Mathematical Modeling of The Challenge to Detect Pancreatic Adenocarcinoma Early with Biomarkers

Abstract
:1. Introduction
2. Results
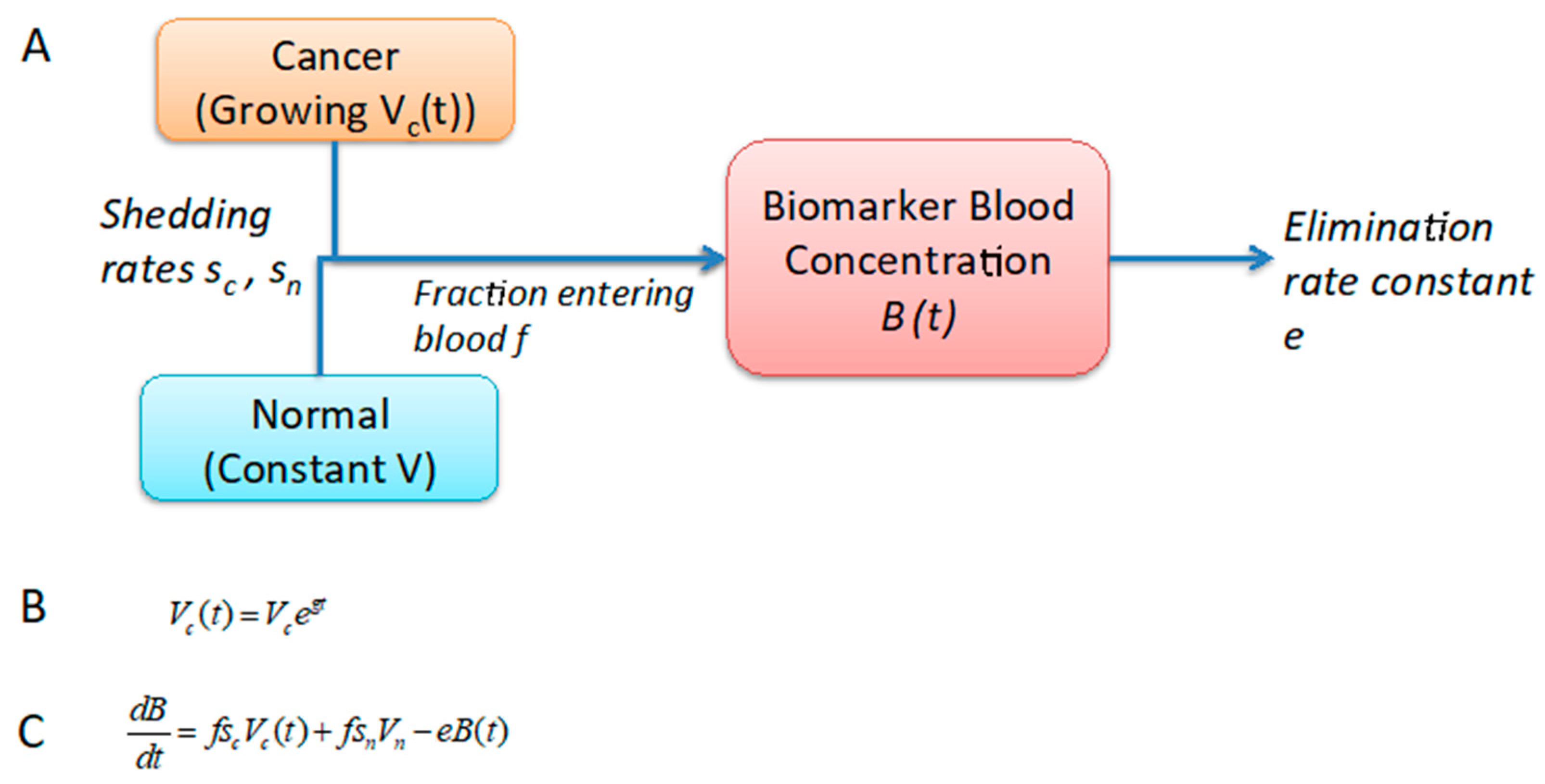
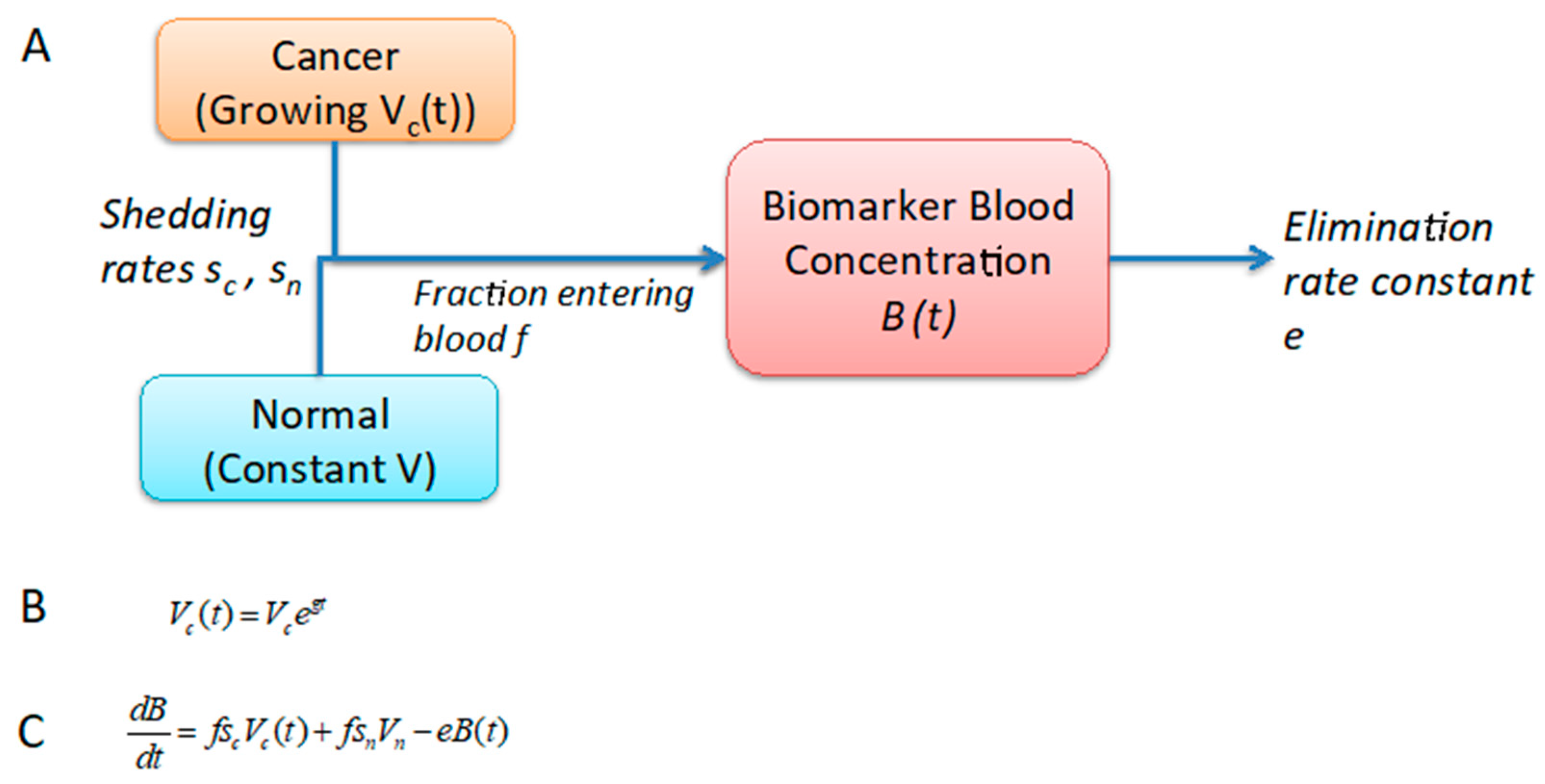
2.1. Development of a One-Compartment Biomarker Model for PDAC
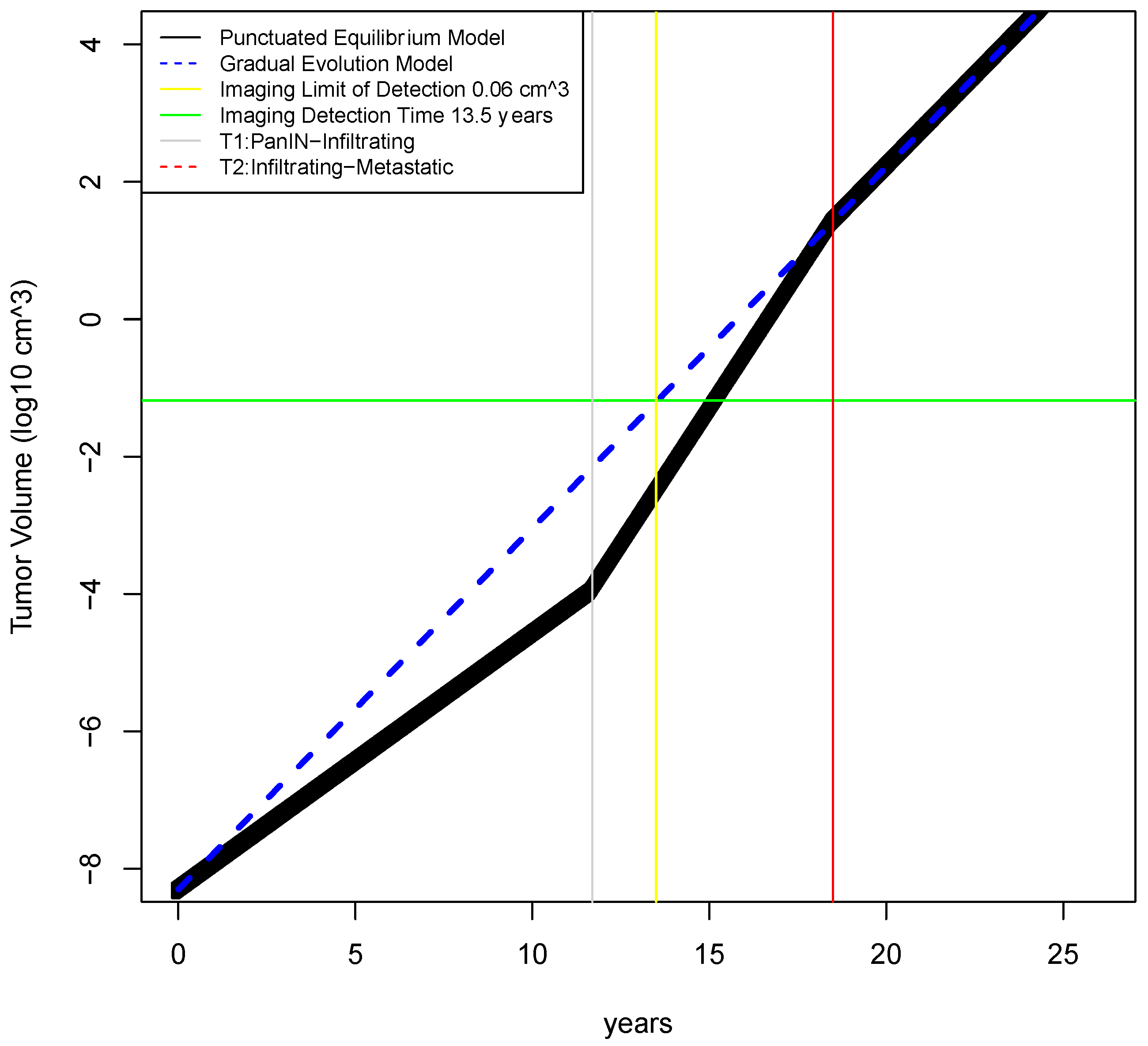
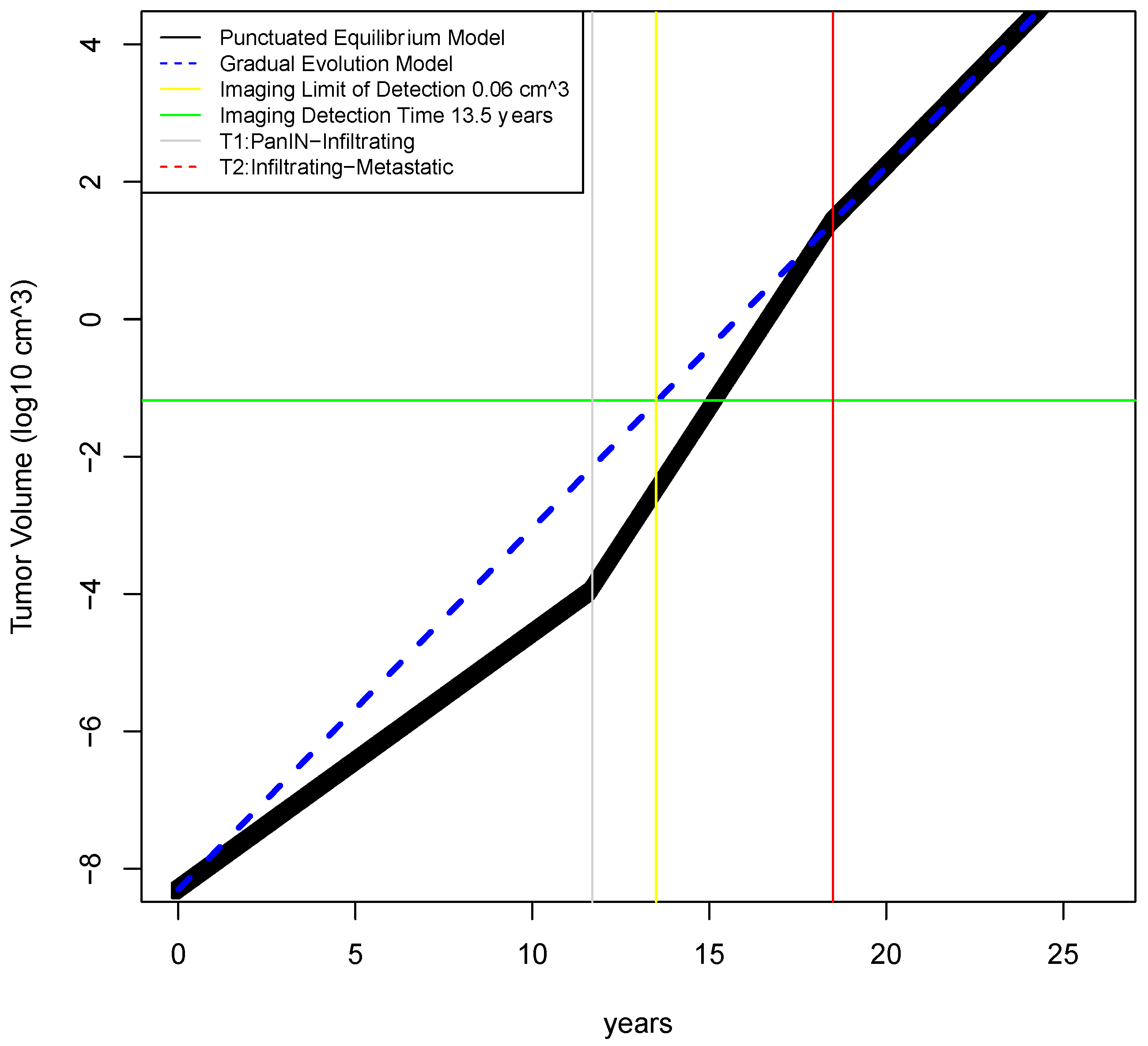
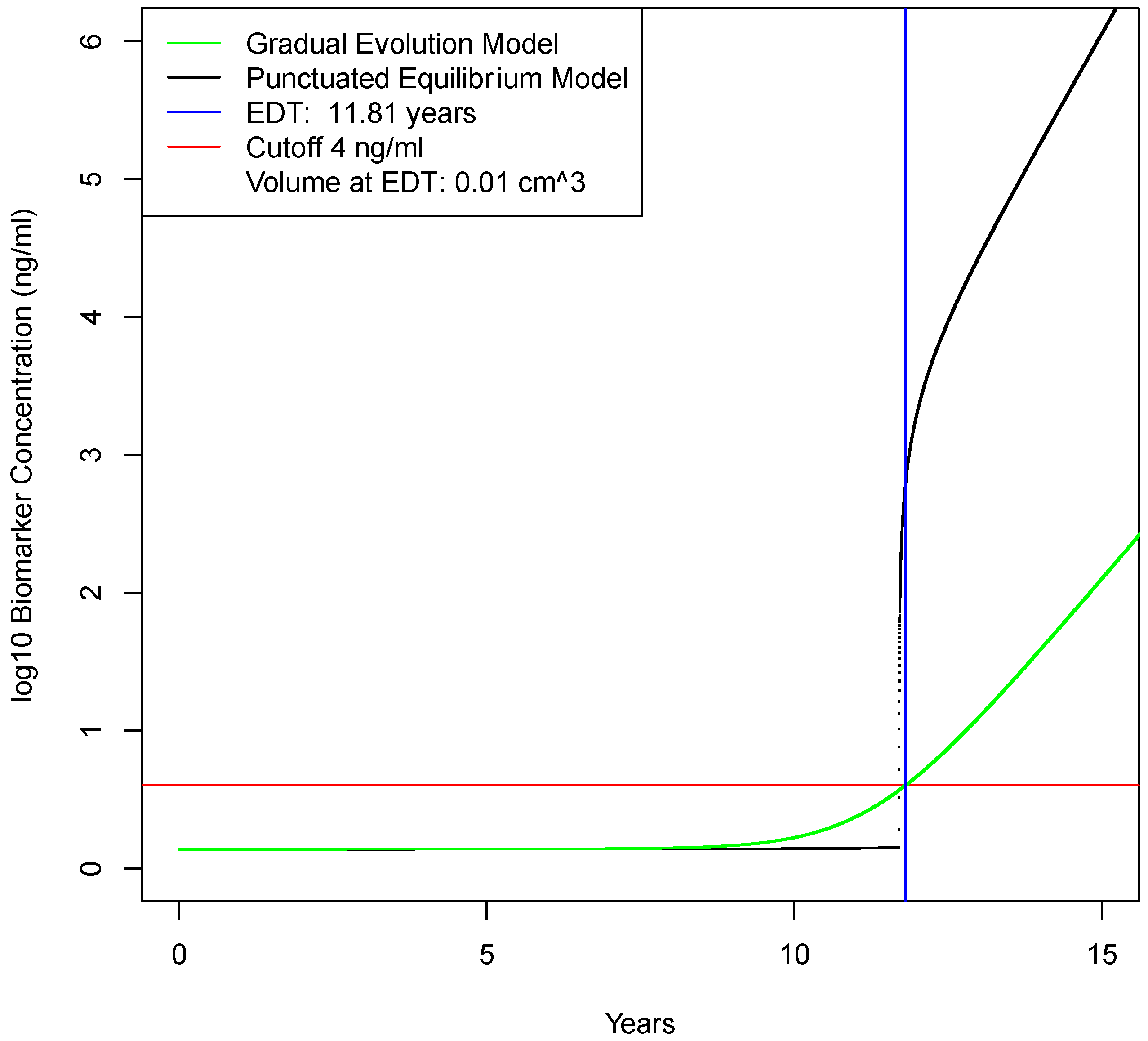
2.2. Determination of Earliest Detection Times for Baseline Parameter Values
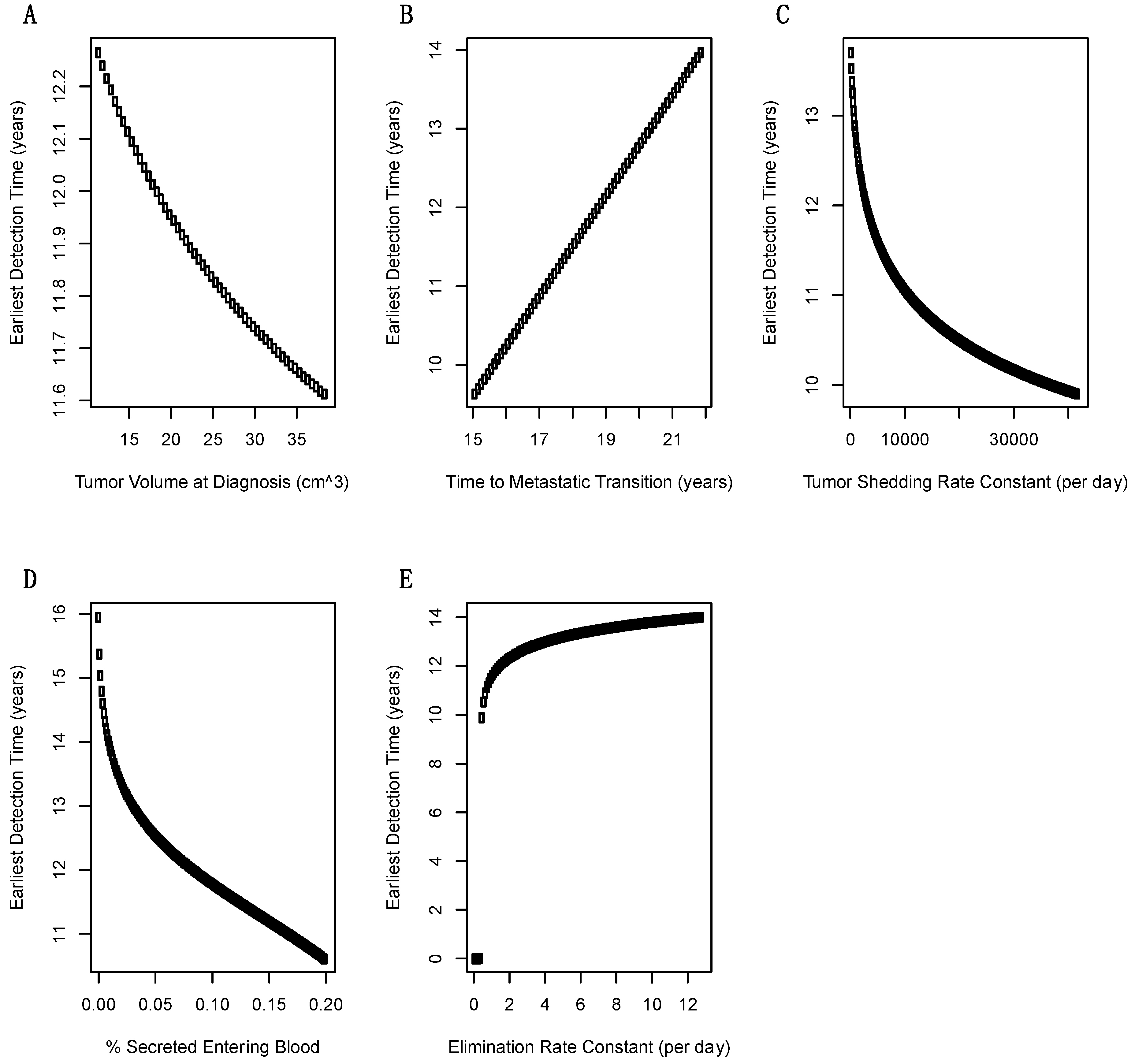
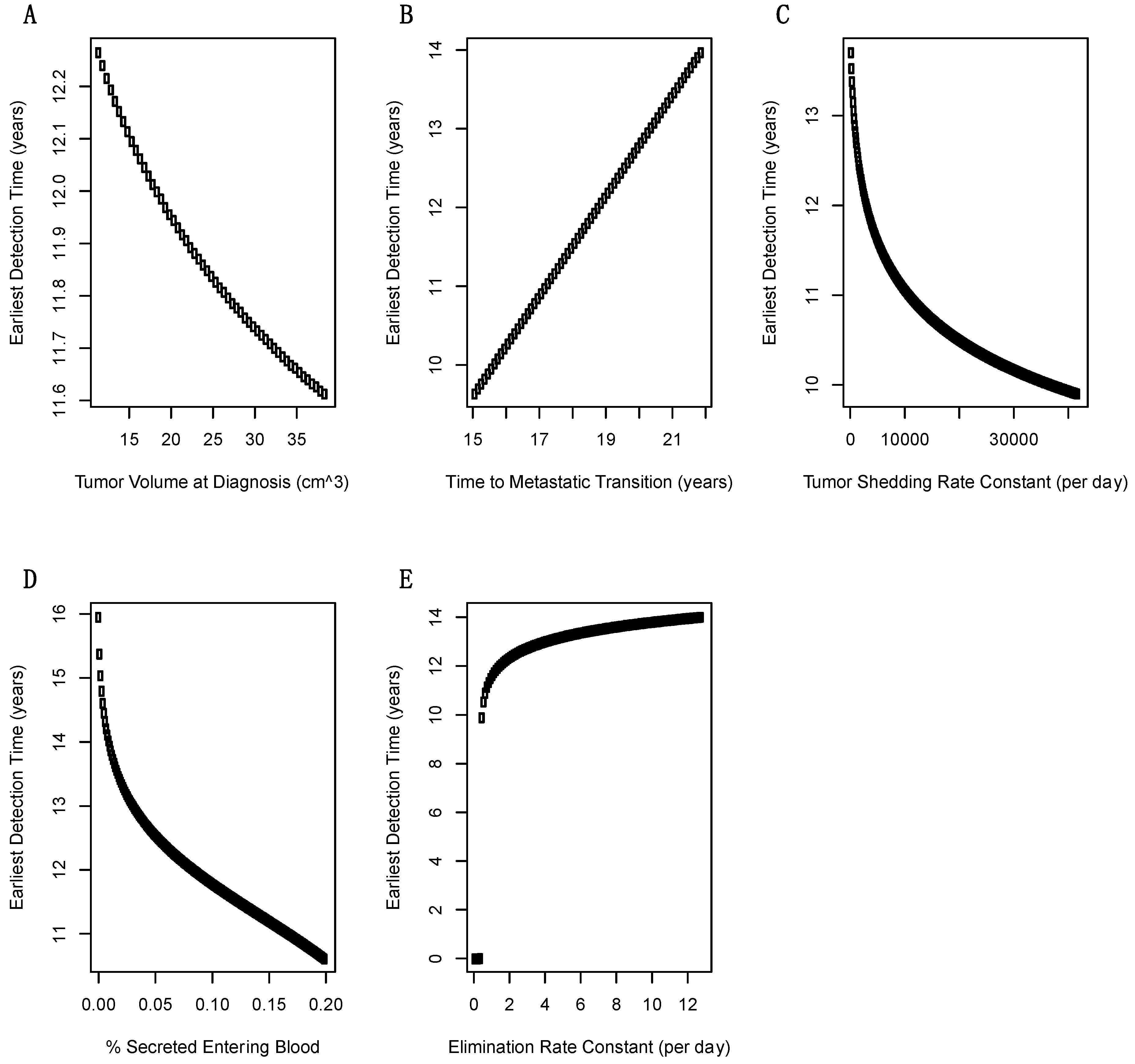
2.3. Sensitivity Analysis of Model Parameters on Earliest Detection Times
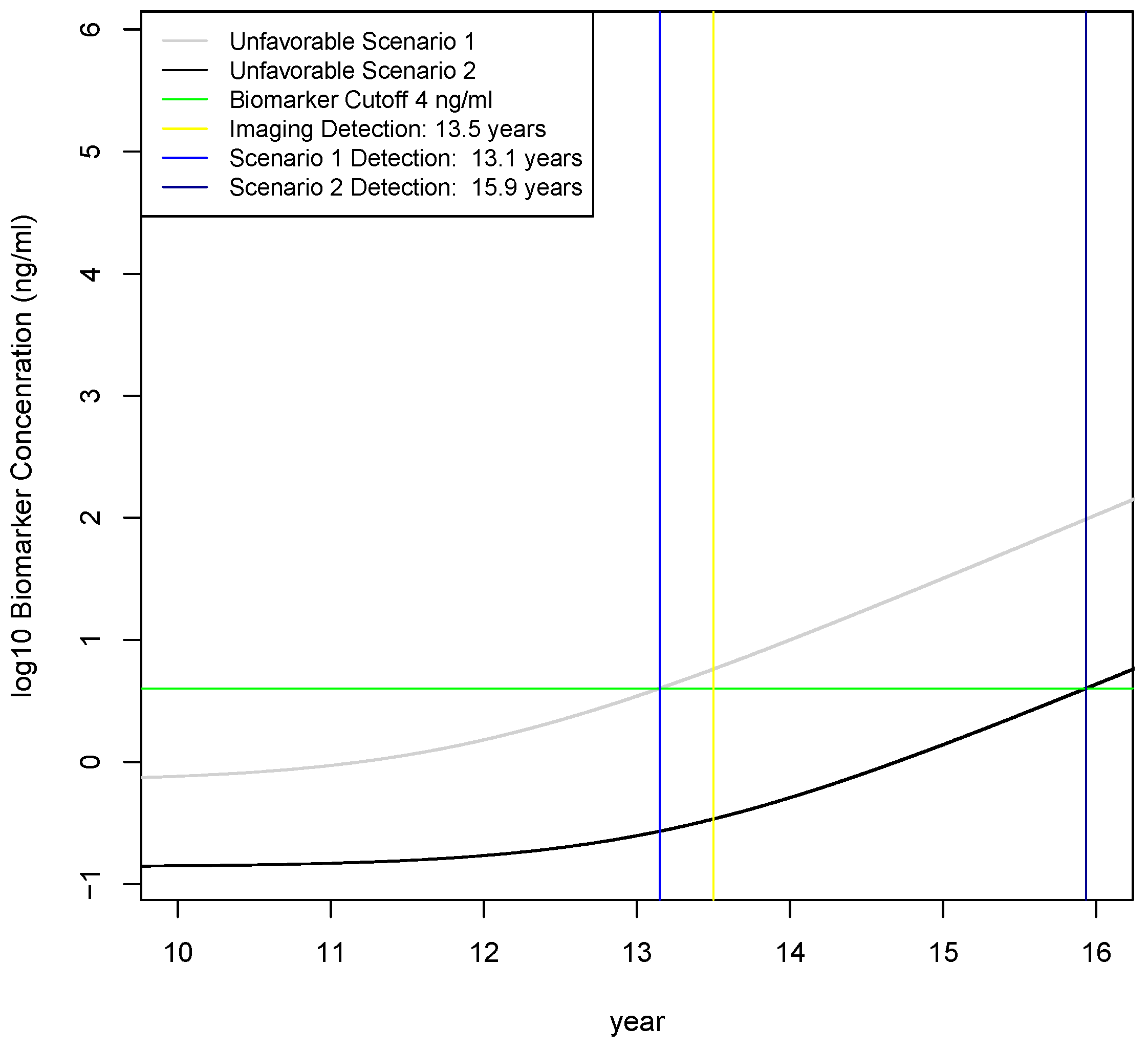
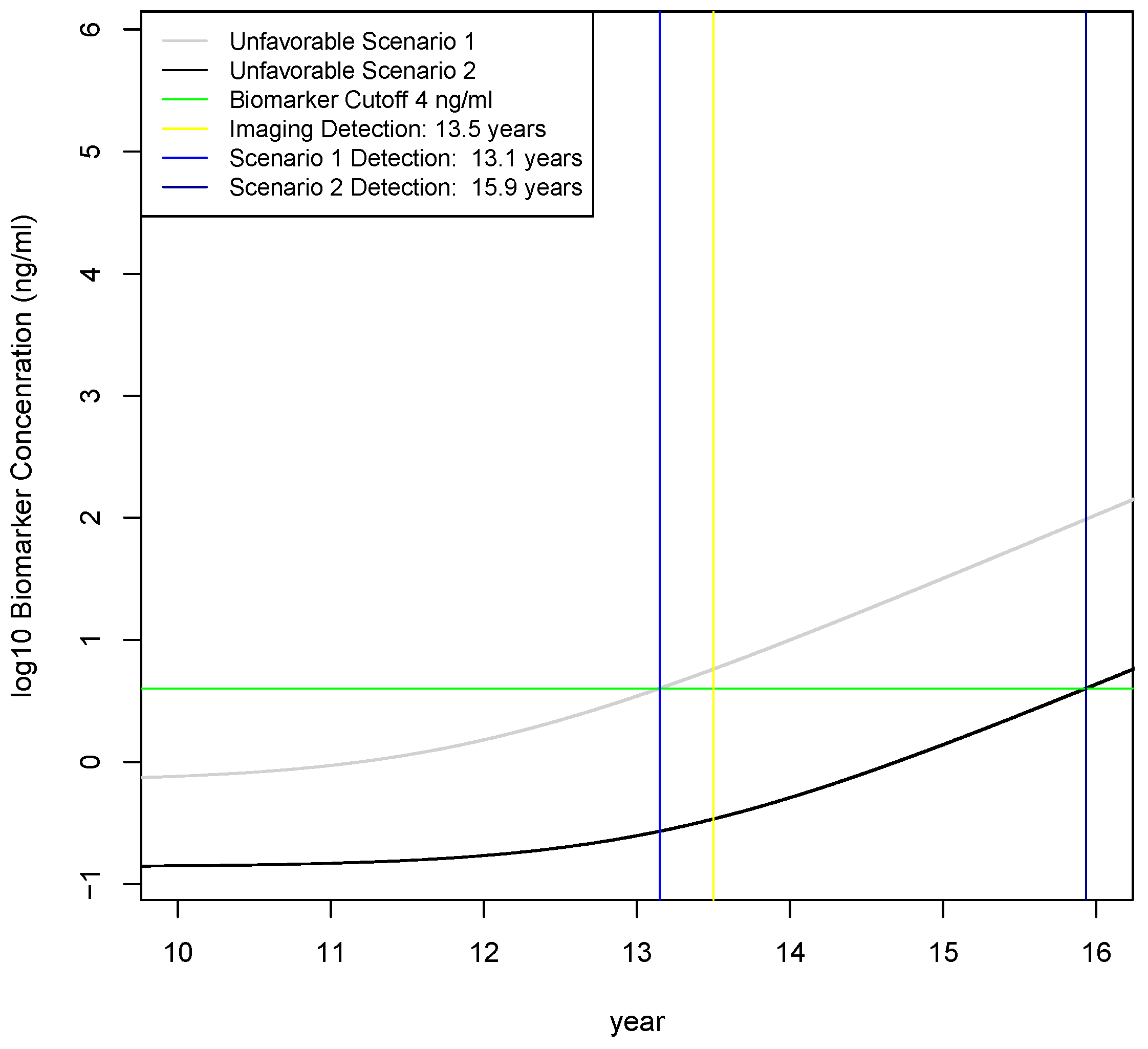
2.4. Calculation of Earliest Detection Times for Two Unfavorable Scenarios
3. Discussion
4. Methods
4.1. Estimation of Tumor Growth Rates
4.2. Estimation of Biomarker Concentration in Blood
4.3. Sensitivity of Earliest Detection Times over Variations in Model Parameters
4.4. Investigation of Three Scenarios on Earliest Detection Times
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Kleeff, J.; Korc, M.; Apte, M.; La Vecchia, C.; Johnson, C.D.; Biankin, A.V.; Neale, R.E.; Tempero, M.; Tuveson, D.A.; Hruban, R.H.; et al. Pancreatic cancer. Nat. Rev. Dis. Primers 2016, 2, 16022. [Google Scholar] [CrossRef]
- Lennon, A.M.; Wolfgang, C.L.; Canto, M.I.; Klein, A.P.; Herman, J.M.; Goggins, M.; Fishman, E.K.; Kamel, I.; Weiss, M.J.; Diaz, L.A.; et al. The early detection of pancreatic cancer: What will it take to diagnose and treat curable pancreatic neoplasia? Cancer Res. 2014, 74, 3381–3389. [Google Scholar] [CrossRef]
- Rifai, N.; Gillette, M.A.; Carr, S.A. Protein biomarker discovery and validation: The long and uncertain path to clinical utility. Nat. Biotechnol. 2006, 24, 971–983. [Google Scholar] [CrossRef]
- Kern, S.E. Why your new cancer biomarker may never work: Recurrent patterns and remarkable diversity in biomarker failures. Cancer Res. 2012, 72, 6097–6101. [Google Scholar] [CrossRef]
- Yachida, S.; Jones, S.; Bozic, I.; Antal, T.; Leary, R.; Fu, B.; Kamiyama, M.; Hruban, R.H.; Eshleman, J.R.; Nowak, M.A.; et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature 2010, 467, 1114–1117. [Google Scholar] [CrossRef] [PubMed]
- Notta, F.; Chan-Seng-Yue, M.; Lemire, M.; Li, Y.; Wilson, G.W.; Connor, A.A.; Denroche, R.E.; Liang, S.B.; Brown, A.M.; Kim, J.C.; et al. A renewed model of pancreatic cancer evolution based on genomic rearrangement patterns. Nature 2016, 538, 378–382. [Google Scholar] [CrossRef] [PubMed]
- Altrock, P.M.; Liu, L.L.; Michor, F. The mathematics of cancer: Integrating quantitative models. Nat. Rev. Cancer 2015, 15, 730–745. [Google Scholar] [CrossRef] [PubMed]
- Swanson, K.R.; True, L.D.; Lin, D.W.; Buhler, K.R.; Vessella, R.; Murray, J.D. A quantitative model for the dynamics of serum prostate-specific antigen as a marker for cancerous growth: An explanation for a medical anomaly. Am. J. Pathol. 2001, 158, 2195–2199. [Google Scholar] [CrossRef]
- Lutz, A.M.; Willmann, J.K.; Cochran, F.V.; Ray, P.; Gambhir, S.S. Cancer screening: A mathematical model relating secreted blood biomarker levels to tumor sizes. PLoS Med. 2008, 5, e170. [Google Scholar] [CrossRef]
- Hori, S.S.; Gambhir, S.S. Mathematical model identifies blood biomarker-based early cancer detection strategies and limitations. Sci. Transl. Med. 2011, 3, 109ra116. [Google Scholar] [CrossRef]
- Hori, S.S.; Lutz, A.M.; Paulmurugan, R.; Gambhir, S.S. A Model-Based Personalized Cancer Screening Strategy for Detecting Early-Stage Tumors Using Blood-Borne Biomarkers. Cancer Res. 2017, 77, 2570–2584. [Google Scholar] [CrossRef] [PubMed]
- Root, A.; Allen, P.; Tempst, P.; Yu, K. Protein Biomarkers for Early Detection of Pancreatic Ductal Adenocarcinoma: Progress and Challenges. Cancers 2018, 10, 67. [Google Scholar] [CrossRef]
- Haeno, H.; Gonen, M.; Davis, M.B.; Herman, J.M.; Iacobuzio-Donahue, C.A.; Michor, F. Computational modeling of pancreatic cancer reveals kinetics of metastasis suggesting optimum treatment strategies. Cell 2012, 148, 362–375. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, H.; Iwata, R.; Moriyama, N. Growth rate of pancreatic adenocarcinoma: Initial clinical experience. Pancreas 2001, 22, 366–369. [Google Scholar] [CrossRef]
- Kisfalvi, K.; Eibl, G.; Sinnett-Smith, J.; Rozengurt, E. Metformin disrupts crosstalk between G protein-coupled receptor and insulin receptor signaling systems and inhibits pancreatic cancer growth. Cancer Res. 2009, 69, 6539–6545. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Garcia-D’Angeli, A.; Brennan, J.P.; Huo, Q. Predicting detection limits of enzyme-linked immunosorbent assay (ELISA) and bioanalytical techniques in general. Analyst 2014, 139, 439–445. [Google Scholar] [CrossRef]
- Matsuno, S.; Egawa, S.; Fukuyama, S.; Motoi, F.; Sunamura, M.; Isaji, S.; Imaizumi, T.; Okada, S.; Kato, H.; Suda, K.; et al. Pancreatic Cancer Registry in Japan: 20 years of experience. Pancreas 2004, 28, 219–230. [Google Scholar] [CrossRef]
- Hart, D.; Shochat, E.; Agur, Z. The growth law of primary breast cancer as inferred from mammography screening trials data. Br. J. Cancer 1998, 78, 382–387. [Google Scholar] [CrossRef] [PubMed]
- Benzekry, S.; Lamont, C.; Beheshti, A.; Tracz, A.; Ebos, J.M.; Hlatky, L.; Hahnfeldt, P. Classical mathematical models for description and prediction of experimental tumor growth. PLoS Comput. Biol. 2014, 10, e1003800. [Google Scholar] [CrossRef] [PubMed]
- Anderson, A.R.; Quaranta, V. Integrative mathematical oncology. Nat. Rev. Cancer 2008, 8, 227–234. [Google Scholar] [CrossRef]
- Wang, Z.; Butner, J.D.; Kerketta, R.; Cristini, V.; Deisboeck, T.S. Simulating cancer growth with multiscale agent-based modeling. Semin. Cancer Biol. 2015, 30, 70–78. [Google Scholar] [CrossRef]
- DuFort, C.C.; DelGiorno, K.E.; Hingorani, S.R. Mounting Pressure in the Microenvironment: Fluids, Solids, and Cells in Pancreatic Ductal Adenocarcinoma. Gastroenterology 2016, 150, 1545–1557. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.J.; Huang, J.; England, C.G.; McNally, L.R.; Frieboes, H.B. Predictive modeling of in vivo response to gemcitabine in pancreatic cancer. PLoS Comput. Biol. 2013, 9, e1003231. [Google Scholar] [CrossRef]
- Commisso, C.; Davidson, S.M.; Soydaner-Azeloglu, R.G.; Parker, S.J.; Kamphorst, J.J.; Hackett, S.; Grabocka, E.; Nofal, M.; Drebin, J.A.; Thompson, C.B.; et al. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 2013, 497, 633–637. [Google Scholar] [CrossRef]
- Levitt, D.G.; Levitt, M.D. Human serum albumin homeostasis: A new look at the roles of synthesis, catabolism, renal and gastrointestinal excretion, and the clinical value of serum albumin measurements. Int. J. Gen. Med. 2016, 9, 229–255. [Google Scholar] [CrossRef] [PubMed]
- Mehrara, E.; Forssell-Aronsson, E.; Johanson, V.; Kolby, L.; Hultborn, R.; Bernhardt, P. A new method to estimate parameters of the growth model for metastatic tumours. Theor. Biol. Med. Model. 2013, 10, 31. [Google Scholar] [CrossRef] [PubMed]
- Hillen, F.; Griffioen, A.W. Tumour vascularization: Sprouting angiogenesis and beyond. Cancer Metastasis Rev. 2007, 26, 489–502. [Google Scholar] [CrossRef]
- Soetaert, K.; Petzoldt, T.; Setzer, R.W. Solving Differential Equations in R: Package deSolve. J. Stat. Softw. 2010, 33, 1–25. [Google Scholar] [CrossRef]
- Marino, S.; Hogue, I.B.; Ray, C.J.; Kirschner, D.E. A methodology for performing global uncertainty and sensitivity analysis in systems biology. J. Theor. Biol. 2008, 254, 178–196. [Google Scholar] [CrossRef]
- Van Riel, N.A. Dynamic modelling and analysis of biochemical networks: Mechanism-based models and model-based experiments. Brief. Bioinform. 2006, 7, 364–374. [Google Scholar] [CrossRef] [PubMed]
- Chun, F.K.; Perrotte, P.; Briganti, A.; Benayoun, S.; Lebeau, T.; Ramirez, A.; Lewinshtein, D.J.; Valiquette, L.; Guay, J.P.; Karakiewicz, P.I. Prostate specific-antigen distribution in asymptomatic Canadian men with no clinical evidence of prostate cancer. BJU Int. 2006, 98, 50–53. [Google Scholar] [CrossRef]
- Cvijovic, M.; Almquist, J.; Hagmar, J.; Hohmann, S.; Kaltenbach, H.M.; Klipp, E.; Krantz, M.; Mendes, P.; Nelander, S.; Nielsen, J.; et al. Bridging the gaps in systems biology. Mol. Genet. Genomics 2014, 289, 727–734. [Google Scholar] [CrossRef] [PubMed]
- Gregori, J.; Mendez, O.; Katsila, T.; Pujals, M.; Salvans, C.; Villarreal, L.; Arribas, J.; Tabernero, J.; Sanchez, A.; Villanueva, J. Enhancing the Biological Relevance of Secretome-Based Proteomics by Linking Tumor Cell Proliferation and Protein Secretion. J. Proteome Res. 2014. [Google Scholar] [CrossRef] [PubMed]
- Vickers, A.J.; Thompson, I.M.; Klein, E.; Carroll, P.R.; Scardino, P.T. A commentary on PSA velocity and doubling time for clinical decisions in prostate cancer. Urology 2014, 83, 592–596. [Google Scholar] [CrossRef] [PubMed]
- Capello, M.; Bantis, L.E.; Scelo, G.; Zhao, Y.; Li, P.; Dhillon, D.S.; Patel, N.J.; Kundnani, D.L.; Wang, H.; Abbruzzese, J.L.; et al. Sequential Validation of Blood-Based Protein Biomarker Candidates for Early-Stage Pancreatic Cancer. J. Natl. Cancer Inst. 2017, 109. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Bamlet, W.R.; Oberg, A.L.; Chaffee, K.G.; Donahue, G.; Cao, X.J.; Chari, S.; Garcia, B.A.; Petersen, G.M.; Zaret, K.S. Detection of early pancreatic ductal adenocarcinoma with thrombospondin-2 and CA19-9 blood markers. Sci. Transl. Med. 2017, 9, eaah5583. [Google Scholar] [CrossRef]
- Cohen, J.D.; Javed, A.A.; Thoburn, C.; Wong, F.; Tie, J.; Gibbs, P.; Schmidt, C.M.; Yip-Schneider, M.T.; Allen, P.J.; Schattner, M.; et al. Combined circulating tumor DNA and protein biomarker-based liquid biopsy for the earlier detection of pancreatic cancers. Proc. Natl. Acad. Sci. USA 2017. [Google Scholar] [CrossRef] [PubMed]
- Kaur, S.; Smith, L.M.; Patel, A.; Menning, M.; Watley, D.C.; Malik, S.S.; Krishn, S.R.; Mallya, K.; Aithal, A.; Sasson, A.R.; et al. A Combination of MUC5AC and CA19-9 Improves the Diagnosis of Pancreatic Cancer: A Multicenter Study. Am. J. Gastroenterol. 2017, 112, 172–183. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Symbol | Unit | Baseline | Range | Source |
|---|---|---|---|---|---|
| Diameter of primary tumor at diagnosis (cohort 1: 69% Stage I–III, 31% StageIV) | 2r | cm | 3.7 | 2.8–4.2 | Haeno [13] |
| Diameter of primary tumor at diagnosis (cohort 2: 100% Stage I–III) | 2r | cm | 3 | 2.5–4 | Haeno [13] |
| Volume of primary tumor at diagnosis (cohort 1: 69% Stage I–III, 31% StageIV) | V | cm3 | 26.5 | 11.5–38.8 | (calculated from Haeno [13]) |
| Volume of primary tumor at diagnosis (cohort 2: 100% Stage I–III) | V | cm3 | 14.1 | 8.2–33.5 | (calclulated from Haeno) |
| Tumor volume doubling time | TVDT | days | 159 | 64–255 | Furukawa [14] |
| Density of cancer cells in solid tumor tissue | dc | cells/cm3 | 2 × 108 | (na) | Lutz [9] |
| Density of pancreatic cancer cells in solid tumor tissue | dc | cells/um3 | 2.85 × 103 | (na) | Kisfalvi [15] |
| Volume of a single cancer cell | vc | mm3 | 5 × 10−6 | (na) | Lutz [9] |
| Time to reach infiltrating capability—gradual model * | T3gm | year | 18.5 * | 12–25 | Yachida [5] |
| Growth rate of gradual model | g | day−1 | 0.003316444 | (na) | Calculated |
| Tumor volume doubling time for gradual model | TVDTgm | days | 209 | (na) | Calculated |
| Time to reach infiltrating capability—punctuated equilibrium model Phase 1 | T3pem | year | 11.7 | 10–23 | (estimated from Notta [6] and Yachida [5]) |
| Tumor diameter at infiltrating capacity punctuated equilibrium model | 2r | cm | 0.01 | (na) | assumption |
| Growth rate of punctuated equilibrium model, phase 1 | g1 | day−1 | 0.001089422 | (na) | Calculated |
| Growth rate of punctuated equilibrium model, phase 2 | g2 | day−1 | 0.007147335 | (na) | Calculated |
| Growth rate of punctuated equilibrium model, phase 3 | g3 | day−1 | 0.003316444 | (na) | Calculated |
| Tumor volume doubling time for punctuated equilibrium model during phase 2 | TVDTpem | days | 97 | (na) | Calculated |
| Average primary tumor volume at autopsy | V | cm3 | 524 | (na) | (calculated from Haeno [13]) |
| Average number and size of metastatic tumors at autopsy | V | n, cm3 | 100, 4.19 | (na) | (calculated from Haeno [13]) |
| Average sum of primary and metastatic tumor volumes at autopsy | V | cm3 | 943 | (na) | (calculated from Haeno [13]) |
| Parameter | Symbol | Unit | Baseline | Range | Source |
|---|---|---|---|---|---|
| Biomarker concentration in blood over time | B(t) | ng/mL | TBC * | TBC * | (na) |
| Volume of blood in a typical male or female person | VM,F | mL | 3150, 3825 | (na) | Lutz [9] |
| Shedding rate of biomarker from cancer cells | sc | ng(105cells)−1 (day)−1(mL)−1 | 2.1 | 2.1–200 | Lutz [9] |
| Shedding rate of biomarkers from tumor | sc | ng/cm3 (day)−1(mL)−1 | 4200 | 420–42,000 | calculated from Lutz |
| Fraction of biomarker that enters blood from interstitium | f | (na) | 10% | (na) | Lutz [9] |
| Steady State biomarker concentration in healthy controls | Bn | ng/ml | 1.38 | (na) | Lutz [9] |
| Influx of biomarker shed from normal cells | VnSn | ng(day)−1(mL)−1 | 17.75 | (na) | calculated |
| Biomarker elimination rate from blood | e | day−1 | 1.286 | 0.129–12.86 | Swanson [8] |
| Limit of detection for molecular assay | LODassay | ng/mL | 0.1 | 0.01–1 | Zhang [16] |
| Limit of detection for imaging | LODimaging | mm3 | 5 mm3 | (na) | Hori [11] |
| Time of detection for imaging assuming gradual evolution model | Td | years | 11.4 | (na) | calculated |
| Parameter | Symbol | Baseline Value | Minimum | Maximum |
|---|---|---|---|---|
| Volume of primary tumor at diagnosis | Vc | 3cm diameter 2.8–4.2 Haeno [13] | 2.8 diameter = 11.49 cm3 | 4.2 diameter = 38.79 cm3 |
| T2: time at invasive-to-metastatic transition | t3 | 18.5 +/− 3.4 years Yachida [5] | 15.1 | 21.9 |
| Shedding rate of biomarkers from tumor | sc | 4200 ng/cm3 (day)−1(mL)−1 over a 100× range Lutz [9] | 420 | 42,000 |
| Fraction of biomarker that enters blood from interstitium | f | 0.10 Lutz [9] | 0.001 | 0.20 |
| Biomarker elimination rate constant from blood | e | 1.286 day−1 over a 100× range Swanson [8] | 0.1286 | 12.86 |
| Early Detection Scenario | Tumor Growth Rate Constant (g) day−1 | Tumor Secretion Rate Constant (Sc) ng/cm3 (day)−1(mL)−1 | Normal Secretion Influx (VnSn) ng/day | Biomarker Elimination Rate Constant (e) day−1 | Fraction of Biomarker Entering Blood (f) |
|---|---|---|---|---|---|
| Baseline | 0.003315953 | 4200 | 17.75 | 1.286 | 10% |
| Scenario 1 | 0.003315953 | 1/2× lower 2100 | unchanged 17.75 | unchanged 1.286 | 1/2× lower 5% |
| Scenario 2 | 0.003325953 | 10× lower 420 | unchanged 17.75 | unchanged 1.286 | 10× lower 1% |
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Root, A. Mathematical Modeling of The Challenge to Detect Pancreatic Adenocarcinoma Early with Biomarkers. Challenges 2019, 10, 26. https://doi.org/10.3390/challe10010026
Root A. Mathematical Modeling of The Challenge to Detect Pancreatic Adenocarcinoma Early with Biomarkers. Challenges. 2019; 10(1):26. https://doi.org/10.3390/challe10010026
Chicago/Turabian StyleRoot, Alex. 2019. "Mathematical Modeling of The Challenge to Detect Pancreatic Adenocarcinoma Early with Biomarkers" Challenges 10, no. 1: 26. https://doi.org/10.3390/challe10010026
APA StyleRoot, A. (2019). Mathematical Modeling of The Challenge to Detect Pancreatic Adenocarcinoma Early with Biomarkers. Challenges, 10(1), 26. https://doi.org/10.3390/challe10010026