Genetic Diversity Analysis and Construction of a Core Germplasm Resource Bank of Xinjiang’s Indigenous Cultivated Grapes

 , and
, and
Abstract
1. Introduction
2. Materials and Methods
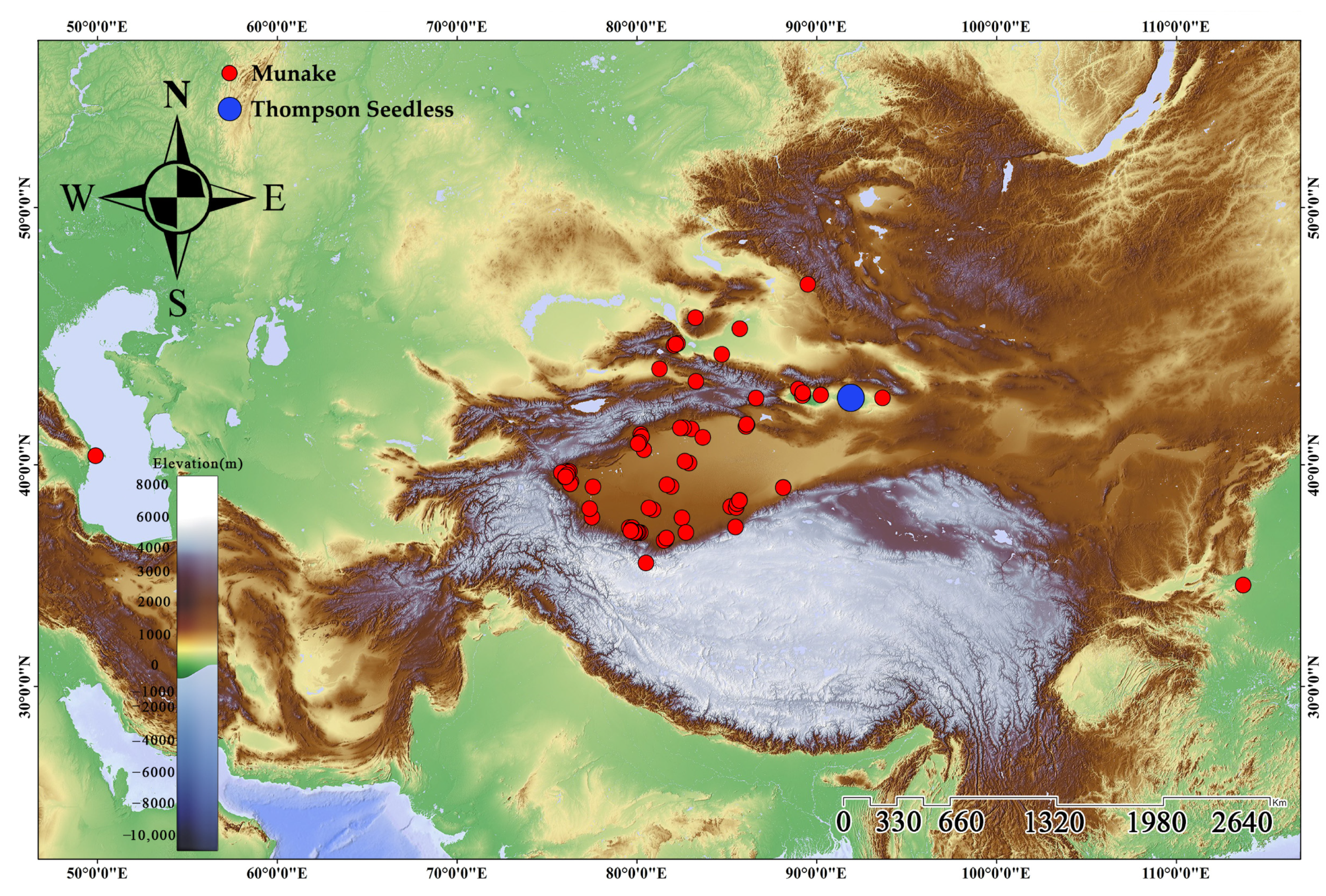
2.1. Plant Material and Sampling
2.2. Genetic Diversity Analysis Based on SSR Molecular Markers
2.2.1. Genotyping and DNA Extraction
2.2.2. Genetic Structure and Genetic Diversity Analysis
2.3. Construction of a Core Germplasm Bank Based on Whole-Genome Resequencing
2.3.1. Sequencing and Sequencing Quality Control
2.3.2. SNP/InDel Detection and Annotation
2.3.3. Genetic Diversity and Population Structure Analysis
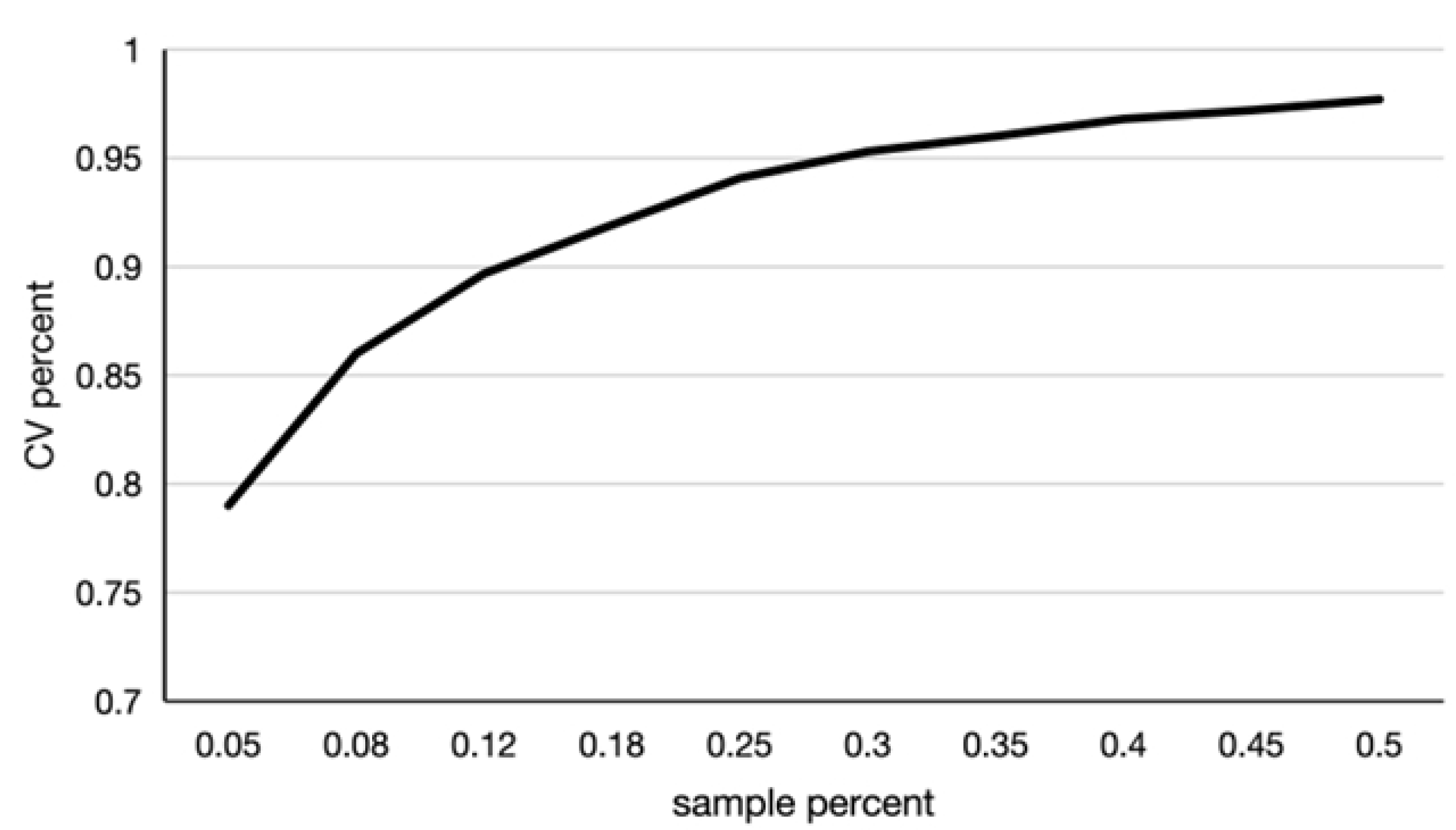
2.3.4. Core Germplasm Collection of Grapes
3. Results
3.1. Mutation Detection in Thompson Seedless Grapes
3.2. Genetic Diversity of Thompson Seedless
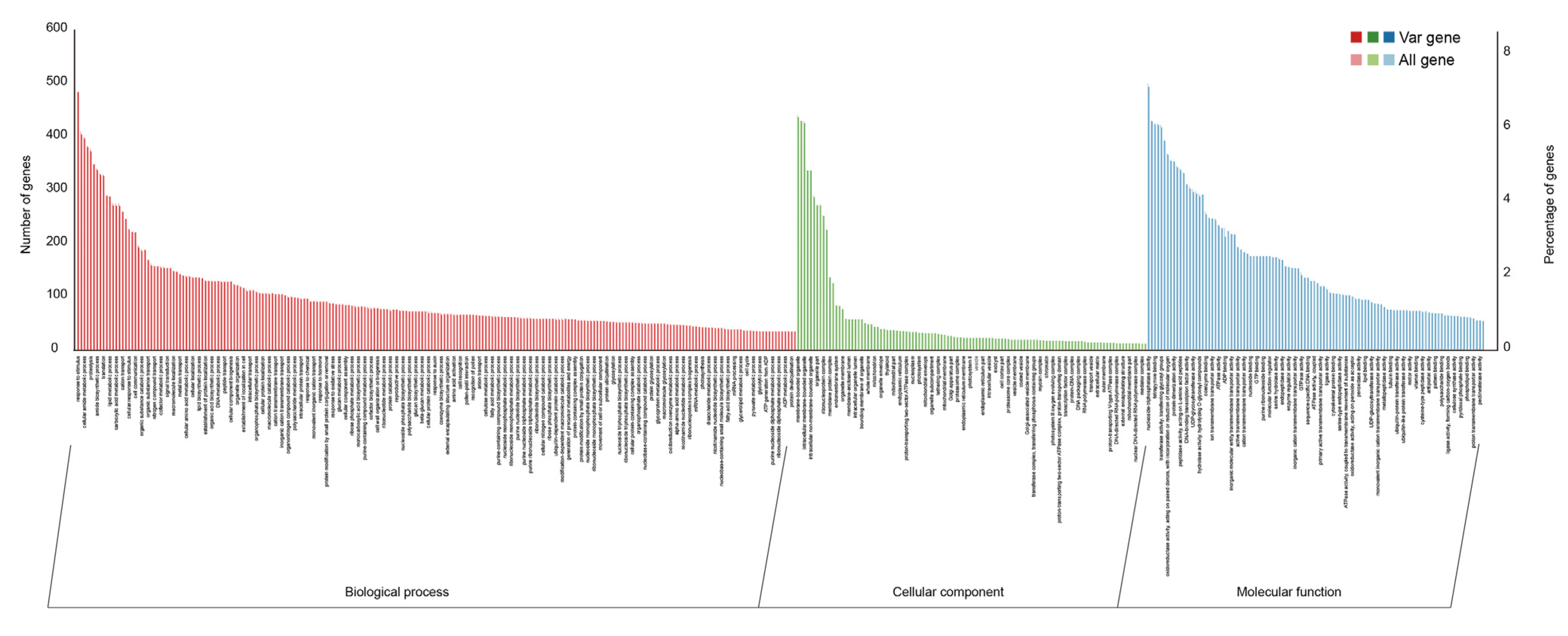
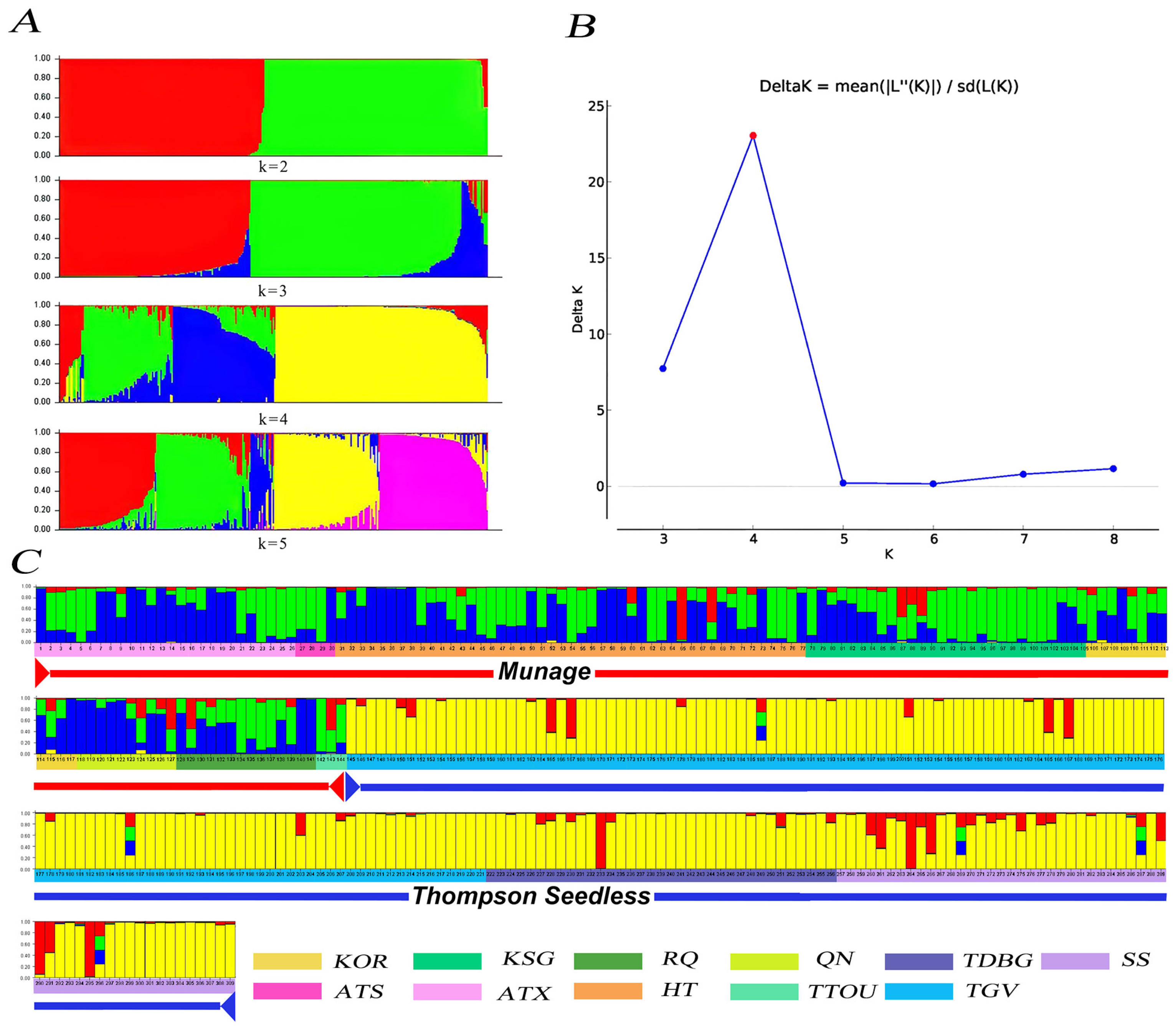
3.3. Whole-Genome Resequencing Elucidates Genetic Diversity and Core Collection of Munake Grape Cultivars
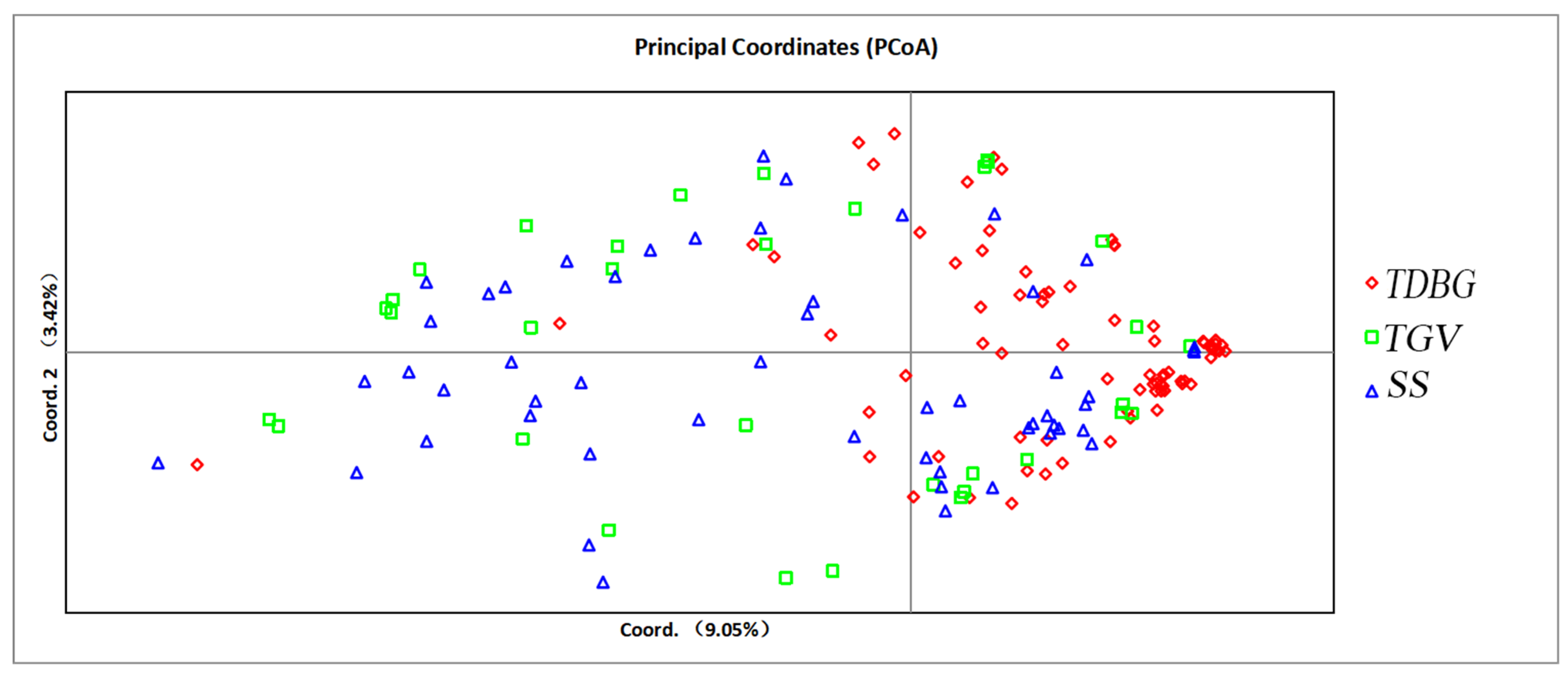
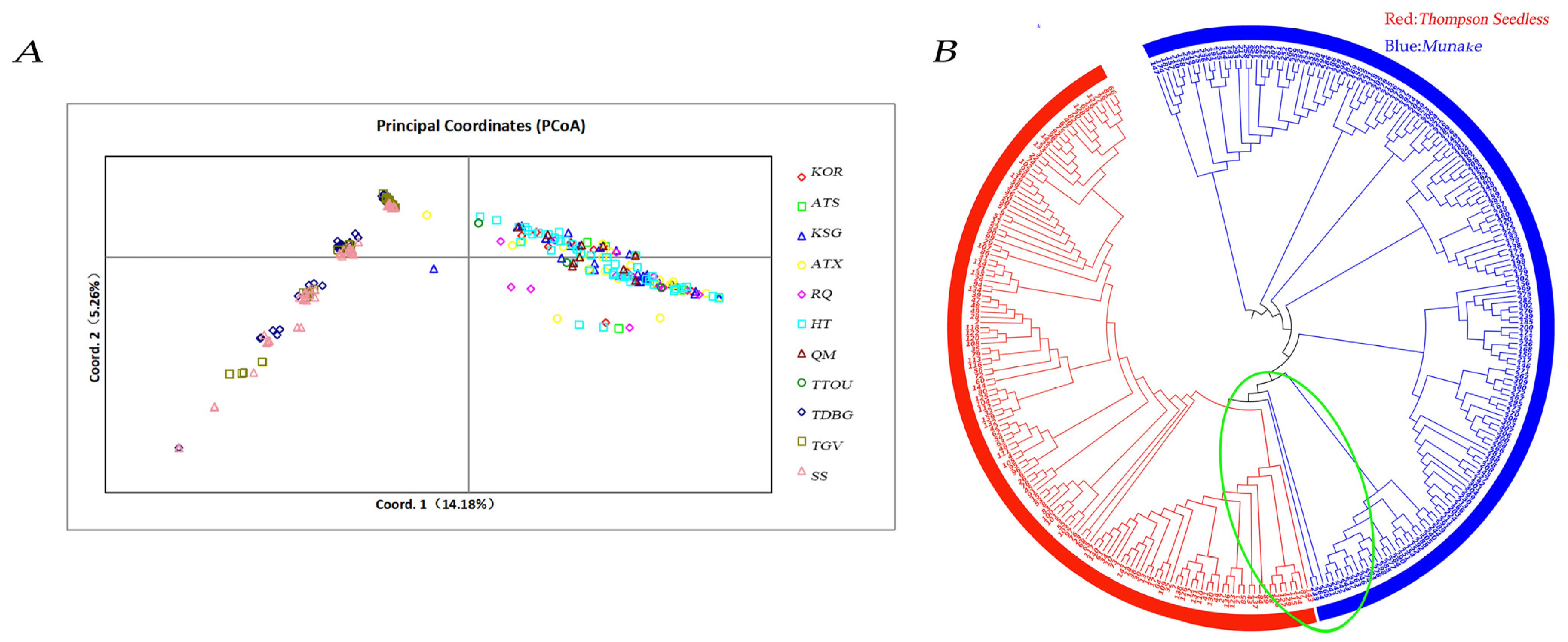
3.4. Analysis of the Relationship Between Thompson Seedless and Munake Grapes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wei, L.Q.; Lu, S.X.; Ma, Z.H.; Guo, R.; Mao, J. Quality Evaluation of 10 Wine Grape Cultivars in Jia yu guan Based on Principal Component Analysis. J. Gansu Agric. Univ. 2020, 55, 90–96. [Google Scholar] [CrossRef]
- Estrada, J.L.; Arboleda, M.D.M.; Dionisio-Sese, M.L. Current status of sea grapes (Caulerpa spp.) farming and wild harvesting in the Philippines. J. Appl. Phycol. 2021, 33, 3215–3223. [Google Scholar] [CrossRef]
- Ye, Q.T.; Li, Y.M.; Zhou, Y.Y.; Li, X.H.; Zhang, W.; Sun, Q.; Han, C.P.; Yan, J.Y. Occurrence and Harm of Grape Trunk Diseases and Pathogen Types at Home and Abroad. Acta Hortic. Sin. 2021, 38, 278–292. [Google Scholar] [CrossRef]
- Swarup, S.; Cargill, E.J.; Crosby, K.; Flagel, L.; Kniskern, J.; Glenn, K.C. Genetic diversity is indispensable for plant breeding to improve crops. Crop Sci. 2021, 61, 839–852. [Google Scholar] [CrossRef]
- Yang, D.; Duan, S.C.; Xia, Q.J.; Liang, Z.; Dong, X.; Margaryan, K.; Musayev, M.; Goryslavets, S.; Zdunić, G.; Bert, P.-F.; et al. Dual domestications and origin of traits in grapevine evolution. Science 2023, 379, 892–901. [Google Scholar] [CrossRef]
- Sousa, B.; Araújo, S.d.S.; Sales, H.; Pontes, R.; Nunes, J. Douro Vineyards: A Perspective for the Valorization and Conservation of Grapevine Genetic Resources. Agronomy 2024, 14, 245. [Google Scholar] [CrossRef]
- Guo, D.-L.; Zhang, J.-Y.; Liu, C.-H. Genetic diversity in some grape varieties revealed by SCoT analyses. Mol. Biol. Rep. 2012, 39, 5307–5313. [Google Scholar] [CrossRef]
- Abudu, M.; Abudukelimu, A.; Liu, Y.S. Challenges and Strategies in the Production of Atush Munake Grapes. Mod. Agric. 2019, 2019, 70–72. [Google Scholar] [CrossRef]
- Zhong, H.X.; Zhang, F.C.; Zhou, X.M.; Pan, M.Q.; Xu, J.; Hao, J.Z.; Han, S.A.; Mei, C.; Xian, H.; Wang, M.; et al. Genome-Wide Identification of Sequence Variations and SSR Marker Development in the Munake Grape Cultivar. Front. Ecol. Evol. 2021, 9, 664835. [Google Scholar] [CrossRef]
- Xi, Z.Y.; Zhang, J.W.; Tang, D.M.; Zhang, Z.Q.; Ping, W.X. Research status and development of grape production around Tarim Basin. Sino-Overseas Grapevine Wine 2008, 50–53. [Google Scholar]
- Luo, Q.W.; Wu, G.H.; Sun, F.; Li, Y.L.; Wang, Y. Innovation and utilization of Grape germplasm resources in Xinjiang. Sino-Overseas Grapevine Wine 2015, 61–63. [Google Scholar]
- Liu, A.H.; Yue, C.Y.; Kong, T.T.; Jiao, S.P.; Zhang, J.W.; Zang, X.P.; Yang, J. Analysis of soil nutrient status of Munake vineyards in Atux city of Xinjiang. Chin. Agric. Sci. Bull. 2015, 31, 199–202. [Google Scholar]
- Yang, C.S. The beginning and evolution of viticulture in China. Sino-Overseas Grapevine Wine 2003, 4–7. [Google Scholar]
- Barnaud, A.; Lacombe, T.; Doligez, A. Linkage disequilibrium in cultivated grapevine, Vitis vinifera L. Theor. Appl. Genet. 2006, 112, 708–716. [Google Scholar] [CrossRef] [PubMed]
- Belaj, A.; de la Rosa, R.; Lorite, I.J.; Mariotti, R.; Cultrera, N.G.M.; Beuzón, C.R.; González-Plaza, J.J.; Muñoz-Mérida, A.; Trelles, O.; Baldoni, L. Usefulness of a New Large Set of High Throughput EST-SNP Markers as a Tool for Olive Germplasm Collection Management. Front. Plant Sci. 2018, 9, 215. [Google Scholar] [CrossRef]
- Palumbo, F.; Galvao, A.C.; Nicoletto, C.; Sambo, P.; Barcaccia, G. Diversity Analysis of Sweet Potato Genetic Resources Using Morphological and Qualitative Traits and Molecular Markers. Genes 2019, 10, 840. [Google Scholar] [CrossRef]
- Chikh-Rouhou, H.; Mezghani, N.; Mnasri, S.; Mezghani, N.; Garcés-Claver, A. Assessing the Genetic Diversity and Population Structure of a Tunisian Melon (Cucumis melo L.) Collection Using Phenotypic Traits and SSR Molecular Markers. Agronomy 2021, 11, 1121. [Google Scholar] [CrossRef]
- Kant, R.; Meena, R.K.; Garg, R.; Vikas; Yadav, A.; Bhandari, M.S. De Novo SSR markers development and genetic diversity assessment in Ailanthus excelsa in India. Physiol. Mol. Biol. Plants 2025, 1–17. [Google Scholar] [CrossRef]
- Emanuelli, F.; Lorenzi, S.; Grzeskowiak, L.; Catalano, V.; Stefanini, M.; Troggio, M.; Myles, S.; Martinez-Zapater, J.M.; Zyprian, E.; Moreira, F.M.; et al. Genetic diversity and population structure assessed by SSR and SNP markers in a large germplasm collection of grapes. BMC Plant Biol. 2013, 13, 39. [Google Scholar] [CrossRef]
- Song, X.F.; Wei, H.C.; Cheng, W.; Yang, S.X.; Zhao, Y.X.; Li, X.; Luo, D.; Zhang, H.; Feng, X.Z. Development of INDEL Markers for Genetic Mapping Based on Whole Genome Resequencing in Soybean. G3-Genes Genomes Genet. 2015, 5, 2793–2799. [Google Scholar] [CrossRef]
- Doligez, A.; Bertrand, Y.; Farnos, M.; Grolier, M.; Romieu, C.; Esnault, F.; Dias, S.; Berger, G.; François, P.; Pons, T.; et al. New stable QTLs for berry weight do not colocalize with QTLs for seed traits in cultivated grapevine (Vitis vinifera L.). BMC Plant Biol. 2013, 13, 217. [Google Scholar] [CrossRef]
- Lijavetzky, D.; Cabezas, J.A.; Ibáñez, A.; Rodríguez, V.; Martínez-Zapater, J.M. High throughput SNP discovery and genotyping in grapevine (Vitis vinifera L.) by combining a re-sequencing approach and SNPlex technology. BMC Genom. 2007, 8, 424. [Google Scholar] [CrossRef] [PubMed]
- Cabezas, J.A.; Ibáñez, J.; Lijavetzky, D.; Vélez, D.; Bravo, G.; Rodríguez, V.; Carreño, I.; Jermakow, A.M.; Carreño, J.; Ruiz-García, L.; et al. A 48 SNP set for grapevine cultivar identification. BMC Plant Biol. 2011, 11, 153. [Google Scholar] [CrossRef]
- Lambert, P.; Campoy, J.A.; Pacheco, I.; Mauroux, J.-B.; Da Silva Linge, C.; Micheletti, D.; Bassi, D.; Rossini, L.; Dirlewanger, E.; Pascal, T.; et al. Identifying SNP markers tightly associated with six major genes in peach [Prunus persica (L.) Batsch] using a high-density SNP array with an objective of marker-assisted selection (MAS). Tree Genet. Genomes 2016, 12, 121. [Google Scholar] [CrossRef]
- Chalbi, A.; Chikh-Rouhou, H.; Mezghani, N.; Slim, A.; Fayos, O.; Bel-Kadhi, M.S.; Garcés-Claver, A. Genetic Diversity Analysis of Onion (Allium cepa L.) from the Arid Region of Tunisia Using Phenotypic Traits and SSR Markers. Horticulturae 2023, 9, 1098. [Google Scholar] [CrossRef]
- Li, S.S.; Sun, C.Y.; Jiang, S.C.; Wang, K.Y.; Wang, Y.; Zhang, M.P. SSR molecular markers and their application in plant genetics and breeding. Jilin Shucai (Jilin Veg.) 2014, 33–38. [Google Scholar]
- Thomas, M.R.; Scott, N.S. Microsatellite repeats in grapevine reveal DNA polymorphisms when analysed as sequence-tagged sites (STSs). Theor. Appl. Genet. 1993, 86, 985–990. [Google Scholar] [CrossRef]
- Wang, Y.L.; Jiang, Y.S.; Qu, Z.C.; Shan, L. Application of SSR Markers in Crop Germplasm Identification. Shandong Nong Ye Ke Xue (Shandong Agric. Sci.) 2012, 44, 11–18. [Google Scholar] [CrossRef]
- Wang, C.X. Application of SSR Molecular Marker Technology in Maize Variety Identification and Protection. Master’s Thesis, Xinjiang Agricultural University, Ürümqi, China, 2007; pp. 1–2. [Google Scholar]
- Bowers, J.E.; Dangl, G.S.; Vignani, R.; Meredith, C.P. Isolation and characterization of new polymorphic simple sequence repeat loci in grape (Vitis vinifera L.). Genome 1996, 39, 628–633. [Google Scholar] [CrossRef]
- Zhang, S.; Li, Y.; Li, Y.; Zhang, Y.; Hao, Y.; Hou, Z.; Qi, J. Development of SSR Markers for and Fingerprinting of Walnut Genetic Resources. Forests 2024, 15, 405. [Google Scholar] [CrossRef]
- Lu, X.; Adedze, Y.M.N.; Chofong, G.N.; Gandeka, M.; Deng, Z.; Teng, L.; Zhang, X.; Sun, G.; Si, L.; Li, W. Identification of high-efficiency SSR markers for assessing watermelon genetic purity. J. Genet. 2018, 97, 1295–1306. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.D.; Guan, L.P.; He, L.L. Universality Identification of Molecular Markers for Grape Seedlessness Genes. Chin. J. Agric. Sci. 2021. [Google Scholar] [CrossRef]
- Boz, Y.; Bakir, M.; Çelikkol, B.P.; Kazan, K.; Yilmaz, F.; Çakir, B.; Aslantas¸, S.¸.; Söylemezoğlu, G.; Yaşasin, A.S.; Özer, C.; et al. Genetic characterization of grape (Vitis vinifera L.) germplasm from Southeast Anatolia by SSR markers. Vitis 2011, 50, 99–106. [Google Scholar] [CrossRef]
- Yang, M.T.; Huang, Z.; Gan, J.P.; Xu, J.C.; Pang, J.L. Research progress of SSR molecular markers. Hangzhou Shifan Da Xue Xue Bao (J. Hangzhou Norm. Univ. Nat. Sci. Ed.) 2019, 18, 429–436. [Google Scholar]
- D’Onofrio, C.; de Lorenzis, G.; Giordani, T.; Natali, L.; Cavallini, A.; Scalabrelli, G. Retrotransposon-based molecular markers for grapevine species and cultivars identification. Tree Genet. Genomes 2010, 6, 451–466. [Google Scholar] [CrossRef]
- Villano, C.; Aiese Cigliano, R.; Esposito, S.; D’Amelia, V.; Iovene, M.; Carputo, D.; Aversano, R. DNA-Based Technologies for Grapevine Biodiversity Exploitation: State of the Art and Future Perspectives. Agronomy 2022, 12, 491. [Google Scholar] [CrossRef]
- Li, J. Current Status of Grape Germplasm Resources Research. Grape Cultiv. Wine Brew. 1998, 61–62. [Google Scholar]
- Zhao, Q.F.; Huang, L.P.; Liu, X.T.; Wang, M.; Xun, Z.L.; Ma, X.H. Advances in the Collection, Preservation, and Research Utilization of Grape Germplasm Resources in China. J. Fruit Resour. 2021, 2, 1–4. [Google Scholar] [CrossRef]
- Wang, Y.; Sun, F.; Li, Y.L.; Sulaiman, A.; Wu, G.; Guo, P. Identification of Xinjiang Local Grape Variety Resources and Their Phylogenetic Relationships Using SSR Markers. Northwestern Hortic. 2023, 40–44. [Google Scholar]
- Li, Z.B.; Ren, T.; Deng, J.J.; Chen, J.; Zhou, S.; Zeng, X.; Ma, J.; Li, F. Comparative Analysis of Chloroplast Genomes Among Three Varieties of Hibiscus mutabilis and Their Close Relatives. Guangxi Plants 2022, 42, 2007–2020. [Google Scholar]
- Aradhya, M.K.; Dangl, G.S.; Prins, B.H.; Boursiquot, J.-M.; Walker, M.A.; Meredith, C.P.; Simon, C.J. Genetic Structure and Differentiation in Cultivated Grape, Vitis vinifera L. Genet. Res. 2003, 81, 179–192. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Zhong, H.; Zhang, F.; Wang, X.; Wu, X.; Wang, J.; Shi, W. Genetic Diversity and Core Germplasm Research of 144 Munake Grape Resources Using 22 Pairs of SSR Markers. Horticulturae 2023, 9, 917. [Google Scholar] [CrossRef]
- Riaz, S.; De Lorenzis, G.; Velasco, D.; Koehmstedt, A.; Maghradze, D.; Bobokashvili, Z.; Musayev, M.; Zdunic, G.; Laucou, V.; Walker, M.A.; et al. Genetic diversity analysis of cultivated and wild grapevine (Vitis vinifera L.) accessions around the Mediterranean basin and Central Asia. BMC Plant Biol. 2018, 18, 137. [Google Scholar] [CrossRef] [PubMed]
- Sabir, A.; Ikten, H.; Mutlu, N.; Sari, D. Genetic Identification and Conservation of Local Turkish Grapevine (Vitis vinifera L.) Genotypes on the Edge of Extinction. Erwerbs-Obstbau 2018, 60, 31–38. [Google Scholar] [CrossRef]
- Di Gaspero, G.; Peterlunger, E.; Testolin, R.; Edwards, K.J.; Cipriani, G. Conservation of microsatellite loci within the genus Vitis. Theor. Appl. Genet. 2000, 101, 301–308. [Google Scholar] [CrossRef]
- Adam-Blondon, A.F.; Roux, C.; Claux, D.; Butterlin, G.; Merdinoglu, D.; This, P. Mapping 245 SSR markers on the Vitis vinifera genome: A tool for grape genetics. Theor. Appl. Genet. 2004, 109, 1017–1027. [Google Scholar] [CrossRef]
- Sefc, K.M.; Regner, F.; Turetschek, E.; Glössl, J.; Steinkellner, H. Identification of microsatellite sequences in Vitis riparia and their applicability for genotyping of different Vitis species. Genome 1999, 42, 367–373. [Google Scholar] [CrossRef]
- Peakall, R.; Smouse, P.E. GENALEX 6.5: Genetic analysis in Excel. Population genetic software for teaching and research. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef]
- Gil, J.; Um, Y.; Kim, S.; Kim, O.T.; Koo, S.C.; Reddy, C.S.; Kim, S.-C.; Hong, C.P.; Park, S.-G.; Kim, H.B.; et al. Development of Genome-Wide SSR Markers from Angelica gigas Nakai Using Next Generation Sequencing. Genes 2017, 8, 238. [Google Scholar] [CrossRef]
- Liu, F.; Hong, Z.; Xu, D.; Jia, H.; Zhang, N.; Liu, X.; Yang, Z.; Lu, M. Genetic Diversity of the Endangered Dalbergia odorifera Revealed by SSR Markers. Forests 2019, 10, 225. [Google Scholar] [CrossRef]
- Sonstebo, J.H.; Tollefsrud, M.M.; Myking, T.; Steffenrem, A.; Nilsen, A.E.; Edvardsen, O.M.; Johnskas, O.R.; El-Kassaby, Y.A. Genetic diversity of Norway spruce (Picea abies (L.) Karst.) seed orchard crops: Effects of number of parents, seed year, and pollen contamination. For. Ecol. Manag 2018, 411, 132–141. [Google Scholar] [CrossRef]
- Tamura, K.; Dudley, J.; Nei, M.; Kumar, S. MEGA4: Molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol. Biol. Evol. 2007, 24, 1596–1599. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data, P. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Donald, F.; Pan, R.; Chen, H.; Li, Z.; Hu, J.; Aldana, R. Dnascope: High Accuracy Small Variant Calling Using Machine Learning. bioRxiv 2022. bioRxiv:2022.05.20.492556. [Google Scholar]
- Pei, S.; Liu, T.; Ren, X.; Li, W.; Chen, C.; Xie, Z. Benchmarking variant callers in next-generation and third-generation sequencing analysis. Brief. Bioinform. 2020, 22, bbaa148. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [PubMed]
- Francis, R.M. POPHELPER: An R package and web app to analyse and visualize population structure. Mol. Ecol. Resour. 2017, 17, 27–32. [Google Scholar] [CrossRef]
- Jombart, T.; Ahmed, I. adegenet 1.3-1: New tools for the analysis of genome-wide SNP data. Bioinformatics 2011, 27, 3070–3071. [Google Scholar] [CrossRef]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, X.H.; Li, Y.W.; Feng, Z.S.; Mu, Z.H.; Wang, J.; Wu, X.Y.; Wang, B.G.; Lu, Z.F.; Li, G.J. Identification and Validation of a Core Single-Nucleotide Polymorphism Marker Set for Genetic Diversity Assessment, Fingerprinting Identification, and Core Collection Development in Bottle Gourd. Front. Plant Sci. 2021, 12, 747940. [Google Scholar] [CrossRef] [PubMed]
- Thachuk, C.; Crossa, J.; Franco, J.; Dreisigacker, S.; Warburton, M.; Davenport, G.F. Core Hunter: An algorithm for sampling genetic resources based on multiple genetic measures. BMC Bioinform. 2009, 10, 243. [Google Scholar] [CrossRef] [PubMed]
- Varshney, R.K.; Graner, A.; Sorrells, M.E. Genic microsatellite markers in plants: Features and applications. Trends Biotechnol. 2005, 23, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Grover, A.; Sharma, P.C. Development and use of molecular markers: Past and present. Crit. Rev. Biotechnol. 2016, 36, 290–302. [Google Scholar] [CrossRef]
- Kantartzi, S.K. Microsatellites: Methods and Protocols; Springer: New York, NY, USA, 2013; pp. 1–13. [Google Scholar]
- O’Hanlon, P.C.; Peakall, R.; Briese, D.T. A review of new PCR-based genetic markers and their utility to weed ecology. Weed Res. 2000, 40, 40–239. [Google Scholar] [CrossRef]
- Powell, W.; Machray, G.C.; Provan, J. Polymorphism revealed by simple sequence repeats. Trends Plant Sci. 1996, 1, 215–222. [Google Scholar] [CrossRef]
- Schlötterer, C. The evolution of molecular markers-just a matter of fashion. Nat. Rev. Genet. 2004, 5, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Wang, N.; Bao, Z.G.; Zhou, Q.; Guarracino, A.; Yang, Y.T.; Wang, P.; Zhang, Z.Y.; Tang, D.; Zhang, P.X.; et al. Leveraging a phased pangenome for haplotype design of hybrid potato. Nature 2025, 640, 408–417. [Google Scholar] [CrossRef]
- Liang, Z.C.; Duan, S.C.; Sheng, J.; Zhu, S.S.; Ni, X.M.; Shao, J.H.; Liu, C.H.; Nick, P.; Du, F.; Fan, P.G.; et al. Whole-genome resequencing of 472 Vitis accessions for grapevine diversity and demographic history analyses. Nat. Commun. 2019, 10, 1190. [Google Scholar] [CrossRef]
- Balfourier, F.; Roussel, V.; Strelchenko, P.; Exbrayat-Vinson, F.; Sourdille, P.; Boutet, G.; Koenig, J.; Ravel, C.; Mitrofanova, O.; Beckert, M.; et al. A worldwide bread wheat core collection arrayed in a 384-well plate. Theor. Appl. Genet. 2007, 114, 1265–1275. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, D.; Wang, M.; Sun, J.; Qi, Y.; Li, J.; Wei, X.; Han, L.; Qiu, Z.; Tang, S.; et al. A core collection, and mini core collection of Oryza sativa L. in China. Theor. Appl. Genet. 2011, 122, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Amatriaín, M.; Cuesta-Marcos, A.; Endelman, J.B.; Comadran, J.; Bonman, J.M.; Bockelman, H.E.; Chao, S.; Russell, J.; Waugh, R.; Hayes, P.M.; et al. The USDA barley core collection: Genetic diversity, population structure, and potential for genome-wide association Studies. PLoS ONE 2014, 9, e94688. [Google Scholar] [CrossRef] [PubMed]
- Allen, A.R.; Taylor, M.; McKeown, B.; Curry, A.I.; Lavery, J.F.; Mitchell, A.; Hartshorne, D.; Fries, R.; Skuce, R.A. Compilation of a panel of informative single nucleotide polymorphisms for bovine identification in the Northern Irish cattle population. BMC Genet. 2010, 11, 5. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Num | Name | Primer Sequence (F) (5′-3′) | Primer Sequence (R) (5′-3′) | MW (Dalton) | TM (°C) |
|---|---|---|---|---|---|
| 1 | VRZAG67 | ACCTGGCCCGACTCCTCTTGTATGC | TCCTGCCGGCGATAACCAAGCTATG | 7627 | 65 |
| 2 | VCHR13a | TGGCAGAGCAAATGAATCAA | TTGGATGGATTGGAATGACC | 6212 | 55 |
| 3 | VVIP31 | TATCCAAGAGACAAATTCCCAC | TTCTCTTGTTTCCTGCAAATGG | 6682 | 56 |
| 4 | VVMD7 | AGAGTTGCGGAGAACAGGAT | CGAACCTTCACACGCTTGAT | 6037 | 57 |
| 5 | VMC4F3-1 | AAAGCACTATGGTGGGTGTAAA | TAACCAATACATGCATCAAGGA | 6720 | 56 |
| 6 | Vchr8a | ACCCACTGCCACTCTCTCAT | AAATCTCCGGGATCCTTTTG | 6083 | 59 |
| 7 | VrZAG29 | ATAACCAGGACAAGTTATTCAAGCC | ACCCAATTGACCATCTTTTATGCTG | 7568 | 56 |
| 8 | VrZAG25 | CTCCACTTCACATCACATGGCATGC | CGGCCAACATTTACTCATCTCTCCC | 7474 | 60 |
| 9 | VVIb66 | CCACTAGTGGTCAGAAAAGAAG | TTGTATTGTGTGCCTCTTCTCA | 6674 | 53 |
| 10 | VVIb01 | TGACCCTCGACCTTAAATCTT | TGGTGAGTGCAATGATAGTAGA | 6879 | 52 |
| 11 | VVMD36 | TAAAATAATAATAGGGGGACACGGG | CAACTGTAAAGGTAAGACACAGTCC | 7769 | 56 |
| 12 | VVS4 | CCATCAGTGATAAAACCTAATGCC | CCCACCTTGCCCTTAGATGTTA | 6622 | 55 |
| 13 | VMC4C6 | CTCCATCCCTATCTCATCAG | CTCTAACACCCAATCTCACA | 5951 | 50 |
| 14 | VVIi51 | ATCCCAAGAGAACCAAGAAACT | GCTGATCTCAGTGCATATGTTG | 6757 | 53 |
| 15 | UDV-015 | TGCACATTTCCCTCCTTAG | CGGGTTACTGGGAAGGGTAT | 6254 | 54 |
| 16 | VMC4H6 | GTATAGAACCACGCATCCAACA | CCCTTAGTTTCCTCGTGCTTTT | 6610 | 54 |
| Locus | Na | Ne | I | Ho | He | uHe | F | Fis | Fst | Nm |
|---|---|---|---|---|---|---|---|---|---|---|
| VVIb01 | 23 | 3.46 | 1.70 | 0.97 | 0.71 | 0.71 | −0.36 | −0.40 | 0.04 | 6.55 |
| VVIb66 | 31 | 6.03 | 2.25 | 0.83 | 0.83 | 0.84 | 0.01 | 0.17 | 0.12 | 1.87 |
| VVMD36 | 34 | 6.48 | 2.38 | 0.90 | 0.85 | 0.85 | −0.07 | −0.17 | 0.11 | 2.04 |
| VVS4 | 31 | 2.83 | 1.63 | 0.24 | 0.65 | 0.65 | 0.62 | 0.41 | 0.24 | 0.78 |
| VrZAG29 | 32 | 3.80 | 1.94 | 0.85 | 0.74 | 0.74 | −0.16 | −0.15 | 0.09 | 2.43 |
| VrZAG25 | 25 | 6.29 | 2.29 | 0.95 | 0.84 | 0.85 | −0.13 | −0.17 | 0.09 | 2.59 |
| VMC4C6 | 26 | 4.77 | 2.07 | 0.88 | 0.79 | 0.79 | −0.11 | −0.10 | 0.07 | 3.14 |
| VVIi51 | 30 | 8.11 | 2.43 | 0.92 | 0.88 | 0.88 | −0.05 | −0.22 | 0.16 | 1.36 |
| UDV-015 | 39 | 13.72 | 2.98 | 0.85 | 0.93 | 0.93 | 0.08 | 0.04 | 0.08 | 2.77 |
| VMC4H6F | 28 | 6.98 | 2.32 | 0.90 | 0.86 | 0.86 | −0.05 | −0.26 | 0.16 | 1.32 |
| VRZAG67 | 19 | 2.58 | 1.29 | 0.77 | 0.61 | 0.61 | −0.26 | −0.31 | 0.03 | 8.68 |
| VCHR13a | 23 | 2.99 | 1.44 | 0.99 | 0.67 | 0.67 | −0.49 | −0.53 | 0.05 | 4.71 |
| VVIP31 | 26 | 5.25 | 1.99 | 1.00 | 0.81 | 0.81 | −0.24 | −0.26 | 0.02 | 10.19 |
| VVMD7 | 21 | 1.95 | 1.21 | 0.28 | 0.49 | 0.49 | 0.43 | 0.37 | 0.03 | 7.55 |
| VMC4F3-1 | 22 | 3.50 | 1.67 | 0.99 | 0.71 | 0.72 | −0.39 | −0.43 | 0.07 | 15.39 |
| Vchr8a | 32 | 7.75 | 2.45 | 1.00 | 0.87 | 0.87 | −0.15 | −0.19 | 0.04 | 6.62 |
| Mean | 27.6 | 5.41 | 2.00 | 0.83 | 0.76 | 0.77 | −0.08 | −0.14 | 0.08 | 1.01 |
| Pop | N | Na | Ne | I | Ho | He | uHe | F | |
|---|---|---|---|---|---|---|---|---|---|
| TDBG | Mean | 65.56 | 10.43 | 3.77 | 1.48 | 0.84 | 0.68 | 0.68 | −0.20 |
| SE | 1.93 | 1.08 | 0.38 | 0.12 | 0.07 | 0.04 | 0.04 | 0.10 | |
| TGV | Mean | 23.38 | 13.25 | 6.07 | 1.92 | 0.84 | 0.77 | 0.78 | −0.13 |
| SE | 1.56 | 1.79 | 0.89 | 0.18 | 0.05 | 0.04 | 0.04 | 0.09 | |
| SS | Mean | 36 | 12.31 | 4.31 | 1.67 | 0.79 | 0.71 | 0.72 | −0.11 |
| SE | 2.27 | 1.22 | 0.61 | 0.12 | 0.06 | 0.03 | 0.03 | 0.08 |
| Index | Total | Min/Max (Ave) |
|---|---|---|
| Observed allele number | 706.000 | 2.000–2.000(2.000) |
| Expected allele number | 682.980 | 1.828–2.000(1.935) |
| Observed heterozygous number | 200.389 | 0.459–0.621(0.568) |
| Expected heterozygous number | 170.420 | 0.453–0.500(0.483) |
| Nei diversity index | 171.068 | 0.454–0.502(0.485) |
| Shannon–Wiener index | 238.542 | 0.645–0.693(0.676) |
| Polymorphism information content | 129.249 | 0.350–0.375(0.366) |
| Pop | Grape Varieties | N | Na | Ne | I | Ho | He | |
|---|---|---|---|---|---|---|---|---|
| KOR | Munake | Mean | 11.833 | 7.500 | 4.635 | 1.638 | 0.847 | 0.741 |
| SE | 0.167 | 1.500 | 0.821 | 0.225 | 0.137 | 0.054 | ||
| ATS | Munake | Mean | 3.833 | 3.333 | 3.188 | 1.029 | 0.833 | 0.599 |
| SE | 0.167 | 0.955 | 0.969 | 0.219 | 0.167 | 0.060 | ||
| KSG | Munake | Mean | 27.833 | 9.833 | 4.204 | 1.672 | 0.786 | 0.737 |
| SE | 0.167 | 0.749 | 0.610 | 0.118 | 0.153 | 0.036 | ||
| ATX | Munake | Mean | 25.667 | 10.167 | 3.775 | 1.624 | 0.801 | 0.705 |
| SE | 0.211 | 1.400 | 0.443 | 0.153 | 0.112 | 0.054 | ||
| RQ | Munake | Mean | 13.500 | 7.667 | 4.299 | 1.580 | 0.771 | 0.707 |
| SE | 0.224 | 0.989 | 0.969 | 0.188 | 0.145 | 0.064 | ||
| HT | Munake | Mean | 46.667 | 15.667 | 4.946 | 1.960 | 0.815 | 0.768 |
| SE | 0.333 | 0.955 | 0.713 | 0.139 | 0.095 | 0.042 | ||
| QN | Munake | Mean | 10.000 | 7.000 | 4.437 | 1.646 | 0.783 | 0.759 |
| SE | 0.000 | 0.775 | 0.517 | 0.119 | 0.101 | 0.027 | ||
| TTOU | Munake | Mean | 3.000 | 4.167 | 3.683 | 1.330 | 0.778 | 0.704 |
| SE | 0.000 | 0.401 | 0.421 | 0.119 | 0.111 | 0.045 | ||
| TDBG | Thompson Seedless | Mean | 67.333 | 9.667 | 3.336 | 1.304 | 0.806 | 0.629 |
| SE | 2.654 | 2.753 | 0.711 | 0.240 | 0.147 | 0.070 | ||
| TGV | Thompson Seedless | Mean | 28.667 | 8.833 | 3.953 | 1.476 | 0.856 | 0.688 |
| SE | 1.145 | 1.922 | 1.020 | 0.212 | 0.121 | 0.049 | ||
| SS | Thompson Seedless | Mean | 39.500 | 14.667 | 4.523 | 1.775 | 0.882 | 0.724 |
| SE | 2.566 | 1.498 | 1.071 | 0.193 | 0.073 | 0.053 |
| All Pops. | Fis | Fst | Gis | Gst | Nm |
|---|---|---|---|---|---|
| VVIP31 | −0.20 | 0.11 | −0.15 | 0.08 | 2.01 |
| VVMD7 | −0.19 | 0.21 | −0.14 | 0.18 | 0.93 |
| Vchr8a | 0.36 | 0.18 | 0.40 | 0.13 | 1.13 |
| VMC4F3 | −0.31 | 0.16 | −0.26 | 0.13 | 1.37 |
| VrZAG67 | −0.18 | 0.17 | −0.13 | 0.14 | 1.22 |
| VCHR13a | −0.37 | 0.20 | −0.33 | 0.18 | 1.01 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, S.; Wang, J.; Li, X.; Wang, X.; Wang, X.; Zhong, H.; Aibibul, I.; Sun, F.; Li, H.; Shi, W. Genetic Diversity Analysis and Construction of a Core Germplasm Resource Bank of Xinjiang’s Indigenous Cultivated Grapes. Agriculture 2025, 15, 871. https://doi.org/10.3390/agriculture15080871
Liu S, Wang J, Li X, Wang X, Wang X, Zhong H, Aibibul I, Sun F, Li H, Shi W. Genetic Diversity Analysis and Construction of a Core Germplasm Resource Bank of Xinjiang’s Indigenous Cultivated Grapes. Agriculture. 2025; 15(8):871. https://doi.org/10.3390/agriculture15080871
Chicago/Turabian StyleLiu, Shiqing, Jiancheng Wang, Xuerong Li, Xianhang Wang, Xiyong Wang, Haixia Zhong, Ilham Aibibul, Feng Sun, Hongbin Li, and Wei Shi. 2025. "Genetic Diversity Analysis and Construction of a Core Germplasm Resource Bank of Xinjiang’s Indigenous Cultivated Grapes" Agriculture 15, no. 8: 871. https://doi.org/10.3390/agriculture15080871
APA StyleLiu, S., Wang, J., Li, X., Wang, X., Wang, X., Zhong, H., Aibibul, I., Sun, F., Li, H., & Shi, W. (2025). Genetic Diversity Analysis and Construction of a Core Germplasm Resource Bank of Xinjiang’s Indigenous Cultivated Grapes. Agriculture, 15(8), 871. https://doi.org/10.3390/agriculture15080871