

A Single-Cell Assessment of Intramuscular and Subcutaneous Adipose Tissue in Beef Cattle

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Tissue Collection
2.2. Adipose Tissue Total RNA Extraction and Bulk Sequencing
2.3. Adipose Tissue Nuclei Isolation and Single-Nuclei RNA Sequencing
3. Results
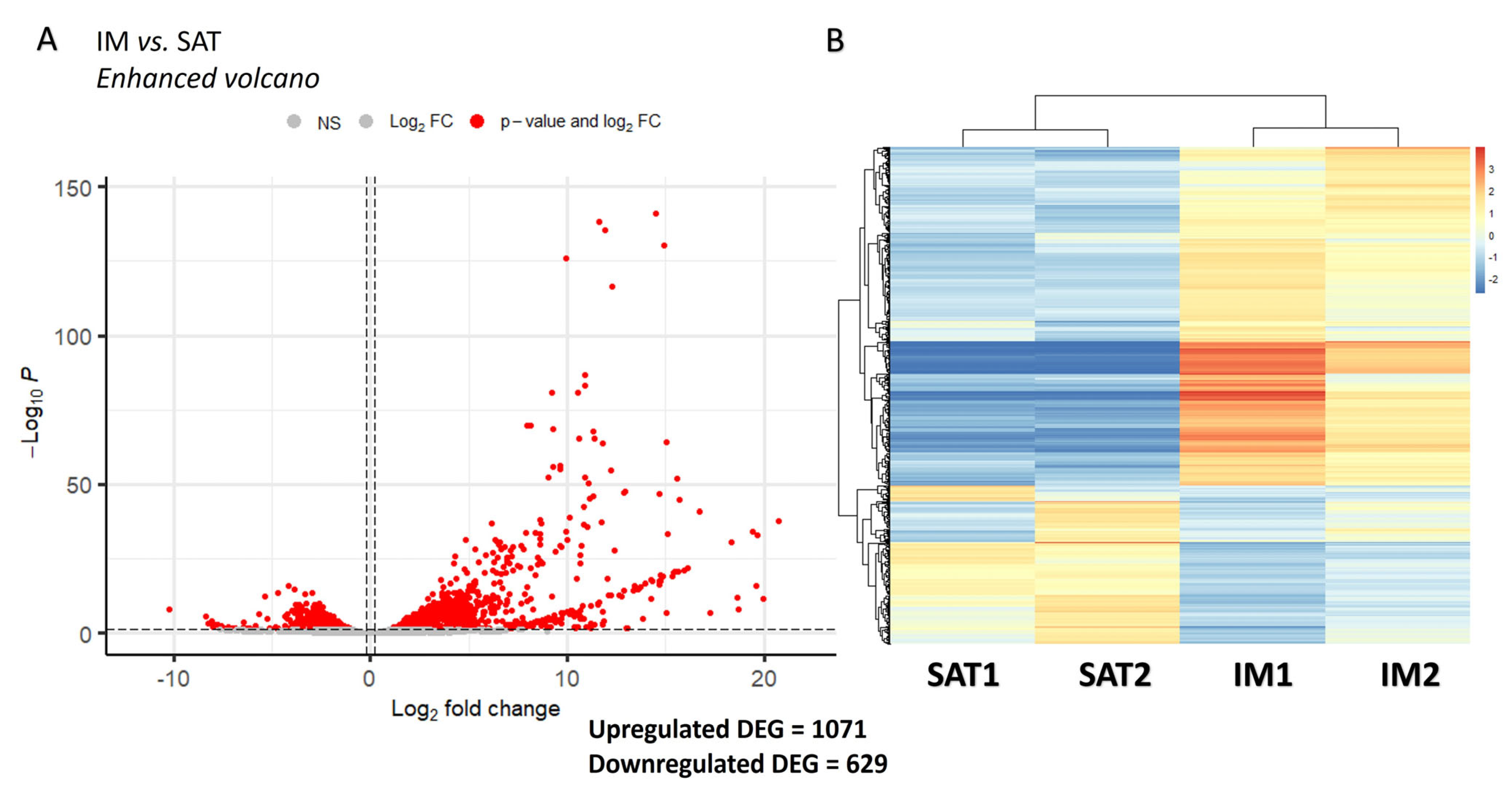
3.1. Bulk RNA Sequencing Analysis Suggests Depot-Specificities Between IM and SAT Gene Profiles in Beef Steers
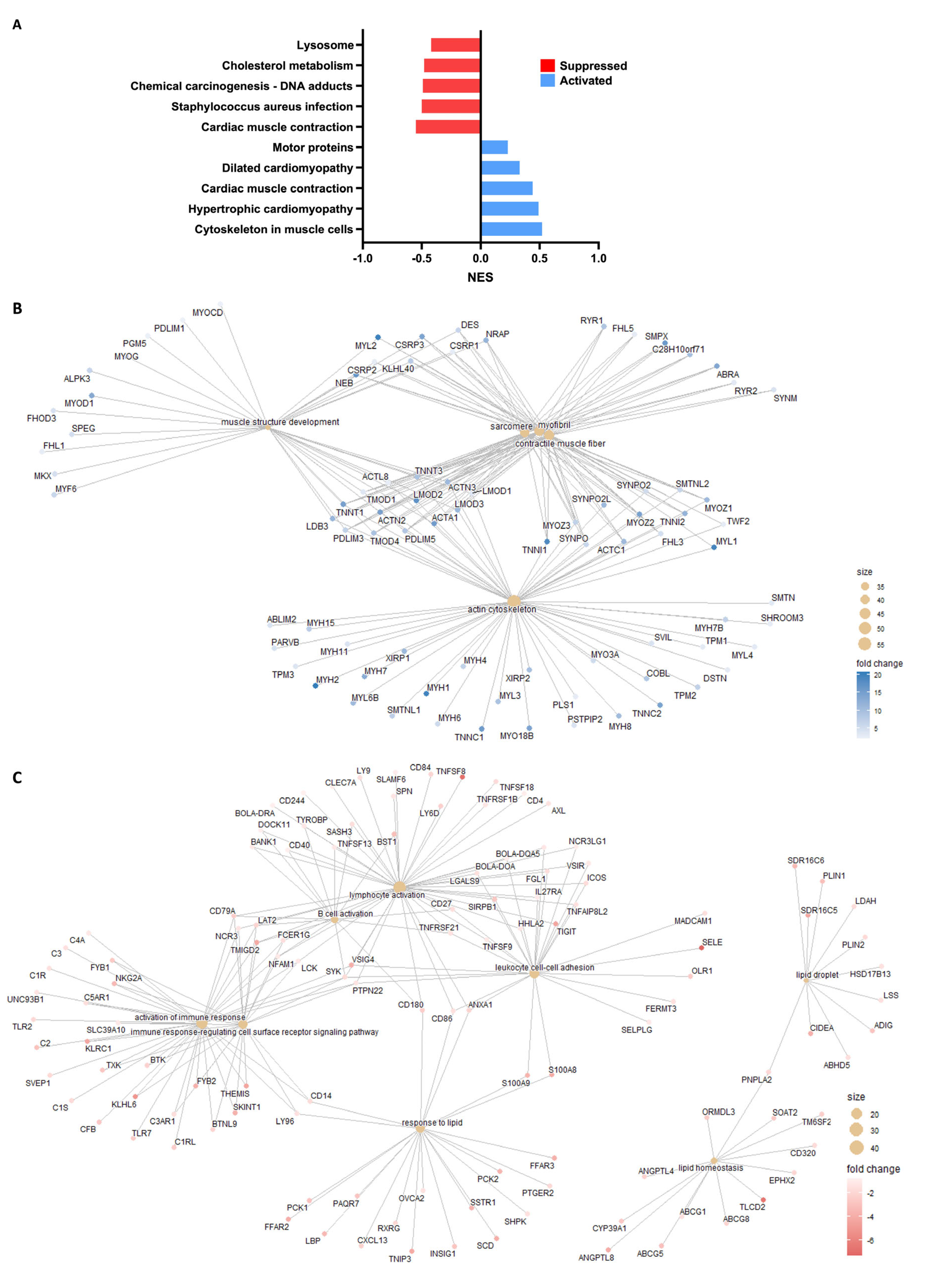
3.2. Functional Analysis Indicates Activation of Lipid Synthesis and Metabolism Pathways in SAT
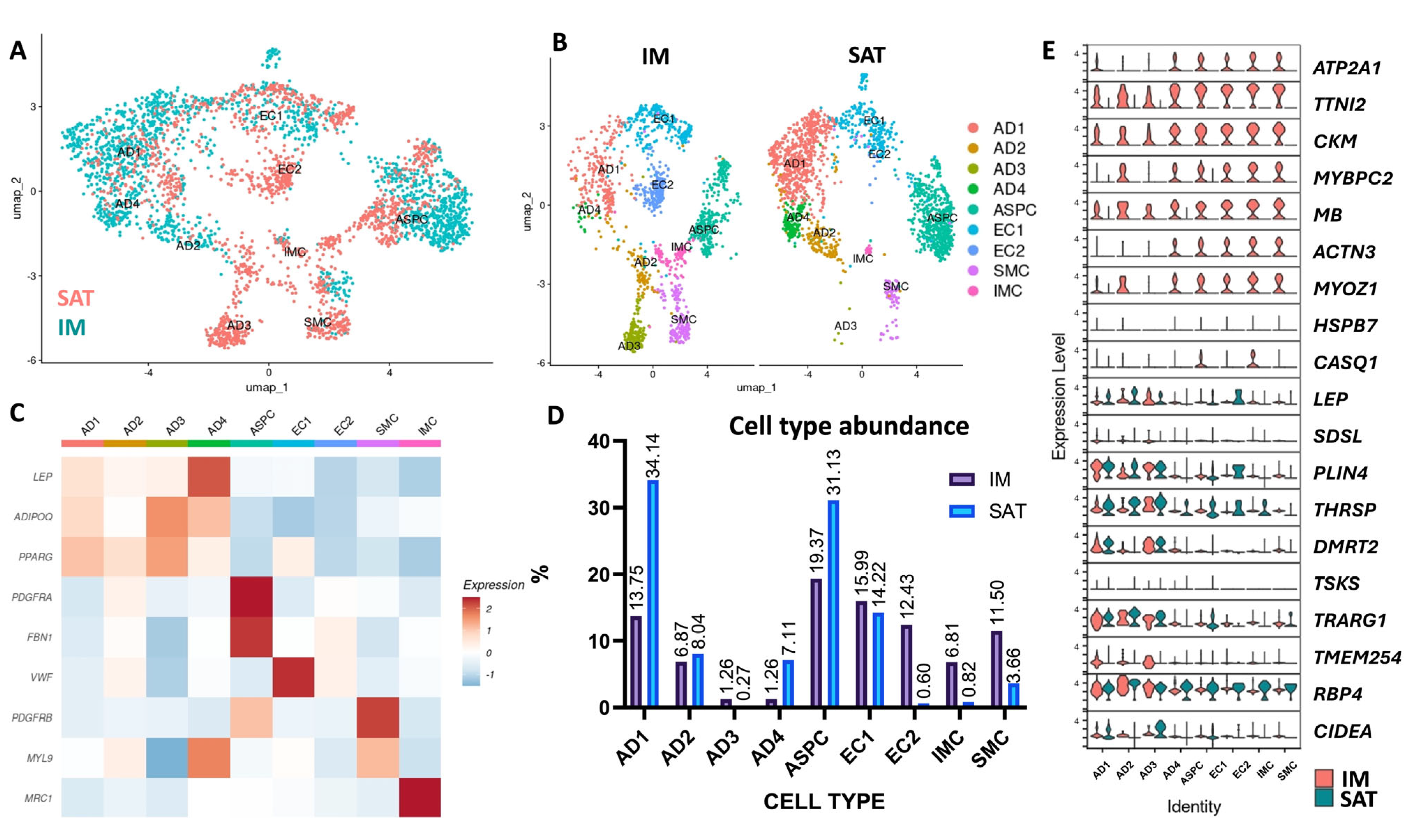
3.3. Single-Nuclei RNA Sequencing Analysis Revealed IM and SAT Cellular Diversity
3.4. Trajectory Analysis Suggests the Presence of Adipocytes in Distinct Differentiation States in IM and SAT
3.5. Endothelial and Smooth Muscle Cell Diversity Reflects Depot-Specific Vascular Architecture
3.6. Immune Cell Enrichment and Macrophage Identity in Intramuscular Fat
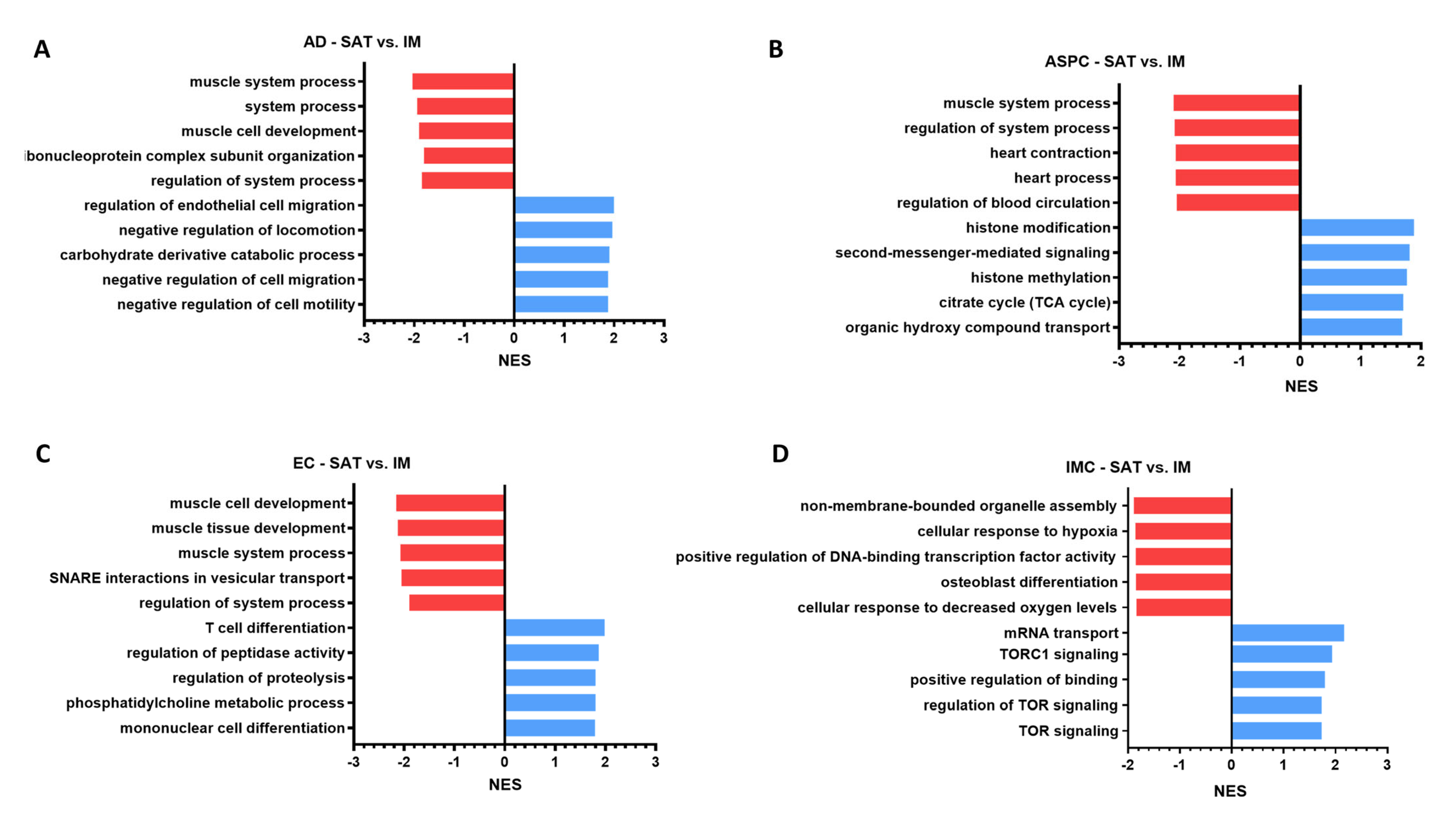
3.7. Single-Cell Functional Analysis Suggests Depot-Specificities Between IM and SAT Gene Profiles in Beef Cattle
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Mature adipocytes |
| ASPC | Adipose stromal and progenitor cells |
| EC | Endothelial cells |
| IM | Intramuscular fat |
| IMC | Immune cells |
| RNAseq | RNA sequencing |
| SAT | Subcutaneous adipose tissue |
| SMC | Smooth muscle cells |
| snRNAseq | Single-nuclei RNA sequencing |
References
- BQA. National Beef Quality Audits; National Beef Packing Company, LLC: Kansas City, MO, USA, 2022. [Google Scholar]
- Mwangi, F.W.; Charmley, E.; Gardiner, C.P.; Malau-Aduli, B.S.; Kinobe, R.T.; Malau-Aduli, A.E. Diet and genetics influence beef cattle performance and meat quality characteristics. Foods 2019, 8, 648. [Google Scholar] [CrossRef]
- Gotoh, T.; Albrecht, E.; Teuscher, F.; Kawabata, K.; Sakashita, K.; Iwamoto, H.; Wegner, J. Differences in muscle and fat accretion in Japanese Black and European cattle. Meat Sci. 2009, 82, 300–308. [Google Scholar] [CrossRef] [PubMed]
- Harvey, I.; Boudreau, A.; Stephens, J.M. Adipose tissue in health and disease. Open Biol. 2020, 10, 200291. [Google Scholar] [CrossRef] [PubMed]
- Horwitz, A.; Birk, R. Adipose tissue hyperplasia and hypertrophy in common and syndromic obesity—The case of BBS obesity. Nutrients 2023, 15, 3445. [Google Scholar] [CrossRef] [PubMed]
- Jiang, N.; Li, Y.; Shu, T.; Wang, J. Cytokines and inflammation in adipogenesis: An updated review. Front. Med. 2019, 13, 314–329. [Google Scholar] [CrossRef]
- Ambele, M.A.; Dhanraj, P.; Giles, R.; Pepper, M.S. Adipogenesis: A complex interplay of multiple molecular determinants and pathways. Int. J. Mol. Sci. 2020, 21, 4283. [Google Scholar] [CrossRef]
- Weidemüller, P.; Kholmatov, M.; Petsalaki, E.; Zaugg, J.B. Transcription factors: Bridge between cell signaling and gene regulation. Proteomics 2021, 21, 2000034. [Google Scholar] [CrossRef]
- Liao, X.; Zhou, H.; Deng, T. The composition, function, and regulation of adipose stem and progenitor cells. J. Genet. Genom. 2022, 49, 308–315. [Google Scholar] [CrossRef]
- Ford, H.; Liu, Q.; Fu, X.; Strieder-Barboza, C. White Adipose Tissue Heterogeneity in the Single-Cell Era: From Mice and Humans to Cattle. Biology 2023, 12, 1289. [Google Scholar] [CrossRef]
- Richard, A.J.; White, U.; Elks, C.M.; Stephens, J.M. Adipose Tissue: Physiology to Metabolic Dysfunction; Endotext [Internet]; MDText.com, Inc.: South Dartmouth, MA, USA, 2020. [Google Scholar]
- Wang, X.; Liang, C.; Li, A.; Cheng, G.; Long, F.; Khan, R.; Wang, J.; Zhang, Y.; Wu, S.; Wang, Y. RNA-Seq and lipidomics reveal different adipogenic processes between bovine perirenal and intramuscular adipocytes. Adipocyte 2022, 11, 448–462. [Google Scholar] [CrossRef]
- Michelotti, T.C.; Kisby, B.R.; Flores, L.S.; Tegeler, A.P.; Fokar, M.; Crasto, C.; Menarim, B.C.; Loux, S.C.; Strieder-Barboza, C. Single-nuclei analysis reveals depot-specific transcriptional heterogeneity and depot-specific cell types in adipose tissue of dairy cows. Front. Cell Dev. Biol. 2022, 10, 1025240. [Google Scholar] [CrossRef]
- Yamada, T.; Kamiya, M.; Higuchi, M.; Nakanishi, N. Fat depot-specific differences of macrophage infiltration and cellular senescence in obese bovine adipose tissues. J. Vet. Med. Sci. 2018, 80, 1495–1503. [Google Scholar] [CrossRef]
- Cinkajzlová, A.; Mráz, M.; Haluzík, M. Adipose tissue immune cells in obesity, type 2 diabetes mellitus and cardiovascular diseases. J. Endocrinol. 2022, 252, R1–R22. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Green, M.M.; Woolums, A.R.; Karisch, B.B.; Harvey, K.M.; Capik, S.F.; Scott, M.A. Influence of the At-Arrival Host Transcriptome on Bovine Respiratory Disease Incidence during Backgrounding. Vet. Sci. 2023, 10, 211. [Google Scholar] [CrossRef] [PubMed]
- Stuart, T.; Butler, A.; Hoffman, P.; Hafemeister, C.; Papalexi, E.; Mauck, W.M.; Hao, Y.; Stoeckius, M.; Smibert, P.; Satija, R. Comprehensive integration of single-cell data. Cell 2019, 177, 1888–1902.e21. [Google Scholar] [CrossRef]
- Loft, A.; Emont, M.P.; Weinstock, A.; Divoux, A.; Ghosh, A.; Wagner, A.; Hertzel, A.V.; Maniyadath, B.; Deplancke, B.; Liu, B.; et al. Towards a consensus atlas of human and mouse adipose tissue at single-cell resolution. Nat. Metab. 2025, 7, 875–894. [Google Scholar] [CrossRef]
- Tan, Z.; Jiang, H. Molecular and Cellular Mechanisms of Intramuscular Fat Development and Growth in Cattle. Int. J. Mol. Sci. 2024, 25, 2520. [Google Scholar] [CrossRef]
- Sheng, X.; Ni, H.; Liu, Y.; Li, J.; Zhang, L.; Guo, Y. RNA-seq analysis of bovine intramuscular, subcutaneous and perirenal adipose tissues. Mol. Biol. Rep. 2014, 41, 1631–1637. [Google Scholar] [CrossRef]
- Ueda, S.; Hosoda, M.; Yoshino, K.-I.; Yamanoue, M.; Shirai, Y. Gene expression analysis provides new insights into the mechanism of intramuscular fat formation in Japanese Black Cattle. Genes 2021, 12, 1107. [Google Scholar] [CrossRef]
- Zhang, Y.; Sun, Y.; Wu, Z.; Xiong, X.; Zhang, J.; Ma, J.; Xiao, S.; Huang, L.; Yang, B. Subcutaneous and intramuscular fat transcriptomes show large differences in network organization and associations with adipose traits in pigs. Sci. China Life Sci. 2021, 64, 1732–1746. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.H.; Lu, J.X.; Chen, Y.; Zhao, Y.Q.; Guo, P.H.; Yang, J.T.; Zang, R.X. Comparison of the adipogenesis in intramuscular and subcutaneous adipocytes from Bamei and Landrace pigs. Biochem. Cell Biol. 2014, 92, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Wan, R.; Du, J.; Ren, L.; Meng, Q. Selective adipogenic effects of propionate on bovine intramuscular and subcutaneous preadipocytes. Meat Sci. 2009, 82, 372–378. [Google Scholar] [CrossRef]
- Emont, M.P.; Jacobs, C.; Essene, A.L.; Pant, D.; Tenen, D.; Colleluori, G.; Di Vincenzo, A.; Jørgensen, A.M.; Dashti, H.; Stefek, A. A single-cell atlas of human and mouse white adipose tissue. Nature 2022, 603, 926–933. [Google Scholar] [CrossRef]
- Wetzels, S.; Bijnen, M.; Wijnands, E.; van de Gaar, J.; Tan, A.; Coort, S.; Biessen, E.A.; Schalkwijk, C.G.; Wouters, K. CD11c− MHC2low Macrophages Are a New Inflammatory and Dynamic Subset in Murine Adipose Tissue. Immunometabolism 2020, 2, e200015. [Google Scholar] [CrossRef]
- Chasapi, A.; Balampanis, K.; Kourea, E.; Kalfarentzos, F.; Lambadiari, V.; Lambrou, G.I.; Melachrinou, M.; Sotiropoulou-Bonikou, G. Can obesity-induced inflammation in skeletal muscle and intramuscular adipose tissue accurately detect liver fibrosis? J. Musculoskelet. Neuronal Interact. 2018, 18, 509. [Google Scholar]
- Yamada, T. Intramuscular adipogenesis in cattle: Effects of body fat distribution and macrophage infiltration. Anim. Sci. J. 2022, 93, e13785. [Google Scholar] [CrossRef]
- Kawai, T.; Autieri, M.V.; Scalia, R. Adipose tissue inflammation and metabolic dysfunction in obesity. Am. J. Physiol. Cell Physiol. 2021, 320, C375–C391. [Google Scholar] [CrossRef]
- Dykier, K.C.; Oltjen, J.W.; Robinson, P.H.; Sainz, R.D. Effects of finishing diet sorting and digestibility on performance and feed efficiency in beef steers. Animal 2020, 14, 59–65. [Google Scholar] [CrossRef]
- Li, M.; Qian, M.; Kyler, K.; Xu, J. Adipose Tissue-Endothelial Cell Interactions in Obesity-Induced Endothelial Dysfunction. Front. Cardiovasc. Med. 2021, 8, 681581. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Green, M.M.; Ford, H.R.; Tegeler, A.P.; Benitez, O.J.; Johnson, B.J.; Strieder-Barboza, C. A Single-Cell Assessment of Intramuscular and Subcutaneous Adipose Tissue in Beef Cattle. Agriculture 2025, 15, 1545. https://doi.org/10.3390/agriculture15141545
Green MM, Ford HR, Tegeler AP, Benitez OJ, Johnson BJ, Strieder-Barboza C. A Single-Cell Assessment of Intramuscular and Subcutaneous Adipose Tissue in Beef Cattle. Agriculture. 2025; 15(14):1545. https://doi.org/10.3390/agriculture15141545
Chicago/Turabian StyleGreen, Mollie M., Hunter R. Ford, Alexandra P. Tegeler, Oscar J. Benitez, Bradley J. Johnson, and Clarissa Strieder-Barboza. 2025. "A Single-Cell Assessment of Intramuscular and Subcutaneous Adipose Tissue in Beef Cattle" Agriculture 15, no. 14: 1545. https://doi.org/10.3390/agriculture15141545
APA StyleGreen, M. M., Ford, H. R., Tegeler, A. P., Benitez, O. J., Johnson, B. J., & Strieder-Barboza, C. (2025). A Single-Cell Assessment of Intramuscular and Subcutaneous Adipose Tissue in Beef Cattle. Agriculture, 15(14), 1545. https://doi.org/10.3390/agriculture15141545