
High Throughput Sequencing Reveals Distinct Bacterial Communities and Functional Diversity in Two Typical Coastal Bays

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Sites and Sample Collection
2.2. DNA Extraction and Sequencing
2.3. Bioinformatic Analyses
2.4. Statistical Analysis
2.5. Functional Prediction
3. Results
3.1. Sample Characteristics
3.2. Alpha Diversity of the Microbial Community
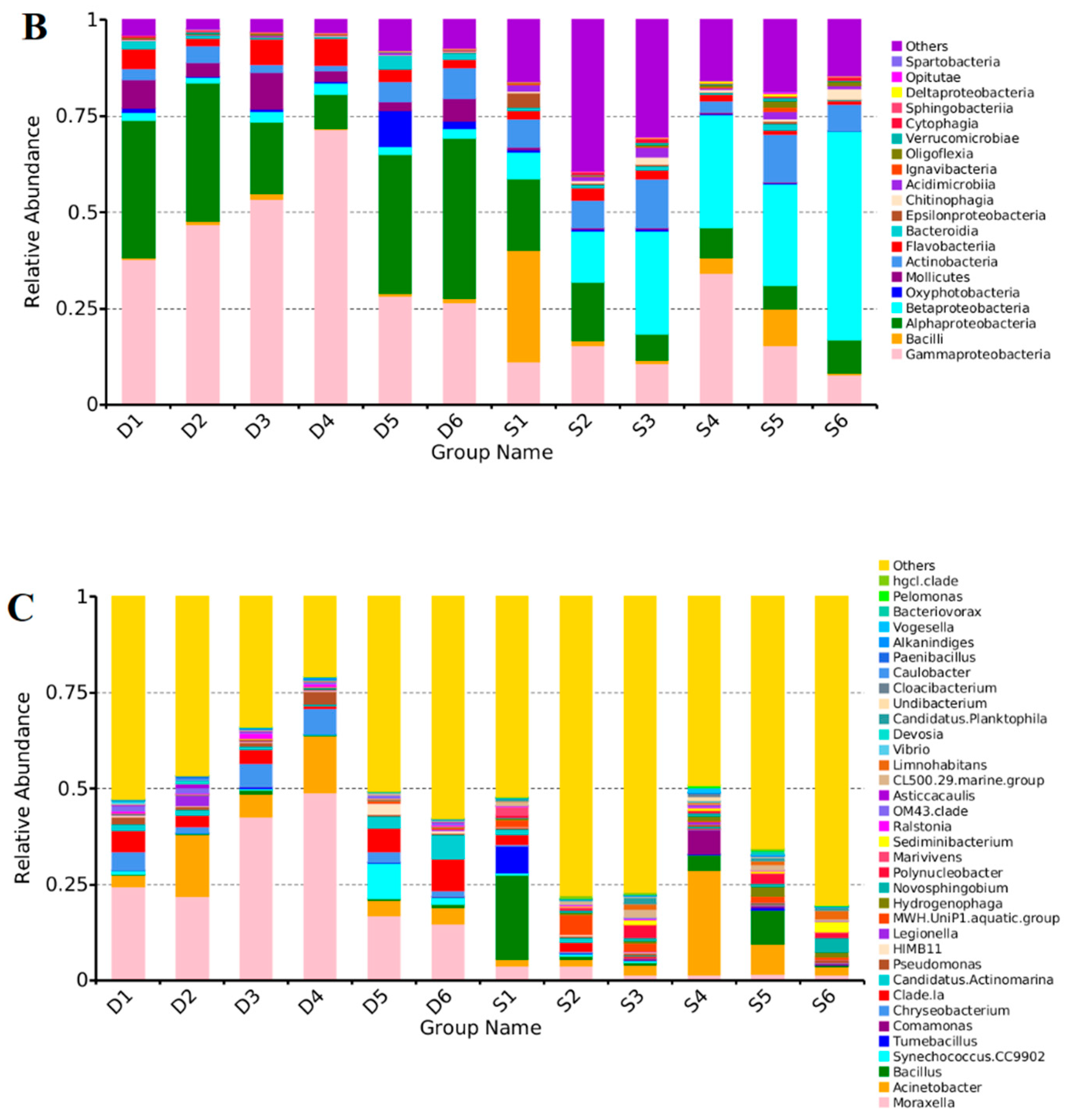
3.3. Distribution Characteristics of the Bacterial Communities
3.4. Correlations between the Microbial Communities and Water Quality
3.5. Functional Predictions of Bacterial Communities
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, Q.Y.; Ren, F.T.; Xiong, X.Y.; Gao, H.J.; Wang, Y.D.; Sun, W.J.; Leng, P.F.; Li, Z.; Bai, Y.W. Spatial distribution and contamination assessment of heavy metal pollution of sediments in coastal reclamation areas: A case study in Shenzhen Bay, China. Environ. Sci. Eur. 2021, 33, 90. [Google Scholar] [CrossRef]
- Howarth, R.; Chan, F.; Conley, D.J.; Garnier, J.; Doney, S.C.; Marino, R.; Billen, G. Coupled biogeochemical cycles: Eutrophication and hypoxia in temperate estuaries and coastal marine ecosystems. Front. Ecol. Environ. 2011, 9, 18–26. [Google Scholar] [CrossRef]
- Galloway, J.N.; Townsend, A.R.; Erisman, J.W.; Bekunda, M.; Cai, Z.; Freney, J.R.; Martinelli, L.A.; Seitzinger, S.P.; Sutton, M.A. Transformation of the nitrogen cycle: Recent trends, questions, and potential solutions. Science 2008, 320, 889–892. [Google Scholar] [CrossRef] [PubMed]
- Genitsaris, S.; Stefanidou, N.; Sommer, U.; Moustaka-Gouni, M. Phytoplankton Blooms, Red Tides and Mucilaginous Aggregates in the Urban Thessaloniki Bay, Eastern Mediterranean. Divers. Basel 2019, 11, 136. [Google Scholar] [CrossRef]
- Madsen, E.L. Microorganisms and their roles in fundamental biogeochemical cycles. Curr. Opin. Biotechnol. 2011, 22, 456–464. [Google Scholar] [CrossRef] [PubMed]
- Panayotova, M. Use of zeolite for cadmium removal from wastewater. J. Environ. Sci. Health Part A-Toxic/Hazard. Subst. Environ. Eng. 2000, 35, 1591–1601. [Google Scholar] [CrossRef]
- Lemonnier, C.; Perennou, M.; Eveillard, D.; Fernandez-Guerra, A.; Leynaert, A.; Marie, L.; Morrison, H.G.; Memery, L.; Paillard, C.; Maignien, L. Linking Spatial and Temporal Dynamic of Bacterioplankton Communities With Ecological Strategies Across a Coastal Frontal Area. Fron. Mar. Sci. 2020, 7, 376. [Google Scholar] [CrossRef]
- Jasna, V.; Nathan, V.K.; Parvathi, A. Comparison of bacterial community structure in coastal and offshore waters of the Bay of Bengal, India. Reg. Stud. Mar. Sci. 2020, 39, 101414. [Google Scholar] [CrossRef]
- Sodha, V.; Shahabuddin, S.; Gaur, R.; Ahmad, I.; Bandyopadhyay, R.; Sridewi, N. Comprehensive Review on Zeolite-Based Nanocomposites for Treatment of Effluents from Wastewater. Nanomaterials 2022, 12, 3199. [Google Scholar] [CrossRef]
- Zhao, W.; Wang, J.J.; Xu, S.; Lei, Y.; Yang, R.; Shi, L.Y.; Wang, X.B.; Huang, Z.Y. Bacterioplankton community variation in Bohai Bay (China) is explained by joint effects of environmental and spatial factors. Microbiologyopen 2020, 9, e997. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, Y.T.; Wang, J.N.; Luo, T.W.; Zhang, R.; Sun, J.; Zheng, Q.; Jiao, N.Z. Seasonal dynamics of bacterial communities in the surface seawater around subtropical Xiamen Island, China, as determined by 16S rRNA gene profiling. Mar. Pollut. Bull. 2019, 142, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Almutairi, A. Spatial-temporal variations and diversity of the bacterioplankton communities in the coastal waters of Kuwait. Mar. Pollut. Bull. 2015, 100, 699–709. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Xu, X.Y.; Wang, B.; Lv, C.P.; Shi, D.Z. Removal of Ammonium from Swine Wastewater Using Synthesized Zeolite from Fly Ash. Sustainability 2020, 12, 3423. [Google Scholar] [CrossRef]
- Sreesai, S.; Sthiannopkao, S. Utilization of zeolite industrial wastewater for removal of copper and zinc from copper-brass pipe industrial wastewater. Can. J. Civ. Eng. 2009, 36, 709–719. [Google Scholar] [CrossRef]
- Sun, F.L.; Wang, Y.S.; Wu, M.L.; Sun, C.C.; Cheng, H. Spatial and vertical distribution of bacterial community in the northern South China Sea. Ecotoxicology 2015, 24, 1478–1485. [Google Scholar] [CrossRef]
- Caruso, G.; Giacobbe, M.G.; Azzaro, F.; Decembrini, F.; Leonardi, M.; Miserocchi, S.; Cao, X.Y.; Song, C.L.; Zhou, Y.Y. All-In-One: Microbial Response to Natural and Anthropogenic Forcings in a Coastal Mediterranean Ecosystem, the Syracuse Bay (Ionian Sea, Italy). J. Mar. Sci. Eng. 2022, 10, 19. [Google Scholar] [CrossRef]
- Ren, Z.; Qu, X.D.; Zhang, M.; Yu, Y.; Peng, W.Q. Distinct Bacterial Communities in Wet and Dry Seasons During a Seasonal Water Level Fluctuation in the Largest Freshwater Lake (Poyang Lake) in China. Front Microbiol 2019, 10, 782456. [Google Scholar] [CrossRef]
- Seidel, L.; Broman, E.; Stahle, M.; Nilsson, E.; Turner, S.; Hendrycks, W.; Sachpazidou, V.; Forsman, A.; Hylander, S.; Dopson, M. Long-Term Warming of Baltic Sea Coastal Waters Affects Bacterial Communities in Bottom Water and Sediments Differently. Front Microbiol 2022, 13, 873281. [Google Scholar] [CrossRef]
- Huang, J.J.; Wang, D.F.; Gong, F.; Bai, Y.; He, X.Q. Changes in Nutrient Concentrations in Shenzhen Bay Detected Using Landsat Imagery between 1988 and 2020. Remote Sens. 2021, 13, 3469. [Google Scholar] [CrossRef]
- Xu, H.L.; Zhang, Y.; Zhu, X.Z.; Zheng, M.F. Effects of rainfall-runoff pollution on eutrophication in coastal zone: A case study in Shenzhen Bay, southern China. Hydrol. Res. 2019, 50, 1062–1075. [Google Scholar] [CrossRef]
- Li, W.J.; Su, H.C.; Cao, Y.C.; Wang, L.L.; Hu, X.J.; Xu, W.J.; Xu, Y.; Li, Z.J.; Wen, G.L. Antibiotic resistance genes and bacterial community dynamics in the seawater environment of Dapeng Cove, South China. Sci. Total Environ. 2020, 723, 138027. [Google Scholar] [CrossRef]
- Zhang, R.; Liu, W.C.; Liu, Y.; Zhang, H.L.; Zhao, Z.H.; Zou, L.Y. Impact of water quality variations on the microbial metagenome across coastal waters in Shenzhen, south China. Ocean Coast. Manage. 2021, 208, 105612. [Google Scholar] [CrossRef]
- Zhang, R.; Liu, Y.; Zhao, X.F.; Zhao, Z.H.; Zhang, H.L.; Huang, X.P.; Xu, W.Q.; Shen, Y.C.; Lan, W.S. High-throughput sequencing reveals the spatial distribution variability of microbial community in coastal waters in Shenzhen. Ecotoxicology 2021, 30, 1429–1436. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.L.; Liu, K.; Ning, T.Z.; Huang, G.; Zhang, Q.M.; Li, Y.L.; Wang, M.Q.; Fan, Y.M.; An, W.L.; Ji, L.B.; et al. Environmental remediation promotes the restoration of biodiversity in the Shenzhen Bay Estuary, South China. Ecosyst. Health Sustain. 2022, 8, 2026250. [Google Scholar] [CrossRef]
- Xu, L.; Wang, L.G.; Wang, X.H.; Van Damme, K.; Ning, J.J.; Li, Y.F.; Huang, D.L.; Liu, S.S.; Li, H.; Du, F.Y. Spatial and temporal variabilities of coastal nekton community structure and phylogenetic diversity in Daya and Dapeng Bay, southern China. Ecol. Indic. 2021, 131, 108226. [Google Scholar] [CrossRef]
- APHA. Standard Methods for the Examination of Water and Wastewater, 21st ed.; American Public Health Association: Washington, DC, USA, 2005. [Google Scholar]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef]
- Li, D.P.; Kang, X.; Chu, L.L.; Wang, Y.F.; Song, X.S.; Zhao, X.X.; Cao, X. Algicidal mechanism of Raoultella ornithinolytica against Microcystis aeruginosa: Antioxidant response, photosynthetic system damage and microcystin degradation. Environ. Pollut. 2021, 287, 117644. [Google Scholar] [CrossRef]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R. Vegan: Community Ecology Package. R Package Version 2.4-3. 2017. Available online: https://CRAN.R-project.org/package=vegan (accessed on 7 April 2017).
- Lozupone, C.; Knight, R. UniFrac: A new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 2005, 71, 8228–8235. [Google Scholar] [CrossRef]
- White, J.R.; Nagarajan, N.; Pop, M. Statistical methods for detecting differentially abundant features in clinical metagenomic samples. PLoS Comp. Biol. 2009, 5, e1000352. [Google Scholar] [CrossRef]
- Reshef, D.N.; Reshef, Y.A.; Finucane, H.K.; Grossman, S.R.; McVean, G.; Turnbaugh, P.J.; Lander, E.S.; Mitzenmacher, M.; Sabeti, P.C. Detecting Novel Associations in Large Data Sets. Science 2011, 334, 1518–1524. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Langille, M.G.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Thurber, R.L.V.; Knight, R. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.C.; Dissard, D.; Douville, E.; Blamart, D.; Bordier, L.; Tribollet, A.; Le Cornec, F.; Pons-Branchu, E.; Dapoigny, A.; Lazareth, C.E. Surface ocean pH variations since 1689 CE and recent ocean acidification in the tropical South Pacific. Nat. Commun. 2018, 9, 2543. [Google Scholar] [CrossRef]
- Yan, Q.; Cheng, T.T.; Song, J.T.; Zhou, J.; Hung, C.C.; Cai, Z.H. Internal nutrient loading is a potential source of eutrophication in Shenzhen Bay, China. Ecol. Indic. 2021, 127, 107736. [Google Scholar] [CrossRef]
- Lu, Z.Y.; Liu, Z.B.; Zhang, C.J.; Wei, Q.Y.; Zhang, S.Y.; Li, M. Spatial and seasonal variations of sediment bacterial communities in a river-bay system in South China. Appl. Microbiol. Biotechnol. 2021, 105, 1979–1989. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Yao, J.; Wang, X.; Zheng, M.; Guo, D.; Chen, Y. Efficient municipal wastewater treatment by oxidation ditch process at low temperature: Bacterial community structure in activated sludge. Sci. Total Environ. 2020, 703, 135031. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Huang, J.M.; Zhou, Y.L.; Almeida, A.; Finn, R.D.; Danchin, A.; He, L.S. Phylogenomics of expanding uncultured environmental Tenericutes provides insights into their pathogenicity and evolutionary relationship with Bacilli. BMC Genom. 2020, 21, 408. [Google Scholar] [CrossRef] [PubMed]
- Cleary, D.F.R.; Huang, Y.S.M. A comparison of the prokaryotic communities associated with seven seaweed species, sediment, and seawater from the Penghu archipelago, Taiwan. Mar. Biol. Res. 2020, 16, 744–761. [Google Scholar] [CrossRef]
- Ismail, N.; Almutairi, A. Bacterioplankton Community Profiling of the Surface Waters of Kuwait. Fron. Mar. Sci. 2022, 9, 838101. [Google Scholar] [CrossRef]
- Laas, P.; Ugarelli, K.; Travieso, R.; Stumpf, S.; Gaiser, E.E.; Kominoski, J.S.; Stingl, U. Water Column Microbial Communities Vary along Salinity Gradients in the Florida Coastal Everglades Wetlands. Microorganisms 2022, 10, 215. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.G.; Dai, T.J.; Tang, Y.S.; Tao, Y.L.; Huang, B.; Mu, Q.L.; Wen, D.H. Sediment bacterial community structures and their predicted functions implied the impacts from natural processes and anthropogenic activities in coastal area. Mar. Pollut. Bull. 2018, 131, 481–495. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Ki, J.-S.; Qian, P.-Y. Microbial diversity in polluted harbor sediments I: Bacterial community assessment based on four clone libraries of 16S rDNA. Estuar. Coast. Shelf Sci. 2008, 76, 668–681. [Google Scholar] [CrossRef]
- Orel, N.; Fadeev, E.; Klun, K.; Licer, M.; Tinta, T.; Turk, V. Bacterial Indicators Are Ubiquitous Members of Pelagic Microbiome in Anthropogenically Impacted Coastal Ecosystem. Front Microbiol 2022, 12, 765091. [Google Scholar] [CrossRef] [PubMed]
- Li, X.H.; You, C.; Qu, L.; Zhou, B.; Tang, X.X.; Xiao, H. Bacterial communities fluctuate in abundance and diversity under simulated oil-contaminated seawater conditions. J. Oceanol. Limnol. 2019, 37, 615–627. [Google Scholar] [CrossRef]
- Li, Y.B.; Huang, D.Y.; Sun, W.M.; Sun, X.X.; Yan, G.; Gao, W.L.; Lin, H.Z. Characterizing sediment bacterial community and identifying the biological indicators in a seawater-freshwater transition zone during the wet and dry seasons. Environ. Sci Pollut R 2022, 29, 41219–41230. [Google Scholar] [CrossRef]
- Chen, Y.; Li, J.; Lyu, Y.J.; Zou, Y.Y.; Li, Q.Q.; Yang, Q.S.; Tang, X.Y.; Yuan, X.C.; Jiang, Z.J.; Zhang, S. Interaction and Assembly of Bacterial Communities in High-Latitude Coral Habitat Associated Seawater. Microorganisms 2022, 10, 558. [Google Scholar] [CrossRef]
- Li, Z.; Yu, E.; Zhang, K.; Gong, W.; Xia, Y.; Tian, J.; Wang, G.; Xie, J. Water Treatment Effect, Microbial Community Structure, and Metabolic Characteristics in a Field-Scale Aquaculture Wastewater Treatment System. Front Microbiol 2020, 11, 930. [Google Scholar] [CrossRef]
- Behnami, A.; Farajzadeh, D.; Isazadeh, S.; Benis, K.Z.; Shakerkhatibi, M.; Shiri, Z.; Ghorghanlu, S.; Yadeghari, A. Diversity of bacteria in a full-scale petrochemical wastewater treatment plant experiencing stable hydrocarbon removal. J. Water Process Eng. 2018, 23, 285–291. [Google Scholar] [CrossRef]
- Sakami, T.; Watanabe, T.; Kakehi, S.; Taniuchi, Y.; Kuwata, A. Spatial variation of bacterial community composition at the expiry of spring phytoplankton bloom in Sendai Bay, Japan. Gene 2016, 576, 610–617. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Site | Latitude | Longitude | Sample | Temperature | pH | Conductivity | Salinity | DO | Ammonium | Nitrite | Nitrate | Phosphate |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (℃) | (ms/cm) | (‰) | (mg/L) | |||||||||
| D1 | 22.59295 | 114.30976 | D1.1 | 31.7 | 7.31 | 33.26 | 24 | 9.58 | 1.116 | 0.001 | 0.501 | 0.087 |
| D1.2 | 31.7 | 7.14 | 28.31 | 24 | 8.43 | 1.135 | 0.005 | 0.405 | 0.081 | |||
| D1.3 | 31.7 | 7.08 | 33.22 | 23 | 8.65 | 0.824 | 0.001 | 0.489 | 0.071 | |||
| D2 | 22.59330 | 114.31009 | D2.1 | 32.2 | 7.18 | 31.29 | 24 | 7.93 | 0.669 | 0.001 | 0.423 | 0.039 |
| D2.2 | 32.2 | 7.17 | 32.8 | 24 | 9.63 | 0.98 | 0 | 0.471 | 0.018 | |||
| D2.3 | 32.2 | 7.21 | 33.06 | 24 | 8.74 | 0.727 | 0.007 | 0.481 | 0.018 | |||
| D3 | 22.59378 | 114.31073 | D3.1 | 32.3 | 7.16 | 34.38 | 24 | 9.15 | 0.416 | 0 | 0.331 | 0.039 |
| D3.2 | 32.3 | 7.17 | 31.04 | 24 | 7.77 | 0.474 | 0.005 | 0.228 | 0.05 | |||
| D3.3 | 32.4 | 7.17 | 33.5 | 25 | 8.33 | 0.533 | 0.003 | 0.507 | 0.029 | |||
| D4 | 22.5955 | 114.31338 | D4.1 | 32.7 | 7.16 | 31.26 | 22 | 7.82 | 0.999 | 0.004 | 0.458 | 0.675 |
| D4.2 | 32.8 | 7.2 | 32.36 | 24 | 8.69 | 0.299 | 0 | 0.513 | 0.675 | |||
| D4.3 | 32.8 | 7.17 | 30.35 | 22 | 8.07 | 1.135 | 0 | 0.447 | 0.717 | |||
| D5 | 22.59499 | 114.31246 | D5.1 | 32.8 | 7.19 | 28.98 | 25 | 9.19 | 1.524 | 0.007 | 0.475 | 0.024 |
| D5.2 | 32.9 | 7.17 | 32.7 | 25 | 8.93 | 1.563 | 0 | 0.486 | 0.024 | |||
| D5.3 | 32.9 | 7.16 | 32.12 | 25 | 9.57 | 1.349 | 0 | 0.382 | 0.034 | |||
| D6 | 22.5944 | 114.31148 | D6.1 | 33 | 7.19 | 30.81 | 22 | 8.03 | 0.338 | 0.001 | 0.51 | 0.024 |
| D6.2 | 32.9 | 7.21 | 30.63 | 22 | 8.29 | 0.377 | 0.004 | 0.5 | 0.024 | |||
| D6.3 | 32.8 | 7.2 | 31.27 | 21 | 8.91 | 0.144 | 0.003 | 0.489 | 0.018 | |||
| S1 | 22.48282 | 113.91925 | S1.1 | 25.4 | 8.3 | 7.231 | 6 | 5.7 | 0.105 | 0.106 | 0.681 | 0.055 |
| S1.2 | 25.9 | 8.31 | 7.461 | 5 | 7.12 | 0.086 | 0.104 | 0.793 | 0.092 | |||
| S1.3 | 25.4 | 8.27 | 7.412 | 5 | 6.54 | 0.105 | 0.113 | 1.572 | 0.039 | |||
| S2 | 22.48885 | 113.93895 | S2.1 | 26 | 8.31 | 7.827 | 5 | 7.2 | 0.124 | 0.225 | 0.589 | 0.06 |
| S2.2 | 25 | 8.36 | 7.834 | 5 | 6.24 | 0.241 | 0.229 | 1.196 | 0.081 | |||
| S2.3 | 24 | 8.37 | 7.857 | 7 | 7.38 | 0.144 | 0.246 | 1.309 | 0.039 | |||
| S3 | 22.52265 | 113.95183 | S3.1 | 25.4 | 8.36 | 1.656 | 1 | 6.86 | 0.299 | 0.129 | 1.441 | 0.05 |
| S3.2 | 26 | 8.36 | 1.506 | 1 | 7.24 | 0.299 | 0.119 | 1.173 | 0.05 | |||
| S3.3 | 24.3 | 8.38 | 1.572 | 1 | 6.73 | 0.299 | 0.093 | 1.637 | 0.045 | |||
| S4 | 22.52448 | 113.99002 | S4.1 | 23.9 | 8.22 | 0.5703 | 1 | 7.37 | 0.008 | 0.011 | 2.604 | 0.024 |
| S4.2 | 24.5 | 8.2 | 0.5717 | 1 | 7.6 | 0.086 | 0.008 | 2.524 | 0.092 | |||
| S4.3 | 24.9 | 8.18 | 0.5716 | 1 | 7.49 | 0.086 | 0.012 | 3.403 | 0.055 | |||
| S5 | 22.52988 | 114.01394 | S5.1 | 25.5 | 8.32 | 1.424 | 1 | 6.95 | 1.174 | 0.364 | 1.644 | 0.024 |
| S5.2 | 24.1 | 8.36 | 1.455 | 1 | 7.06 | 0.883 | 0.371 | 1.488 | 0.087 | |||
| S5.3 | 23.1 | 8.38 | 1.41 | 1 | 7 | 0.96 | 0.414 | 2.065 | 0.039 | |||
| S6 | 22.50845 | 114.03359 | S6.1 | 25.4 | 8.28 | 0.5617 | 1 | 5.38 | 0.338 | 0.098 | 1.936 | 0.029 |
| S6.2 | 26 | 8.28 | 0.5652 | 1 | 6.36 | 0.494 | 0.075 | 1.995 | 0.102 | |||
| S6.3 | 25 | 8.3 | 0.5598 | 1 | 6.22 | 0.299 | 0.093 | 2.573 | 0.029 | |||
| Group | R-Value | p-Value |
|---|---|---|
| D vs. S | 0.8155 | 0.001 |
| D (between) | 0.046 | 0.349 |
| S (between) | 0.737 | 0.001 |
| Factor | r | p |
|---|---|---|
| Temperature | 0.6186 | 0.001 |
| pH | 0.6510 | 0.001 |
| Conductivity | 0.6877 | 0.001 |
| DO | 0.4593 | 0.001 |
| salinity | 0.6713 | 0.001 |
| Ammonium | 0.0771 | 0.049 |
| Nitrite | 0.2613 | 0.001 |
| Nitrate | 0.5120 | 0.001 |
| Phosphate | 0.0535 | 0.151 |
| Physicochemical factors | 0.6807 | 0.001 |
| Nutrients | 0.4968 | 0.001 |
| Environmental Factor | Average MIC a | Degree b | Positive | Negative |
|---|---|---|---|---|
| Nitrate | 0.826318 | 14 | 10 | 4 |
| Nitrite | 0.790931 | 8 | 8 | 0 |
| DO | 0.77123 | 15 | 4 | 11 |
| Salinity | 0.764808 | 20 | 6 | 14 |
| Conductivity | 0.749791 | 20 | 5 | 15 |
| Temperature | 0.712552 | 20 | 6 | 14 |
| PH | 0.709486 | 19 | 14 | 5 |
| Phosphate | 0.312937 | 3 | 3 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ouyang, L.; Chen, X.; Zhang, W.; Li, S.; Huang, Q.; Zhang, Y.; Yan, C.; Li, S. High Throughput Sequencing Reveals Distinct Bacterial Communities and Functional Diversity in Two Typical Coastal Bays. J. Mar. Sci. Eng. 2022, 10, 1878. https://doi.org/10.3390/jmse10121878
Ouyang L, Chen X, Zhang W, Li S, Huang Q, Zhang Y, Yan C, Li S. High Throughput Sequencing Reveals Distinct Bacterial Communities and Functional Diversity in Two Typical Coastal Bays. Journal of Marine Science and Engineering. 2022; 10(12):1878. https://doi.org/10.3390/jmse10121878
Chicago/Turabian StyleOuyang, Liao, Xianglan Chen, Wenxuan Zhang, Shuangfei Li, Qiang Huang, Yi Zhang, Chengwei Yan, and Shaofeng Li. 2022. "High Throughput Sequencing Reveals Distinct Bacterial Communities and Functional Diversity in Two Typical Coastal Bays" Journal of Marine Science and Engineering 10, no. 12: 1878. https://doi.org/10.3390/jmse10121878
APA StyleOuyang, L., Chen, X., Zhang, W., Li, S., Huang, Q., Zhang, Y., Yan, C., & Li, S. (2022). High Throughput Sequencing Reveals Distinct Bacterial Communities and Functional Diversity in Two Typical Coastal Bays. Journal of Marine Science and Engineering, 10(12), 1878. https://doi.org/10.3390/jmse10121878