
Organic Emitters Showing Excited-States Energy Inversion: An Assessment of MC-PDFT and Correlation Energy Functionals Beyond TD-DFT



Abstract
:1. Introduction
2. Systems, Methods, and Computational Details
2.1. Choice of the Target Systems
2.2. Physical Meaning of Reduced Density Matrices
2.3. Theories Going beyond (TD-)DFT
2.4. Computational Details
3. Results and Discussion
3.1. Reference Results Available
| MAP: | () value of 1.110 (1.334) eV, with eV |
| 4AP: | () value of 2.258 (2.342) eV, with eV |
| 5AP: | () value of 2.308 (2.541) eV, with eV |
| 7AP: | () value of 2.847 (3.226) eV, with eV |
3.2. TD-DFT Calculations
3.3. MC-PDFT Calculations
3.4. Lie–Clementi (LC) and Colle–Salvetti (CS) Calculations
4. Conclusions
5. Concluding Remarks: A Personal Note
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Liu, S.; Langenaeker, W. Hund’s multiplicity rule: A unified interpretation. Theor. Chem. Acc. 2003, 110, 338–344. [Google Scholar] [CrossRef]
- Köhler, A.; Beljonne, D. The singlet–triplet exchange energy in conjugated polymers. Adv. Funct. Mater. 2004, 14, 11–18. [Google Scholar] [CrossRef]
- Gierschner, J.; Cornil, J.; Egelhaaf, H.J. Optical bandgaps of π-conjugated organic materials at the polymer limit: Experiment and theory. Adv. Mater. 2007, 19, 173–191. [Google Scholar] [CrossRef]
- Köhler, A.; Bässler, H. Triplet states in organic semiconductors. Mater. Sci. Eng. R Rep. 2009, 66, 71–109. [Google Scholar] [CrossRef]
- Becke, A.D. Singlet-triplet splittings from the virial theorem and single-particle excitation energies. J. Chem. Phys. 2018, 148, 044112. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Communication: Optical gap in polyacetylene from a simple quantum chemistry exciton model. J. Chem. Phys. 2018, 149, 081102. [Google Scholar] [CrossRef]
- Kollmar, H.; Staemmler, V. Violation of Hund’s rule by spin polarization in molecules. Theor. Chim. Acta 1978, 48, 223–239. [Google Scholar] [CrossRef]
- Olivier, Y.; Yurash, B.; Muccioli, L.; D’Avino, G.; Mikhnenko, O.; Sancho-García, J.C.; Adachi, C.; Nguyen, T.Q.; Beljonne, D. Nature of the singlet and triplet excitations mediating thermally activated delayed fluorescence. Phys. Rev. Mater. 2017, 1, 075602. [Google Scholar] [CrossRef]
- Dhali, R.; Phan Huu, D.A.; Terenziani, F.; Sissa, C.; Painelli, A. Thermally activated delayed fluorescence: A critical assessment of environmental effects on the singlet–triplet energy gap. J. Chem. Phys. 2021, 154, 134112. [Google Scholar] [CrossRef]
- Audebert, P.; Kroke, E.; Posern, C.; Lee, S.H. State of the Art in the Preparation and Properties of Molecular Monomeric s-Heptazines: Syntheses, Characteristics, and Functional Applications. Chem. Rev. 2021, 121, 2515–2544. [Google Scholar] [CrossRef]
- Leupin, W.; Wirz, J. Low-lying electronically excited states of cycl[3.3.3]azine, a bridged 12π-perimeter. J. Am. Chem. Soc. 1980, 102, 6068–6075. [Google Scholar] [CrossRef]
- Leupin, W.; Magde, D.; Persy, G.; Wirz, J. 1,4,7-Triazacycl[3.3.3]azine: Basicity, photoelectron spectrum, photophysical properties. J. Am. Chem. Soc. 1986, 108, 17–22. [Google Scholar] [CrossRef]
- Pollice, R.; Friederich, P.; Lavigne, C.; dos Passos Gomes, G.; Aspuru-Guzik, A. Organic molecules with inverted gaps between first excited singlet and triplet states and appreciable fluorescence rates. Matter 2021, 4, 1654–1682. [Google Scholar] [CrossRef]
- de Silva, P. Inverted singlet–triplet gaps and their relevance to thermally activated delayed fluorescence. J. Phys. Chem. Lett. 2019, 10, 5674–5679. [Google Scholar] [CrossRef]
- Ehrmaier, J.; Rabe, E.J.; Pristash, S.R.; Corp, K.L.; Schlenker, C.W.; Sobolewski, A.L.; Domcke, W. Singlet–triplet inversion in heptazine and in polymeric carbon nitrides. J. Phys. Chem. A 2019, 123, 8099–8108. [Google Scholar] [CrossRef]
- Sandoval-Salinas, M.E.; Carreras, A.; Casanova, D. Triangular graphene nanofragments: Open-shell character and doping. Phys. Chem. Chem. Phys. 2019, 21, 9069–9076. [Google Scholar] [CrossRef]
- Ricci, G.; San-Fabián, E.; Olivier, Y.; Sancho-García, J.C. Singlet-triplet excited-state inversion in heptazine and related molecules: Assessment of TD-DFT and ab initio methods. ChemPhysChem 2021, 22, 553–560. [Google Scholar] [CrossRef] [PubMed]
- Sanz-Rodrigo, J.; Ricci, G.; Olivier, Y.; Sancho-Garcia, J.C. Negative Singlet–Triplet Excitation Energy Gap in Triangle-Shaped Molecular Emitters for Efficient Triplet Harvesting. J. Phys. Chem. A 2021, 125, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Pios, S.; Huang, X.; Sobolewski, A.L.; Domcke, W. Triangular boron carbon nitrides: An unexplored family of chromophores with unique properties for photocatalysis and optoelectronics. Phys. Chem. Chem. Phys. 2021, 23, 12968–12975. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, K. Can TDDFT render the electronic excited states ordering of Azine derivative? A closer investigation with DLPNO-STEOM-CCSD. Chem. Phys. Lett. 2021, 779, 138827. [Google Scholar] [CrossRef]
- Sobolewski, A.L.; Domcke, W. Are Heptazine-Based Organic Light-Emitting Diode Chromophores Thermally Activated Delayed Fluorescence or Inverted Singlet–Triplet Systems? J. Phys. Chem. Lett. 2021, 12, 6852–6860. [Google Scholar] [CrossRef]
- Li, J.; Nakagawa, T.; MacDonald, J.; Zhang, Q.; Nomura, H.; Miyazaki, H.; Adachi, C. Highly efficient organic light-emitting diode based on a hidden thermally activated delayed fluorescence channel in a heptazine derivative. Adv. Mater. 2013, 25, 3319–3323. [Google Scholar] [CrossRef]
- Li, J.; Zhang, Q.; Nomura, H.; Miyazaki, H.; Adachi, C. Thermally activated delayed fluorescence from 3nπ* to 1nπ* up-conversion and its application to organic light-emitting diodes. Appl. Phys. Lett. 2014, 105, 98. [Google Scholar] [CrossRef]
- Moscardó, F.; San-Fabián, E. Density-functional formalism and the two-body problem. Phys. Rev. A 1991, 44, 1549. [Google Scholar] [CrossRef]
- Moscardó, F.; San-Fabián, E. A density functional for the correlation energy, deduced in the framework of the correlation factor approach. Int. J. Quantum Chem. 1991, 40, 23–32. [Google Scholar] [CrossRef]
- Moscardó, F.; Pérez-Jiménez, Á.J. Self-consistent field calculations using two-body density functionals for correlation energy component: I. Atomic systems. J. Comput. Chem. 1998, 19, 1887–1898. [Google Scholar] [CrossRef]
- Pastorczak, E.; Pernal, K. Electronic excited states from the adiabatic-connection formalism with complete active space wave functions. J. Phys. Chem. Lett. 2018, 9, 5534–5538. [Google Scholar] [CrossRef]
- Pastorczak, E.; Hapka, M.; Veis, L.; Pernal, K. Capturing the dynamic correlation for arbitrary spin-symmetry CASSCF reference with adiabatic connection approaches: Insights into the electronic structure of the tetramethyleneethane diradical. J. Phys. Chem. Lett. 2019, 10, 4668–4674. [Google Scholar] [CrossRef]
- Hoyer, C.E.; Ghosh, S.; Truhlar, D.G.; Gagliardi, L. Multiconfiguration pair-density functional theory is as accurate as CASPT2 for electronic excitation. J. Phys. Chem. Lett. 2016, 7, 586–591. [Google Scholar] [CrossRef] [PubMed]
- Gagliardi, L.; Truhlar, D.G.; Li Manni, G.; Carlson, R.K.; Hoyer, C.E.; Bao, J.L. Multiconfiguration pair-density functional theory: A new way to treat strongly correlated systems. Acc. Chem. Res. 2017, 50, 66–73. [Google Scholar] [CrossRef]
- Stoneburner, S.J.; Truhlar, D.G.; Gagliardi, L. Transition metal spin-state energetics by MC-PDFT with high local exchange. J. Phys. Chem. A 2020, 124, 1187–1195. [Google Scholar] [CrossRef] [PubMed]
- Lykhin, A.O.; Truhlar, D.G.; Gagliardi, L. Dipole Moment Calculations Using Multiconfiguration Pair-Density Functional Theory and Hybrid Multiconfiguration Pair-Density Functional Theory. J. Chem. Theory Comput. 2021, 17, 7586–7601. [Google Scholar] [CrossRef]
- Colle, R.; Salvetti, O. Approximate calculation of the correlation energy for the closed shells. Theor. Chim. Acta 1975, 37, 329–334. [Google Scholar] [CrossRef]
- Colle, R.; Salvetti, O. Approximate calculation of the correlation energy for the closed and open shells. Theor. Chim. Acta 1979, 53, 55–63. [Google Scholar] [CrossRef]
- Colle, R.; Salvetti, O. Generalization of the Colle–Salvetti correlation energy method to a many-determinant wave function. J. Chem. Phys. 1990, 93, 534–544. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle–Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785. [Google Scholar] [CrossRef] [Green Version]
- Lie, G.C.; Clementi, E. Study of the electronic structure of molecules. XXI. Correlation energy corrections as a functional of the Hartree-Fock density and its application to the hydrides of the second row atoms. J. Chem. Phys. 1974, 60, 1275–1287. [Google Scholar] [CrossRef]
- Lie, G.C.; Clementi, E. Study of the electronic structure of molecules. XXII. Correlation energy corrections as a functional of the Hartree-Fock type density and its application to the homonuclear diatomic molecules of the second row atoms. J. Chem. Phys. 1974, 60, 1288–1296. [Google Scholar] [CrossRef]
- Brandenburg, J.G.; Bannwarth, C.; Hansen, A.; Grimme, S. B97-3c: A revised low-cost variant of the B97-D density functional method. J. Chem. Phys. 2018, 148, 064104. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Weigend, F. Hartree–Fock exchange fitting basis sets for H to Rn. J. Comput. Chem. 2008, 29, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Chai, J.D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chai, J.D.; Head-Gordon, M. Long-range corrected double-hybrid density functionals. J. Chem. Phys. 2009, 131, 174105. [Google Scholar] [CrossRef]
- Neese, F. Software update: The ORCA program system, version 4.0. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2018, 8, e1327. [Google Scholar] [CrossRef]
- Barca, G.M.J.; Bertoni, C.; Carrington, L.; Datta, D.; De Silva, N.; Deustua, J.E.; Fedorov, D.G.; Gour, J.R.; Gunina, A.O.; Guidez, E.; et al. Recent developments in the general atomic and molecular electronic structure system. J. Chem. Phys. 2020, 152, 154102. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Jordá, J.M.; San-Fabián, E.; Moscardó, F. Spectroscopic constants of diatomic molecules computed correcting Hartree-Fock or general-valence-bond potential-energy curves with correlation-energy functionals. Phys. Rev. A 1992, 45, 4407. [Google Scholar] [CrossRef] [PubMed]
- Moscardó, F.; Muñoz-Fraile, F.; Pérez-Jiménez, A.J.; Pérez-Jordá, J.M.; San-Fabián, E. Improvement of multiconfigurational wave functions and energies by correlation energy functionals. J. Phys. Chem. A 1998, 102, 10900–10902. [Google Scholar] [CrossRef]
- Halpern, A.M.; Rossman, M.A.; Hosmane, R.S.; Leonard, N.J. Photophysics of the S1 tautm. SO transition in tri-s-triazine. J. Phys. Chem. 1984, 88, 4324–4326. [Google Scholar] [CrossRef]
- Penfold, T.J. On predicting the excited-state properties of thermally activated delayed fluorescence emitters. J. Phys. Chem. C 2015, 119, 13535–13544. [Google Scholar] [CrossRef]
- Grimme, S.; Neese, F. Double-hybrid density functional theory for excited electronic states of molecules. J. Chem. Phys. 2007, 127, 154116. [Google Scholar] [CrossRef]
- Ottochian, A.; Morgillo, C.; Ciofini, I.; Frisch, M.J.; Scalmani, G.; Adamo, C. Double hybrids and time-dependent density functional theory: An implementation and benchmark on charge transfer excited states. J. Comput. Chem. 2020, 41, 1242–1251. [Google Scholar] [CrossRef] [PubMed]
- Sancho-Garcia, J.C.; Bremond, E.; Ricci, G.; Pérez-Jiménez, Á.J.; Olivier, Y.; Adamo, C. Violation of Hund’s Rule in Molecules: Predicting the Excited-State Energy Inversion by TD-DFT with Double-Hybrid Methods. J. Chem. Phys. 2021, 156. [Google Scholar] [CrossRef]
- Sancho-García, J.; Moscardó, F. Usefulness of the Colle–Salvetti model for the treatment of the nondynamic correlation. J. Chem. Phys. 2003, 118, 1054–1058. [Google Scholar] [CrossRef]
- Moscardó, F.; San-Fabián, E.; Pastor-Abia, L. The Colle–Salvetti wavefunction revisited: A comparison between three approaches for obtaining the correlation energy. Theor. Chem. Acc. 2006, 115, 334–342. [Google Scholar] [CrossRef]
- Blaha, P.; Schwarz, K.; Tran, F.; Laskowski, R.; Madsen, G.K.; Marks, L.D. WIEN2k: An APW+ lo program for calculating the properties of solids. J. Chem. Phys. 2020, 152, 074101. [Google Scholar] [CrossRef]

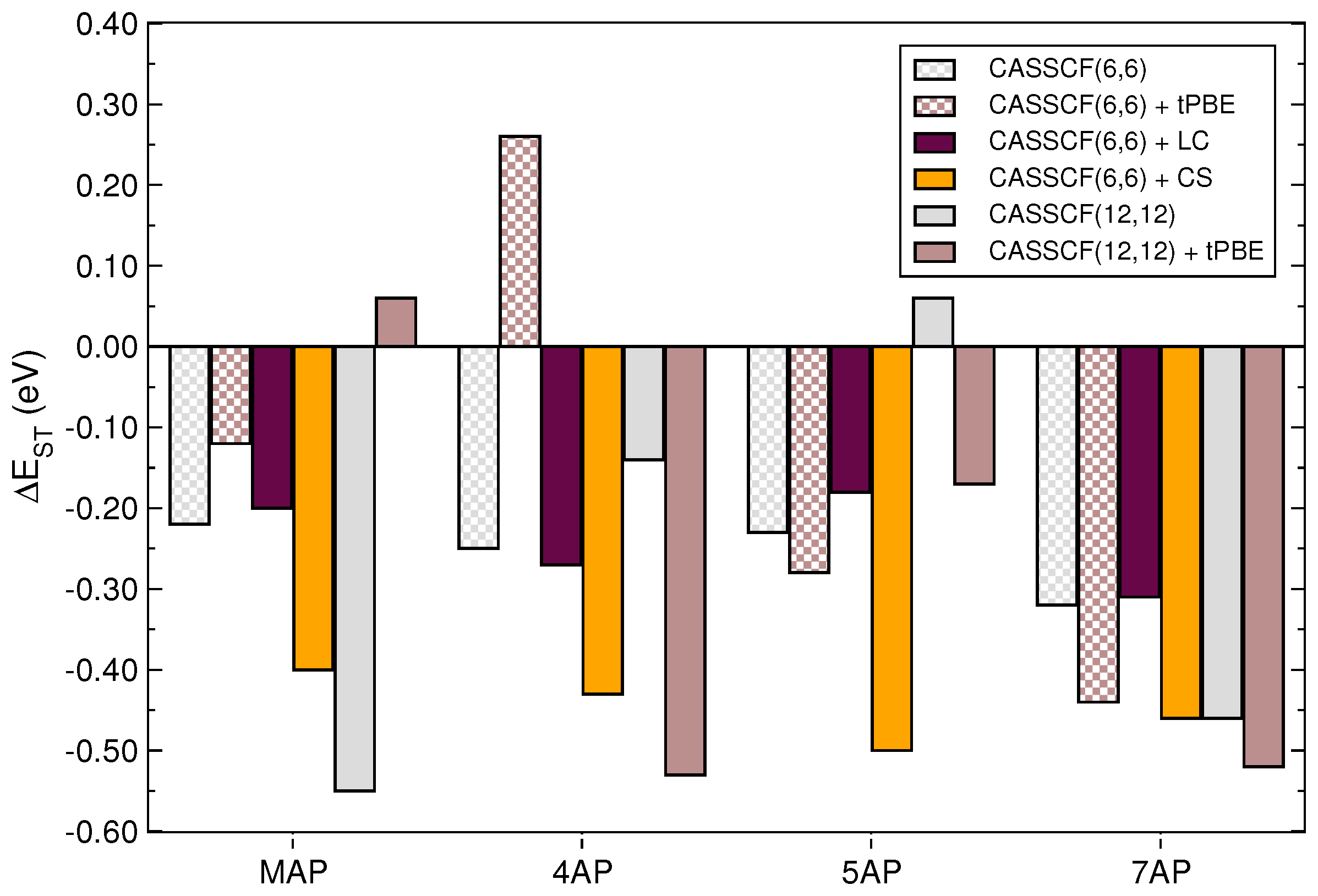


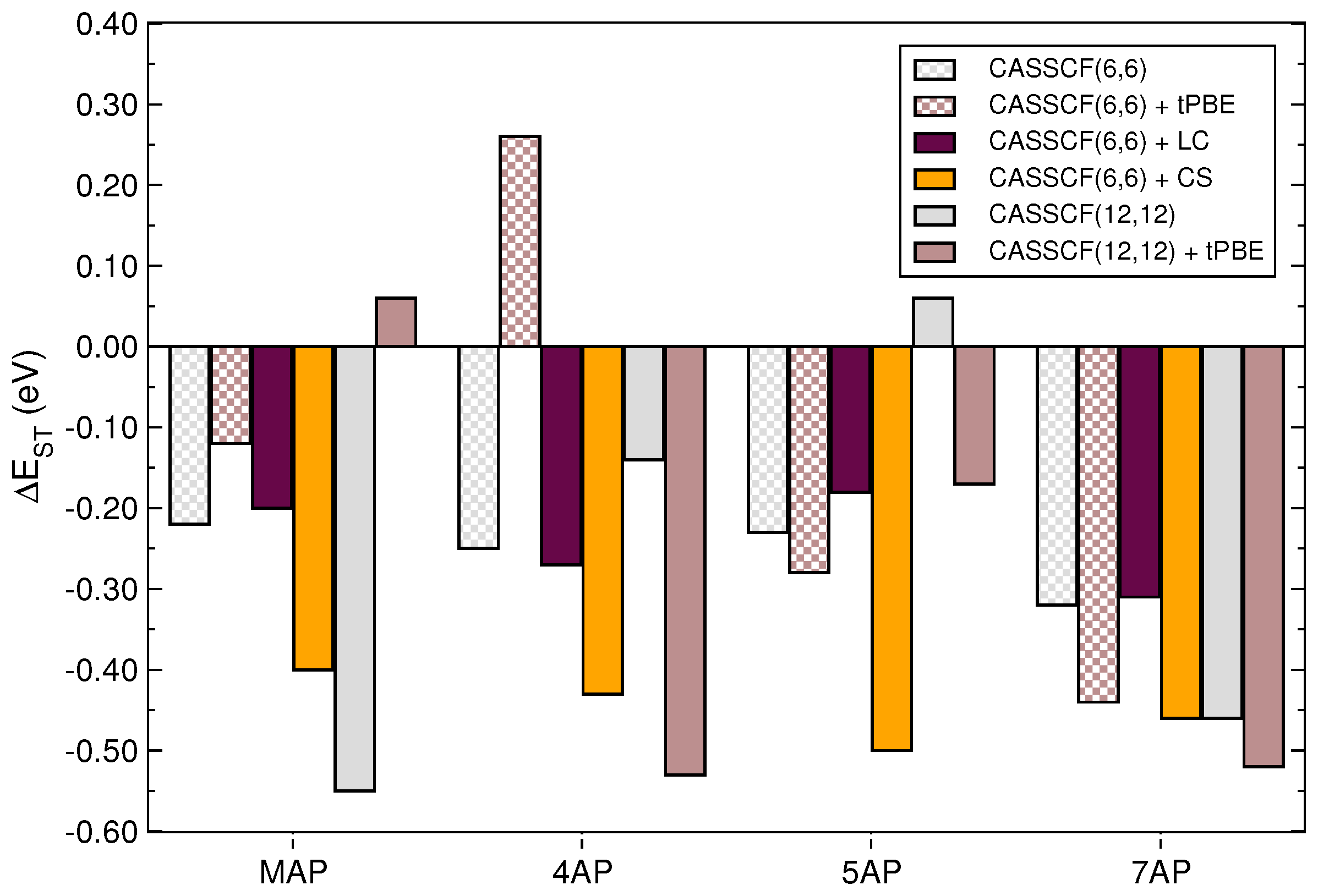
{kind=link}
{kind=link}
{kind=link}
| Molecule | Method | |||
|---|---|---|---|---|
| B97 | 1.420 | 1.195 | 0.26 | |
| MAP | B97X | 1.385 | 1.167 | 0.22 |
| B97X-2 | 0.791 | 1.166 | −0.38 | |
| B97 | 2.695 | 2.199 | 0.41 | |
| 4AP | B97X | 2.531 | 2.150 | 0.38 |
| B97X-2 | 1.642 | 2.092 | −0.45 | |
| B97 | 2.752 | 2.381 | 0.37 | |
| 5AP | B97X | 2.687 | 2.335 | 0.35 |
| B97X-2 | 1.650 | 2.298 | −0.65 | |
| B97 | 3.391 | 3.132 | 0.26 | |
| 7AP | B97X | 3.310 | 3.059 | 0.26 |
| B97X-2 | 1.933 | 2.592 | −0.66 |
| Basis Set | Molecule | Method | |||
|---|---|---|---|---|---|
| def2-SVP | MAP | CASSCF(6,6) | 1.218 | 1.436 | −0.22 |
| 4AP | CASSCF(6,6) | 2.554 | 2.803 | −0.25 | |
| 5AP | CASSCF(6,6) | 2.686 | 2.916 | −0.23 | |
| 7AP | CASSCF(6,6) | 3.896 | 4.217 | −0.32 | |
| MAP | CASSCF(12,12) | 0.145 | 0.696 | −0.55 | |
| 4AP | CASSCF(12,12) | 2.214 | 2.358 | −0.14 | |
| 5AP | CASSCF(12,12) | 2.581 | 2.519 | 0.06 | |
| 7AP | CASSCF(12,12) | 2.752 | 3.210 | −0.46 | |
| def2-TZVP | MAP | CASSCF(6,6) | 1.256 | 1.427 | −0.17 |
| 4AP | CASSCF(6,6) | 2.964 | 2.864 | 0.10 | |
| 5AP | CASSCF(6,6) | 2.995 | 3.068 | −0.07 | |
| 7AP | CASSCF(6,6) | 5.237 | 4.437 | 0.80 | |
| MAP | CASSCF(12,12) | 0.179 | 0.722 | −0.54 | |
| 4AP | CASSCF(12,12) | 1.977 | 2.171 | −0.19 | |
| 5AP | CASSCF(12,12) | 2.762 | 2.688 | 0.07 | |
| 7AP | CASSCF(12,12) | 4.334 | 4.637 | −0.30 |
| Basis Set | Molecule | Method | |||
|---|---|---|---|---|---|
| def2-SVP | MAP | CASSCF(6,6) + tPBE | 1.168 | 1.284 | −0.12 |
| 4AP | CASSCF(6,6) + tPBE | 2.135 | 1.871 | 0.26 | |
| 5AP | CASSCF(6,6) + tPBE | 2.153 | 2.437 | −0.28 | |
| 7AP | CASSCF(6,6) + tPBE | 2.715 | 3.155 | −0.44 | |
| MAP | CASSCF(12,12) + tPBE | 1.523 | 1.463 | 0.06 | |
| 4AP | CASSCF(12,12) + tPBE | 2.181 | 2.713 | −0.53 | |
| 5AP | CASSCF(12,12) + tPBE | 2.720 | 2.889 | −0.17 | |
| 7AP | CASSCF(12,12) + tPBE | 2.849 | 3.373 | −0.52 | |
| def2-TZVP | MAP | CASSCF(6,6) + tPBE | 1.191 | 1.209 | −0.02 |
| 4AP | CASSCF(6,6) + tPBE | 2.038 | 1.865 | 0.17 | |
| 5AP | CASSCF(6,6) + tPBE | 1.140 | 2.769 | −1.63 | |
| 7AP | CASSCF(6,6) + tPBE | 4.641 | 3.325 | 1.32 | |
| MAP | CASSCF(12,12) + tPBE | 1.515 | 1.438 | 0.08 | |
| 4AP | CASSCF(12,12) + tPBE | 2.420 | 2.572 | −0.15 | |
| 5AP | CASSCF(12,12) + tPBE | 2.304 | 2.579 | −0.17 | |
| 7AP | CASSCF(12,12) + tPBE | 1.906 | 2.148 | −0.24 |
| Basis Set | Molecule | Method | |||
|---|---|---|---|---|---|
| def2-SVP | MAP | CASSCF(6,6) + LC | 1.737 | 1.941 | −0.20 |
| CASSCF(6,6) + CS | 0.968 | 1.364 | −0.40 | ||
| 4AP | CASSCF(6,6) + LC | 3.116 | 3.386 | −0.27 | |
| CASSCF(6,6) + CS | 2.501 | 2.931 | −0.43 | ||
| 5AP | CASSCF(6,6) + LC | 3.273 | 3.453 | −0.18 | |
| CASSCF(6,6) + CS | 2.543 | 3.043 | −0.50 | ||
| 7AP | CASSCF(6,6) + LC | 4.568 | 4.892 | −0.32 | |
| CASSCF(6,6) + CS | 3.554 | 4.017 | −0.46 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sancho-García, J.-C.; San-Fabián, E. Organic Emitters Showing Excited-States Energy Inversion: An Assessment of MC-PDFT and Correlation Energy Functionals Beyond TD-DFT. Computation 2022, 10, 13. https://doi.org/10.3390/computation10020013
Sancho-García J-C, San-Fabián E. Organic Emitters Showing Excited-States Energy Inversion: An Assessment of MC-PDFT and Correlation Energy Functionals Beyond TD-DFT. Computation. 2022; 10(2):13. https://doi.org/10.3390/computation10020013
Chicago/Turabian StyleSancho-García, Juan-Carlos, and Emilio San-Fabián. 2022. "Organic Emitters Showing Excited-States Energy Inversion: An Assessment of MC-PDFT and Correlation Energy Functionals Beyond TD-DFT" Computation 10, no. 2: 13. https://doi.org/10.3390/computation10020013
APA StyleSancho-García, J.-C., & San-Fabián, E. (2022). Organic Emitters Showing Excited-States Energy Inversion: An Assessment of MC-PDFT and Correlation Energy Functionals Beyond TD-DFT. Computation, 10(2), 13. https://doi.org/10.3390/computation10020013