–5481. [Google Scholar] [CrossRef]
- Fu, T. Sensing Behavior of CdS Nanoparticles to SO2, H2S and NH3 at Room Temperature. Mater. Res. Bull. 2013, 48, 1784–1790. [Google Scholar] [CrossRef]
- Zhang, J.; Pang, J.; Chen, H.; Wei, G.; Wei, S.; Yan, J.; Jin, S. Study on SO2 and Cl2 Sensor Application of 2D PbSe Based on First Principles Calculations. RSC Adv. 2022, 12, 8530–8535. [Google Scholar] [CrossRef]
- Zhang, D.; Wu, D.; Zong, X.; Yang, Z. Enhanced SO2 Gas Sensing Properties of Metal Organic Frameworks-Derived Titanium Dioxide/Reduced Graphene Oxide Nanostructure. J. Mater. Sci. Mater. Electron. 2019, 30, 11070–11078. [Google Scholar] [CrossRef]
- Zheng, W.; Liu, X.; Xie, J.; Lu, G.; Zhang, J. Emerging van Der Waals Junctions Based on TMDs Materials for Advanced Gas Sensors. Coord. Chem. Rev. 2021, 447, 214151. [Google Scholar] [CrossRef]
- Liu, H.; Zhou, J.; Yu, L.; Wang, Q.; Liu, B.; Li, P.; Zhang, Y. High-Sensitivity SO2 Gas Sensor Based on Noble Metal Doped WO3 Nanomaterials. Int. J. Electrochem. Sci. 2021, 16, 211240. [Google Scholar] [CrossRef]
- Zhou, X.; Zhao, C.; Wu, G.; Chen, J.; Li, Y. DFT Study on the Electronic Structure and Optical Properties of N, Al, and N-Al Doped Graphene. Appl. Surf. Sci. 2018, 459, 354–362. [Google Scholar] [CrossRef]
- Wang, C.; Fang, Y.; Duan, H.; Liang, G.; Li, W.; Chen, D.; Long, M. DFT Study of CO2 Adsorption Properties on Pristine, Vacancy and Doped Graphenes. Solid State Commun. 2021, 337, 114436. [Google Scholar] [CrossRef]
- Khudair, S.A.M.; Jappor, H.R. Adsorption of Gas Molecules on Graphene Doped with Mono and Dual Boron as Highly Sensitive Sensors and Catalysts. J. Nanostructures 2020, 10, 217–229. [Google Scholar] [CrossRef]
- Xiao, M.; Zhang, B.; Song, H.; Lv, Y.; Xiao, B. Effects of External Electric Field on Adsorption Behavior of Organic Molecules on Stanene: Highly Sensitive Sensor Devices. Solid State Commun. 2021, 338, 114459. [Google Scholar] [CrossRef]
- Peng, C.; Zhang, X. Chemical Functionalization of Graphene Nanoplatelets with Hydroxyl, Amino, and Carboxylic Terminal Groups. Chemistry 2021, 3, 873–888. [Google Scholar] [CrossRef]
- Liu, J.; Li, B.; Li, Q. Two-Dimensional Doped Materials. Magnetochemistry 2022, 8, 172. [Google Scholar] [CrossRef]
- Lin, P.C.; Villarreal, R.; Achilli, S.; Bana, H.; Nair, M.N.; Tejeda, A.; Verguts, K.; De Gendt, S.; Auge, M.; Hofsäss, H.; et al. Doping Graphene with Substitutional Mn. ACS Nano 2021, 15, 5449–5458. [Google Scholar] [CrossRef] [PubMed]
- Canales, M.; Ramírez-De-arellano, J.M.; Arellano, J.S.; Magaña, L.F. Ab Initio Study of the Interaction of a Graphene Surface Decorated with a Metal-Doped C30 with Carbon Monoxide, Carbon Dioxide, Methane, and Ozone. Int. J. Mol. Sci. 2022, 23, 4933. [Google Scholar] [CrossRef] [PubMed]
- Losurdo, M.; Yi, C.; Suvorova, A.; Rubanov, S.; Kim, T.H.; Giangregorio, M.M.; Jiao, W.; Bergmair, I.; Bruno, G.; Brown, A.S. Demonstrating the Capability of the High-Performance Plasmonic Gallium-Graphene Couple. ACS Nano 2014, 8, 3031–3041. [Google Scholar] [CrossRef]
- Sharma, S.; Verma, A.S. A Theoretical Study of H2S Adsorption on Graphene Doped with B, Al and Ga. Phys. B Condens. Matter 2013, 427, 12–16. [Google Scholar] [CrossRef]
- Capelle, K. A Bird’s-Eye View of Density-Functional Theory. Braz. J. Phys. 2006, 36, 1318–1343. [Google Scholar] [CrossRef]
- Lewars, E.G. Computational Chemistry; Springer International Publishing: Cham, Switzerland, 2016; ISBN 978-3-319-30914-9. [Google Scholar]
- Khosravi, M.; Murthy, V.; Mackinnon, I.D.R. Evaluation of DFT Methods to Calculate Structure and Partial Atomic Charges for Zeolite N. Comput. Mater. Sci. 2020, 171, 109225. [Google Scholar] [CrossRef]
- Liang, X.Y.; Ding, N.; Ng, S.P.; Wu, C.M.L. Adsorption of Gas Molecules on Ga-Doped Graphene and Effect of Applied Electric Field: A DFT Study. Appl. Surf. Sci. 2017, 411, 11–17. [Google Scholar] [CrossRef]
- Lv, Z.; Xu, H.; Xu, W.; Peng, B.; Zhao, C.; Xie, M.; Lv, X.; Gao, Y.; Hu, K.; Fang, Y.; et al. Quasi-Topological Intercalation Mechanism of Bi0.67NbS2 Enabling 100 C Fast-Charging for Sodium-Ion Batteries. Adv. Energy Mater. 2023, 13, 2300790. [Google Scholar] [CrossRef]
- Xu, H.; Zhu, J.Z.; Zou, C.; Zhang, F.; Ming, D.; Guan, D.; Ma, L. Theoretical Design of Core–Shell 3d-Metal Nanoclusters for Efficient Hydrogen-Evolving Reaction. Energy Fuels 2023, 37, 16781–16789. [Google Scholar] [CrossRef]
- Xu, H.; Guan, D.; Ma, L. The Bio-Inspired Heterogeneous Single-Cluster Catalyst Ni100–Fe4 S4 for Enhanced Electrochemical CO2 Reduction to CH4. Nanoscale 2023, 15, 2756–2766. [Google Scholar] [CrossRef]
- Ye, N.; Yang, Z.; Liu, Y. Applications of Density Functional Theory in COVID-19 Drug Modeling. Drug Discov. Today 2022, 27, 1411–1419. [Google Scholar] [CrossRef]
- Rasukkannu, M.; Velauthapillai, D.; Bianchini, F.; Vajeeston, P. Properties of Novel Non-Silicon Materials for Photovoltaic Applications: A First-Principle Insight. Materials 2018, 11, 2006. [Google Scholar] [CrossRef]
- Zheng, Z.; Wang, H. Different Elements Doped Graphene Sensor for CO2 Greenhouse Gases Detection: The DFT Study. Chem. Phys. Lett. 2019, 721, 33–37. [Google Scholar] [CrossRef]
- Shokuhi Rad, A.; Pouralijan Foukolaei, V. Density Functional Study of Al-Doped Graphene Nanostructure towards Adsorption of CO, CO2 and H2O. Synth. Met. 2015, 210, 171–178. [Google Scholar] [CrossRef]
- Jiang, M.; Zhang, W.; Zhao, K.; Guan, F.; Wang, Y. Investigations on the Electronic Structure and Optical Properties of (Ga, N, Ga-N) Doped Graphene by First-Principle Calculations. Int. J. Mod. Phys. B 2021, 35, 2150067. [Google Scholar] [CrossRef]
- Xie, T.; Wang, P.; Tian, C.; Zhao, G.; Jia, J.; Zhao, C.; Wu, H. The Adsorption Behavior of Gas Molecules on Co/N Co–Doped Graphene. Molecules 2021, 26, 7700. [Google Scholar] [CrossRef]
- Chen, G.; Gan, L.; Xiong, H.; Zhang, H. Density Functional Theory Study of B, N, and Si Doped Penta-Graphene as the Potential Gas Sensors for NH3 Detection. Membranes 2022, 12, 77. [Google Scholar] [CrossRef] [PubMed]
- Delley, B. From Molecules to Solids with the DMol3 Approach. J. Chem. Phys. 2000, 113, 7756–7764. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Askari Ardehjani, N.; Farmanzadeh, D. DFT Investigation of Metal Doped Graphene Capacity for Adsorbing of Ozone, Nitrogen Dioxide and Sulfur Dioxide Molecules. Adsorption 2019, 25, 661–667. [Google Scholar] [CrossRef]
- Salih, E.; Ayesh, A.I. Sensitive SO2 Gas Sensor Utilizing Pt-Doped Graphene Nanoribbon: First Principles Investigation. Mater. Chem. Phys. 2021, 267, 124695. [Google Scholar] [CrossRef]
- Mulliken, R.S. Electronic Population Analysis on LCAO-MO Molecular Wave Functions. I. J. Chem. Phys. 1955, 23, 1833–1840. [Google Scholar] [CrossRef]
- Xie, T.; Wang, P.; Tian, C.; Zhao, G.; Jia, J.; He, C.; Zhao, C.; Wu, H. Adsorption Characteristics of Gas Molecules Adsorbed on Graphene Doped with Mn: A First Principle Study. Molecules 2022, 27, 2315. [Google Scholar] [CrossRef]
- Zhang, H.P.; Luo, X.G.; Lin, X.Y.; Lu, X.; Leng, Y.; Song, H.T. Density Functional Theory Calculations on the Adsorption of Formaldehyde and Other Harmful Gases on Pure, Ti-Doped, or N-Doped Graphene Sheets. Appl. Surf. Sci. 2013, 283, 559–565. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gas Molecule | Bond | Length (Å) | Angle (°) |
|---|---|---|---|
| SO2 | S-O | 1.47 | 119.9 |
| System | Adsorption Distance (Å) | Bond Length (Å) | Bond Angle (°) | (eV) | QT (e) |
|---|---|---|---|---|---|
| Graphene + SO2 | 3.06 | 1.48 (S-O1) 1.48 (S-O2) | 119.2 | −0.32 | −0.095 |
| System | Adsorption Distance (Å) | Bond Length (Å) | Bond Angle (°) | (eV) | QT (e) |
|---|---|---|---|---|---|
| N-doped graphene + SO2 | 3.22 | 1.51 (S-O1) 1.51 (S-O2) | 117.6 | −0.48 | −0.236 |
| System | Adsorption Distance (Å) | Bond Length (Å) | Bond Angle (°) | (eV) | QT (e) |
|---|---|---|---|---|---|
| Ga-doped graphene + SO2 | 2.45 | 1.49 (S-O1) 1.49 (S-O2) | 121.3 | −2.61 | −0.148 |
| System | Adsorption Site | Adsorption Distance (Å) | Bond Types (Å) | Bond Length (Å) | Bond Angle (°) | QT (e) | |
|---|---|---|---|---|---|---|---|
| -Ga-N- co-doped graphene + SO2 | B | 2.447 | 1.903 (Ga-N) | 1.507 (S-O1) 1.507 (S-O2) | 120.8 | −2.75 | −0.258 |
| H | 2.449 | 1.898 (Ga-N) | 1.504 (S-O1) 1.497 (S-O2) | 120.6 | −2.55 | −0.214 | |
| T | 2.450 | 1.901 (Ga-N) | 1.504 (S-O1) 1.498 (S-O2) | 120.5 | −2.53 | −0.218 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Akhmetsadyk, D.; Ilyin, A.; Guseinov, N.; Beall, G. Adsorption of SO2 Molecule on Pristine, N, Ga-Doped and -Ga-N- co-Doped Graphene: A DFT Study. Computation 2023, 11, 235. https://doi.org/10.3390/computation11120235
Akhmetsadyk D, Ilyin A, Guseinov N, Beall G. Adsorption of SO2 Molecule on Pristine, N, Ga-Doped and -Ga-N- co-Doped Graphene: A DFT Study. Computation. 2023; 11(12):235. https://doi.org/10.3390/computation11120235
Chicago/Turabian StyleAkhmetsadyk, Dinara, Arkady Ilyin, Nazim Guseinov, and Gary Beall. 2023. "Adsorption of SO2 Molecule on Pristine, N, Ga-Doped and -Ga-N- co-Doped Graphene: A DFT Study" Computation 11, no. 12: 235. https://doi.org/10.3390/computation11120235
APA StyleAkhmetsadyk, D., Ilyin, A., Guseinov, N., & Beall, G. (2023). Adsorption of SO2 Molecule on Pristine, N, Ga-Doped and -Ga-N- co-Doped Graphene: A DFT Study. Computation, 11(12), 235. https://doi.org/10.3390/computation11120235