Spectrum and Frequency of Tumors, Cancer Risk and Survival in Chilean Families with Lynch Syndrome: Experience of the Implementation of a Registry

 , , , and
, , , and
Abstract
1. Introduction
2. Methods
2.1. Hereditary Colorectal Cancer Registry from Clinica Las Condes
2.2. Genetic Analysis
2.3. MMR Variant Nomenclature and Classification
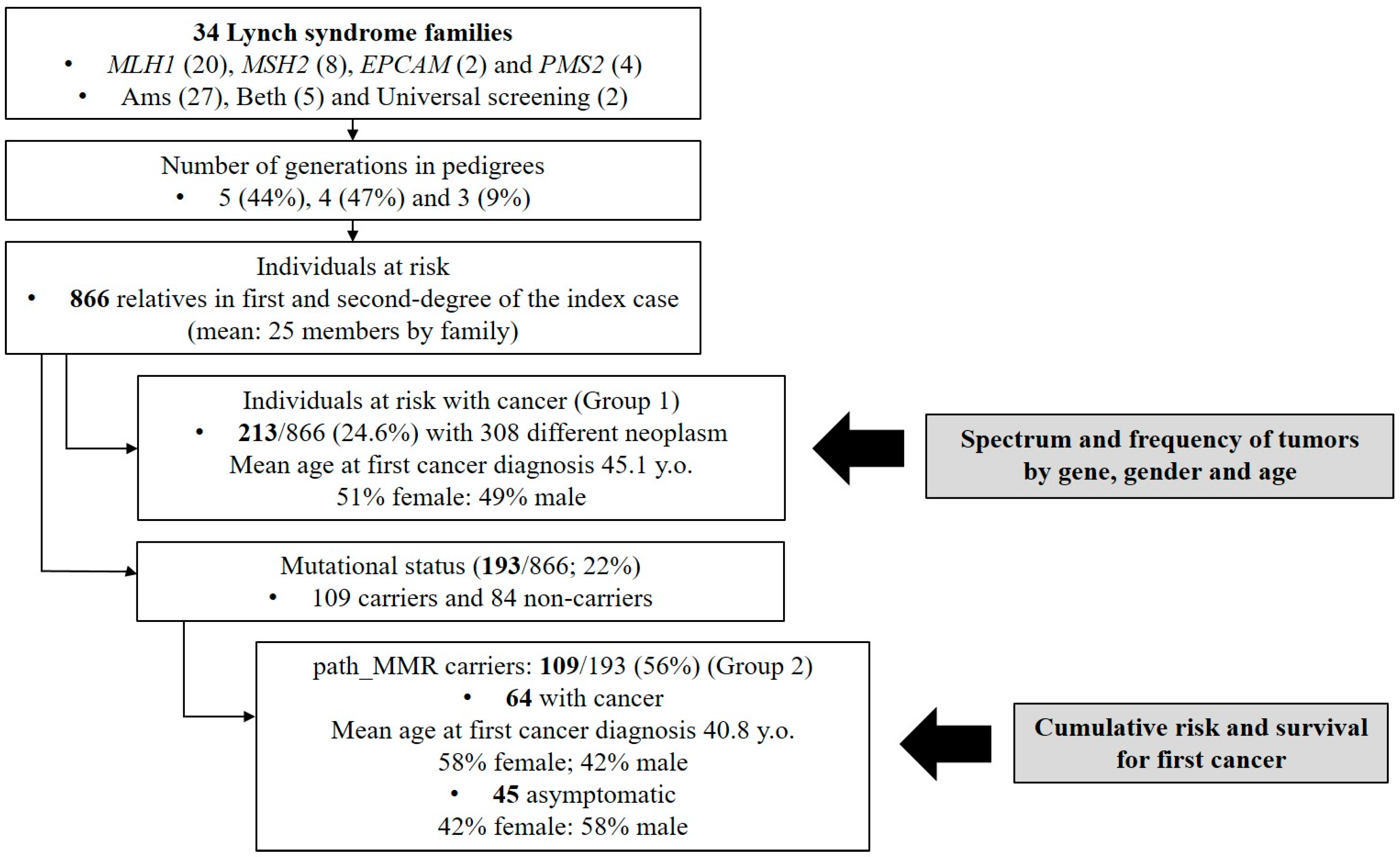
2.4. Study Population
2.5. Statistical Analysis
3. Results
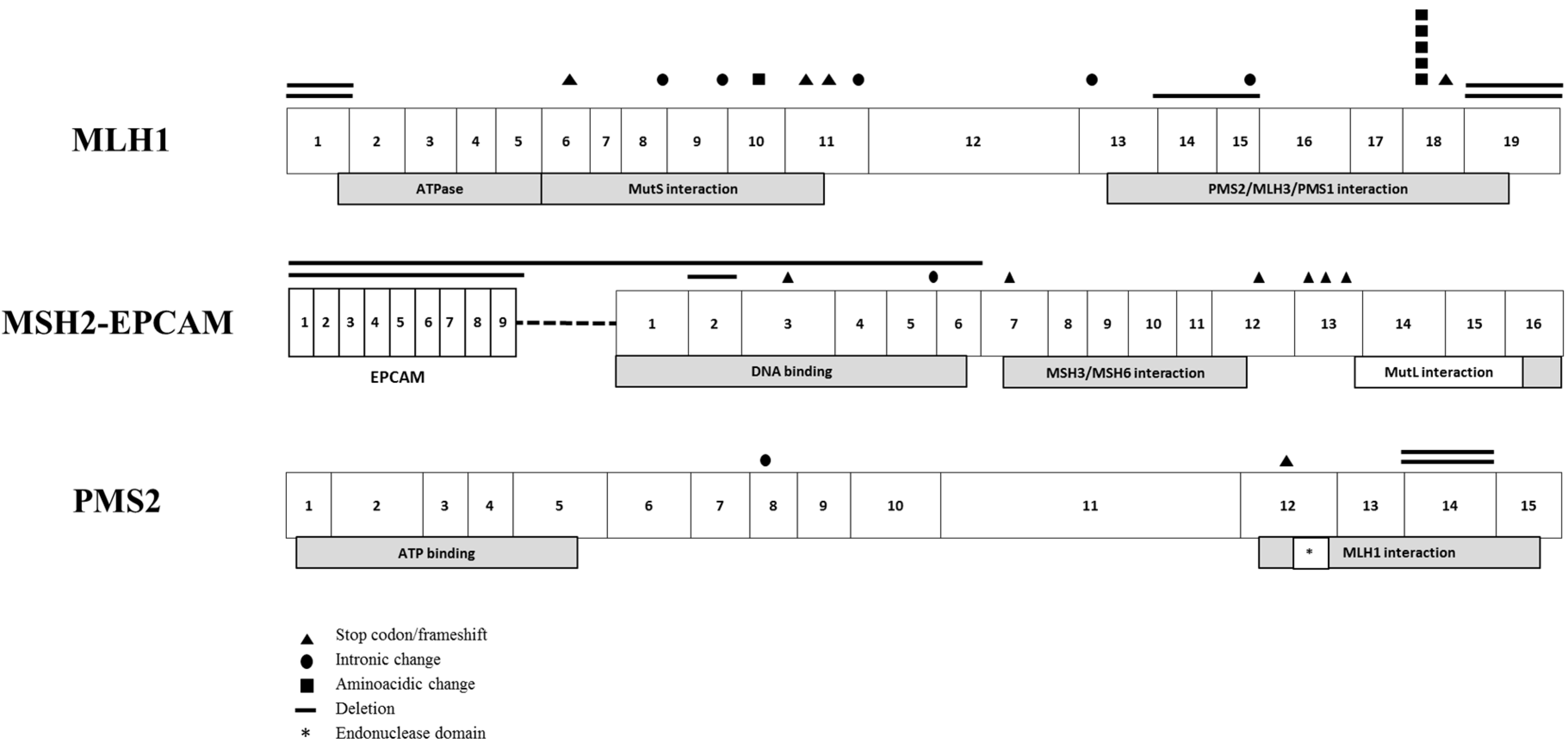
3.1. Genetic Characterization of LS Families
3.2. Clinicopathologic Features of LS Families
3.3. Spectrum and Frequency of Tumors by Gene, Gender and Age
3.4. Characterization of Path_MMR Carriers
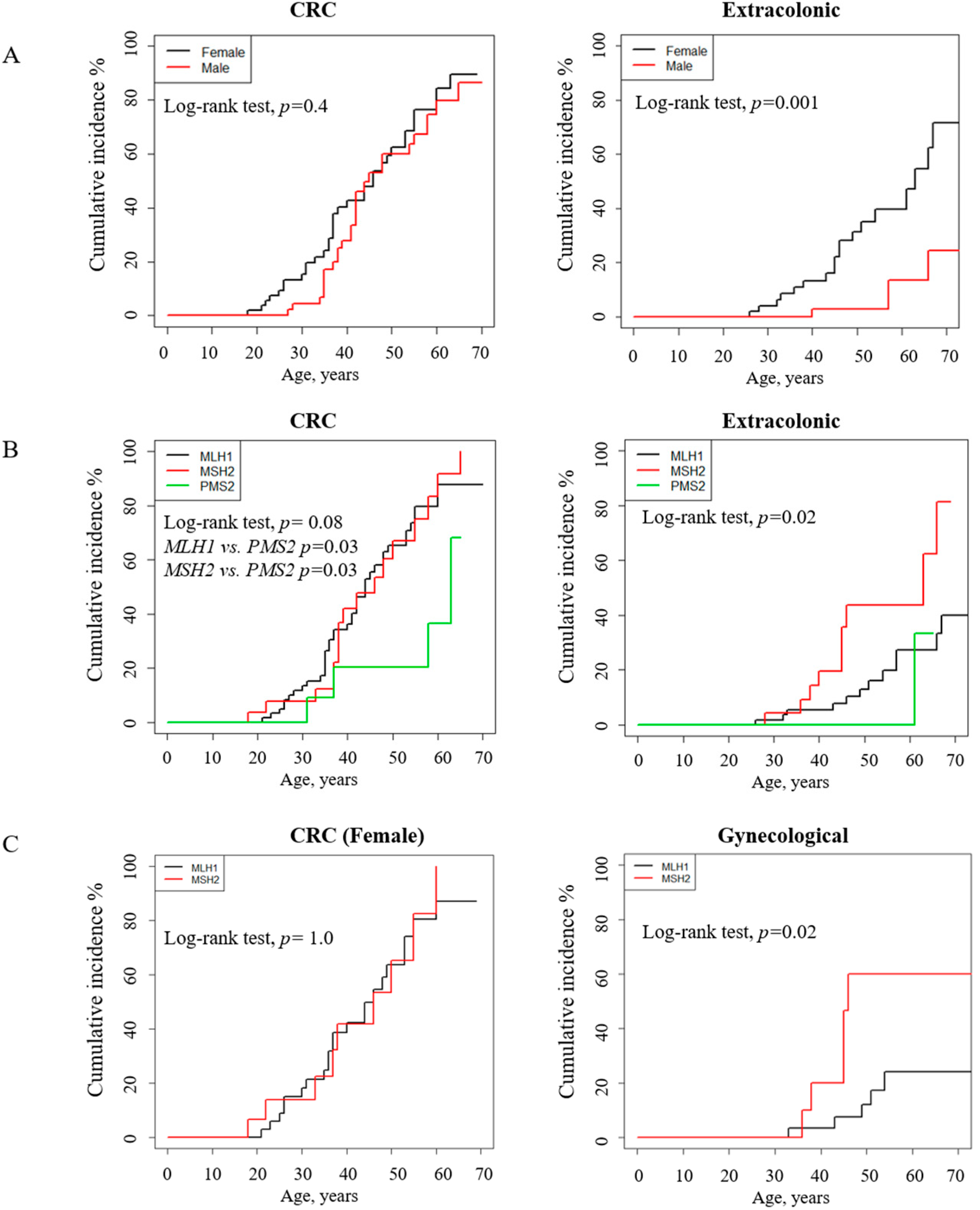
3.5. Path_ MMR Carriers: Tumor Spectrum, Cumulative Incidence and Survival Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AMS | Amsterdam |
| CRC | colorectal cancer |
| HGMD | Human Gene Mutation Database |
| HGVS | Human Genome Variation Society |
| InSIGHT | International Society of Gastrointestinal Hereditary Tumors |
| LS | Lynch syndrome |
| LOVD | Leiden Open Variation Database |
| MLPA | Multiplex ligation dependent-probes amplification |
| MMR | mismatch-repair gene |
| MSI | microsatellite instability |
| NGS | Next generation sequencing |
| Path_MMR | Pathogenic (disease-causing) variant of an MMR gene. |
| path_MLH1 | Pathogenic (disease-causing) variant of the MLH1 gene |
| path_MSH2 | Pathogenic (disease-causing) variant of the MSH2 gene |
| path_MSH6 | Pathogenic (disease-causing) variant of the MSH6 gene |
| path_PMS2 | Pathogenic (disease-causing) variant of the PMS2 gene |
| path_EPCAM | Pathogenic (disease-causing) variant of the EPCAM gene |
| UMD | Universal Mutation Database |
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Departamento de Estadísticas de Información de Salud. Available online: www.deis.minsal.cl (accessed on 14 May 2020).
- Zarate, A.J.; Alonso, F.T.; Garmendia, M.L.; Lopez-Kostner, F. Increasing crude and adjusted mortality rates for colorectal cancer in a developing South American country. Colorectal Dis. 2013, 15, 47–51. [Google Scholar] [CrossRef]
- Pinol, V.; Castells, A.; Andreu, M.; Castellvi-Bel, S.; Alenda, C.; Llor, X.; Xicola, R.M.; Rodriguez-Moranta, F.; Paya, A.; Jover, R.; et al. Accuracy of revised Bethesda guidelines, microsatellite instability, and immunohistochemistry for the identification of patients with hereditary nonpolyposis colorectal cancer. JAMA 2005, 293, 1986–1994. [Google Scholar] [CrossRef] [PubMed]
- Balmana, J.; Castells, A.; Cervantes, A.; Group, E.G.W. Familial colorectal cancer risk: ESMO Clinical Practice Guidelines. Ann. Oncol. 2010, 21, v78–v81. [Google Scholar] [CrossRef] [PubMed]
- Thompson, B.A.; Spurdle, A.B.; Plazzer, J.P.; Greenblatt, M.S.; Akagi, K.; Al-Mulla, F.; Bapat, B.; Bernstein, I.; Capella, G.; den Dunnen, J.T.; et al. Application of a 5-tiered scheme for standardized classification of 2360 unique mismatch repair gene variants in the InSiGHT locus-specific database. Nat. Genet. 2014, 46, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.G.; Ahnen, D.J. Familial colon cancer syndromes: An update of a rapidly evolving field. Curr. Gastroenterol. Rep. 2012, 14, 428–438. [Google Scholar] [CrossRef] [PubMed]
- Lynch, H.T.; de la Chapelle, A. Hereditary colorectal cancer. N. Engl. J. Med. 2003, 348, 919–932. [Google Scholar] [CrossRef] [PubMed]
- Kuiper, R.P.; Vissers, L.E.; Venkatachalam, R.; Bodmer, D.; Hoenselaar, E.; Goossens, M.; Haufe, A.; Kamping, E.; Niessen, R.C.; Hogervorst, F.B.; et al. Recurrence and variability of germline EPCAM deletions in Lynch syndrome. Hum. Mutat. 2011, 32, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Moller, P.; Seppala, T.; Bernstein, I.; Holinski-Feder, E.; Sala, P.; Evans, D.G.; Lindblom, A.; Macrae, F.; Blanco, I.; Sijmons, R.; et al. Cancer incidence and survival in Lynch syndrome patients receiving colonoscopic and gynaecological surveillance: First report from the prospective Lynch syndrome database. Gut 2017, 66, 464–472. [Google Scholar] [CrossRef] [PubMed]
- Moller, P.; Seppala, T.; Bernstein, I.; Holinski-Feder, E.; Sala, P.; Evans, D.G.; Lindblom, A.; Macrae, F.; Blanco, I.; Sijmons, R.; et al. Incidence of and survival after subsequent cancers in carriers of pathogenic MMR variants with previous cancer: A report from the prospective Lynch syndrome database. Gut 2017, 66, 1657–1664. [Google Scholar] [CrossRef] [PubMed]
- Gudbjartsson, D.F.; Helgason, H.; Gudjonsson, S.A.; Zink, F.; Oddson, A.; Gylfason, A.; Besenbacher, S.; Magnusson, G.; Halldorsson, B.V.; Hjartarson, E.; et al. Large-scale whole-genome sequencing of the Icelandic population. Nat. Genet. 2015, 47, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Moller, P.; Seppala, T.T.; Bernstein, I.; Holinski-Feder, E.; Sala, P.; Gareth Evans, D.; Lindblom, A.; Macrae, F.; Blanco, I.; Sijmons, R.H.; et al. Cancer risk and survival in path_MMR carriers by gene and gender up to 75 years of age: A report from the Prospective Lynch Syndrome Database. Gut 2018, 67, 1306–1316. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Valentin, M.; Joost, P.; Therkildsen, C.; Jonsson, M.; Rambech, E.; Nilbert, M. Frequent mismatch-repair defects link prostate cancer to Lynch syndrome. BMC Urol. 2016, 16, 15. [Google Scholar] [CrossRef] [PubMed]
- Joost, P.; Therkildsen, C.; Dominguez-Valentin, M.; Jonsson, M.; Nilbert, M. Urinary Tract Cancer in Lynch Syndrome; Increased Risk in Carriers of MSH2 Mutations. Urology 2015, 86, 1212–1217. [Google Scholar] [CrossRef] [PubMed]
- Rosty, C.; Walsh, M.D.; Lindor, N.M.; Thibodeau, S.N.; Mundt, E.; Gallinger, S.; Aronson, M.; Pollett, A.; Baron, J.A.; Pearson, S.; et al. High prevalence of mismatch repair deficiency in prostate cancers diagnosed in mismatch repair gene mutation carriers from the colon cancer family registry. Fam. Cancer 2014, 13, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Walsh, M.D.; Buchanan, D.D.; Cummings, M.C.; Pearson, S.A.; Arnold, S.T.; Clendenning, M.; Walters, R.; McKeone, D.M.; Spurdle, A.B.; Hopper, J.L.; et al. Lynch syndrome-associated breast cancers: Clinicopathologic characteristics of a case series from the colon cancer family registry. Clin. Cancer Res. 2010, 16, 2214–2224. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, K.; Hurtado, C.; Hevia, M.A.; Wielandt, A.M.; de la Fuente, M.; Church, J.; Carvallo, P.; Lopez-Kostner, F. Spectrum of MLH1 and MSH2 mutations in Chilean families with suspected Lynch syndrome. Dis. Colon Rectum 2010, 53, 450–459. [Google Scholar] [CrossRef] [PubMed]
- Vaccaro, C.A.; Bonadeo, F.; Roverano, A.V.; Peltomaki, P.; Bala, S.; Renkonen, E.; Redal, M.A.; Mocetti, E.; Mullen, E.; Ojea-Quintana, G.; et al. Hereditary nonpolyposis colorectal cancer (Lynch Syndrome) in Argentina: Report from a referral hospital register. Dis. Colon Rectum 2007, 50, 1604–1611. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Valentin, M.; Nilbert, M.; Wernhoff, P.; Lopez-Kostner, F.; Vaccaro, C.; Sarroca, C.; Palmero, E.I.; Giraldo, A.; Ashton-Prolla, P.; Alvarez, K.; et al. Mutation spectrum in South American Lynch syndrome families. Hered. Cancer Clin. Pract. 2013, 11, 18. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Carneiro da Silva, F.; Ferreira, J.R.; Torrezan, G.T.; Figueiredo, M.C.; Santos, E.M.; Nakagawa, W.T.; Brianese, R.C.; Petrolini de Oliveira, L.; Begnani, M.D.; Aguiar-Junior, S.; et al. Clinical and molecular characterization of brazilian patients suspected to have lynch syndrome. PLoS ONE 2015, 10, e0139753. [Google Scholar] [CrossRef]
- Da Silva, F.C.; de Oliveira, L.P.; Santos, E.M.; Nakagawa, W.T.; Aguiar Junior, S.; Valentin, M.D.; Rossi, B.M.; de Oliveira Ferreira, F. Frequency of extracolonic tumors in Brazilian families with Lynch syndrome: Analysis of a hereditary colorectal cancer institutional registry. Fam. Cancer 2010, 9, 563–570. [Google Scholar] [CrossRef] [PubMed]
- Oliveira Ferreira, F.; Napoli Ferreira, C.C.; Rossi, B.M.; Toshihiko Nakagawa, W.; Aguilar, S., Jr.; Monteiro Santos, E.M.; Vierira Costa, M.L.; Lopes, A. Frequency of extra-colonic tumors in hereditary nonpolyposis colorectal cancer (HNPCC) and familial colorectal cancer (FCC) Brazilian families: An analysis by a Brazilian Hereditary Colorectal Cancer Institutional Registry. Fam. Cancer 2004, 3, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Vasen, H.F.; Blanco, I.; Aktan-Collan, K.; Gopie, J.P.; Alonso, A.; Aretz, S.; Bernstein, I.; Bertario, L.; Burn, J.; Capella, G.; et al. Revised guidelines for the clinical management of Lynch syndrome (HNPCC): Recommendations by a group of European experts. Gut 2013, 62, 812–823. [Google Scholar] [CrossRef] [PubMed]
- Giardiello, F.M.; Allen, J.I.; Axilbund, J.E.; Boland, C.R.; Burke, C.A.; Burt, R.W.; Church, J.M.; Dominitz, J.A.; Johnson, D.A.; Kaltenbach, T.; et al. Guidelines on genetic evaluation and management of Lynch syndrome: A consensus statement by the US Multi-Society Task Force on colorectal cancer. Gastroenterology 2014, 147, 502–526. [Google Scholar] [CrossRef] [PubMed]
- Vasen, H.F.; Mecklin, J.P.; Khan, P.M.; Lynch, H.T. The International Collaborative Group on Hereditary Non-Polyposis Colorectal Cancer (ICG-HNPCC). Dis. Colon Rectum 1991, 34, 424–425. [Google Scholar] [CrossRef] [PubMed]
- Vasen, H.F.; Watson, P.; Mecklin, J.P.; Lynch, H.T. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology 1999, 116, 1453–1456. [Google Scholar] [CrossRef]
- Rodriguez-Bigas, M.A.; Boland, C.R.; Hamilton, S.R.; Henson, D.E.; Jass, J.R.; Khan, P.M.; Lynch, H.; Perucho, M.; Smyrk, T.; Sobin, L.; et al. A National cancer institute workshop on hereditary nonpolyposis colorectal cancer syndrome: Meeting highlights and Bethesda guidelines. J. Natl. Cancer Inst. 1997, 89, 1758–1762. [Google Scholar] [CrossRef] [PubMed]
- Umar, A.; Boland, C.R.; Terdiman, J.P.; Syngal, S.; de la Chapelle, A.; Ruschoff, J.; Fishel, R.; Lindor, N.M.; Burgart, L.J.; Hamelin, R.; et al. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J. Natl. Cancer Inst. 2004, 96, 261–268. [Google Scholar] [CrossRef]
- Wielandt, A.M.; Zarate, A.J.; Hurtado, C.; Orellana, P.; Alvarez, K.; Pinto, E.; Contreras, L.; Corvalan, A.; Kronberg, U.; Lopez-Kostner, F. Lynch syndrome: Selection of families by microsatellite instability and immunohistochemistry. Rev. Med. Chil. 2012, 140, 1132–1139. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rossi, B.M.; Palmero, E.I.; Lopez-Kostner, F.; Sarroca, C.; Vaccaro, C.A.; Spirandelli, F.; Ashton-Prolla, P.; Rodriguez, Y.; de Campos Reis Galvao, H.; Reis, R.M.; et al. A survey of the clinicopathological and molecular characteristics of patients with suspected Lynch syndrome in Latin America. BMC Cancer 2017, 17, 623. [Google Scholar] [CrossRef] [PubMed]
- Senter, L.; Clendenning, M.; Sotamaa, K.; Hampel, H.; Green, J.; Potter, J.D.; Lindblom, A.; Lagerstedt, K.; Thibodeau, S.N.; Lindor, N.M.; et al. The clinical phenotype of Lynch syndrome due to germ-line PMS2 mutations. Gastroenterology 2008, 135, 419–428. [Google Scholar] [CrossRef] [PubMed]
- Della Valle, A.; Rossi, B.M.; Palmero, E.I.; Antelo, M.; Vaccaro, C.A.; Lopez-Kostner, F.; Alvarez, K.; Cruz-Correa, M.; Bruno, L.I.; Forones, N.M.; et al. A snapshot of current genetic testing practice in Lynch syndrome: The results of a representative survey of 33 Latin American existing centres/registries. Eur. J. Cancer 2019, 119, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Peltomaki, P. Update on Lynch syndrome genomics. Fam. Cancer 2016, 15, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Valentin, M.; Sampson, J.R.; Seppala, T.T.; Ten Broeke, S.W.; Plazzer, J.P.; Nakken, S.; Engel, C.; Aretz, S.; Jenkins, M.A.; Sunde, L.; et al. Cancer risks by gene, age, and gender in 6350 carriers of pathogenic mismatch repair variants: Findings from the Prospective Lynch Syndrome Database. Genet. Med. 2020, 22, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Kastrinos, F.; Stoffel, E.M.; Balmana, J.; Steyerberg, E.W.; Mercado, R.; Syngal, S. Phenotype comparison of MLH1 and MSH2 mutation carriers in a cohort of 1914 individuals undergoing clinical genetic testing in the United States. Cancer Epidemiol. Biomark. Prev. 2008, 17, 2044–2051. [Google Scholar] [CrossRef] [PubMed]
- Lin-Hurtubise, K.M.; Yheulon, C.G.; Gagliano, R.A., Jr.; Lynch, H.T. Excess of extracolonic non-endometrial multiple primary cancers in MSH2 germline mutation carriers over MLH1. J. Surg. Oncol. 2013, 108, 433–437. [Google Scholar] [CrossRef]
- Pande, M.; Wei, C.; Chen, J.; Amos, C.I.; Lynch, P.M.; Lu, K.H.; Lucio, L.A.; Boyd-Rogers, S.G.; Bannon, S.A.; Mork, M.E.; et al. Cancer spectrum in DNA mismatch repair gene mutation carriers: Results from a hospital based Lynch syndrome registry. Fam. Cancer 2012, 11, 441–447. [Google Scholar] [CrossRef]
- Vasen, H.F.; Stormorken, A.; Menko, F.H.; Nagengast, F.M.; Kleibeuker, J.H.; Griffioen, G.; Taal, B.G.; Moller, P.; Wijnen, J.T. MSH2 mutation carriers are at higher risk of cancer than MLH1 mutation carriers: A study of hereditary nonpolyposis colorectal cancer families. J. Clin. Oncol. 2001, 19, 4074–4080. [Google Scholar] [CrossRef] [PubMed]
- Barrow, E.; Robinson, L.; Alduaij, W.; Shenton, A.; Clancy, T.; Lalloo, F.; Hill, J.; Evans, D.G. Cumulative lifetime incidence of extracolonic cancers in Lynch syndrome: A report of 121 families with proven mutations. Clin. Genet. 2009, 75, 141–149. [Google Scholar] [CrossRef]
- Win, A.K.; Lindor, N.M.; Young, J.P.; Macrae, F.A.; Young, G.P.; Williamson, E.; Parry, S.; Goldblatt, J.; Lipton, L.; Winship, I.; et al. Risks of primary extracolonic cancers following colorectal cancer in lynch syndrome. J. Natl. Cancer Inst. 2012, 104, 1363–1372. [Google Scholar] [CrossRef]
- Roberts, M.E.; Jackson, S.A.; Susswein, L.R.; Zeinomar, N.; Ma, X.; Marshall, M.L.; Stettner, A.R.; Milewski, B.; Xu, Z.; Solomon, B.D.; et al. MSH6 and PMS2 germ-line pathogenic variants implicated in Lynch syndrome are associated with breast cancer. Genet. Med. 2018, 20, 1167–1174. [Google Scholar] [CrossRef] [PubMed]
- Win, A.K.; Lindor, N.M.; Winship, I.; Tucker, K.M.; Buchanan, D.D.; Young, J.P.; Rosty, C.; Leggett, B.; Giles, G.G.; Goldblatt, J.; et al. Risks of colorectal and other cancers after endometrial cancer for women with Lynch syndrome. J. Natl. Cancer Inst. 2013, 105, 274–279. [Google Scholar] [CrossRef] [PubMed]
- Harkness, E.F.; Barrow, E.; Newton, K.; Green, K.; Clancy, T.; Lalloo, F.; Hill, J.; Evans, D.G. Lynch syndrome caused by MLH1 mutations is associated with an increased risk of breast cancer: A cohort study. J. Med. Genet. 2015, 52, 553–556. [Google Scholar] [CrossRef] [PubMed]
- Lotsari, J.E.; Gylling, A.; Abdel-Rahman, W.M.; Nieminen, T.T.; Aittomaki, K.; Friman, M.; Pitkanen, R.; Aarnio, M.; Jarvinen, H.J.; Mecklin, J.P.; et al. Breast carcinoma and Lynch syndrome: Molecular analysis of tumors arising in mutation carriers, non-carriers, and sporadic cases. Breast Cancer Res. 2012, 14, R90. [Google Scholar] [CrossRef] [PubMed]
- Porkka, N.K.; Olkinuora, A.; Kuopio, T.; Ahtiainen, M.; Eldfors, S.; Almusa, H.; Mecklin, J.P.; Peltomaki, P. Does breast carcinoma belong to the Lynch syndrome tumor spectrum?—Somatic mutational profiles vs. ovarian and colorectal carcinomas. Oncotarget 2020, 11, 1244–1256. [Google Scholar] [CrossRef] [PubMed]
- Ferreccio, C.; Rollan, A.; Harris, P.R.; Serrano, C.; Gederlini, A.; Margozzini, P.; Gonzalez, C.; Aguilera, X.; Venegas, A.; Jara, A. Gastric cancer is related to early Helicobacter pylori infection in a high-prevalence country. Cancer Epidemiol. Biomark. Prev. 2007, 16, 662–667. [Google Scholar] [CrossRef] [PubMed]
- Saita, C.; Yamaguchi, T.; Horiguchi, S.I.; Yamada, R.; Takao, M.; Iijima, T.; Wakaume, R.; Aruga, T.; Tabata, T.; Koizumi, K. Tumor development in Japanese patients with Lynch syndrome. PLoS ONE 2018, 13, e0195572. [Google Scholar] [CrossRef] [PubMed]
- Kastrinos, F.; Mukherjee, B.; Tayob, N.; Wang, F.; Sparr, J.; Raymond, V.M.; Bandipalliam, P.; Stoffel, E.M.; Gruber, S.B.; Syngal, S. Risk of pancreatic cancer in families with Lynch syndrome. JAMA 2009, 302, 1790–1795. [Google Scholar] [CrossRef] [PubMed]
- Bujanda, L.; Herreros-Villanueva, M. Pancreatic Cancer in Lynch Syndrome Patients. J. Cancer 2017, 8, 3667–3674. [Google Scholar] [CrossRef]
- Watson, P.; Vasen, H.F.A.; Mecklin, J.P.; Bernstein, I.; Aarnio, M.; Jarvinen, H.J.; Myrhoj, T.; Sunde, L.; Wijnen, J.T.; Lynch, H.T. The risk of extra-colonic, extra-endometrial cancer in the Lynch syndrome. Int. J. Cancer 2008, 123, 444–449. [Google Scholar] [CrossRef]
- Grindedal, E.M.; Moller, P.; Eeles, R.; Stormorken, A.T.; Bowitz-Lothe, I.M.; Landro, S.M.; Clark, N.; Kvale, R.; Shanley, S.; Maehle, L. Germ-line mutations in mismatch repair genes associated with prostate cancer. Cancer Epidemiol. Biomark. Prev. 2009, 18, 2460–2467. [Google Scholar] [CrossRef] [PubMed]
- Van der Post, R.S.; Kiemeney, L.A.; Ligtenberg, M.J.; Witjes, J.A.; Hulsbergen-van de Kaa, C.A.; Bodmer, D.; Schaap, L.; Kets, C.M.; van Krieken, J.H.; Hoogerbrugge, N. Risk of urothelial bladder cancer in Lynch syndrome is increased, in particular among MSH2 mutation carriers. J. Med. Genet. 2010, 47, 464–470. [Google Scholar] [CrossRef] [PubMed]
- Engel, C.; Loeffler, M.; Steinke, V.; Rahner, N.; Holinski-Feder, E.; Dietmaier, W.; Schackert, H.K.; Goergens, H.; von Knebel Doeberitz, M.; Goecke, T.O.; et al. Risks of less common cancers in proven mutation carriers with lynch syndrome. J. Clin. Oncol. 2012, 30, 4409–4415. [Google Scholar] [CrossRef] [PubMed]
- De Jong, A.E.; Hendriks, Y.M.; Kleibeuker, J.H.; de Boer, S.Y.; Cats, A.; Griffioen, G.; Nagengast, F.M.; Nelis, F.G.; Rookus, M.A.; Vasen, H.F. Decrease in mortality in Lynch syndrome families because of surveillance. Gastroenterology 2006, 130, 665–671. [Google Scholar] [CrossRef] [PubMed]
- Watson, P.; Lin, K.M.; Rodriguez-Bigas, M.A.; Smyrk, T.; Lemon, S.; Shashidharan, M.; Franklin, B.; Karr, B.; Thorson, A.; Lynch, H.T. Colorectal carcinoma survival among hereditary nonpolyposis colorectal carcinoma family members. Cancer 1998, 83, 259–266. [Google Scholar] [CrossRef]
- Maxwell, G.L.; Risinger, J.I.; Alvarez, A.A.; Barrett, J.C.; Berchuck, A. Favorable survival associated with microsatellite instability in endometrioid endometrial cancers. Obstet. Gynecol. 2001, 97, 417–422. [Google Scholar] [CrossRef] [PubMed]
- Pinero, T.A.; Soukarieh, O.; Rolain, M.; Alvarez, K.; Lopez-Kostner, F.; Torrezan, G.T.; Carraro, D.M.; De Oliveira Nascimento, I.L.; Bomfim, T.F.; Machado-Lopes, T.M.B.; et al. MLH1 intronic variants mapping to + 5 position of splice donor sites lead to deleterious effects on RNA splicing. Fam. Cancer 2020. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family | Criteria # | Gene | Nucleotide Change ## | Protein Change |
|---|---|---|---|---|
| HNPCC056 | Ams | MLH1 | Deletion exon 1 b | p.0? |
| HNPCC008 | Ams | MLH1 | Deletion exon 1 b | p.0? |
| HNPCC006 | Ams | MLH1 | c.503dupA a | p.N168Kfs*4 |
| HNPCC002 | Ams | MLH1 | c.677 + 5G > A b | p.Q197Rfs*8/p.E53Ffs*8 |
| HNPCC029 | Ams | MLH1 | c.790 + 1G > A a | p.E227_S295del |
| HNPCC043 | Ams | MLH1 | c.794G > C c | p.R265P |
| HNPCC009 | Ams | MLH1 | c.901C > T a | p.Q301* |
| HNPCC080 | Ams | MLH1 | c.997_1000delAAGC c | p.K333Sfs*33 |
| HNPCC037 | Ams | MLH1 | c.1038 + 1G > T b | p.T347Ffs*14 |
| HNPCC062 | Ams | MLH1 | c.1559 − 2A > C c | p.L521Kfs*34 |
| HNPCC018 | Ams | MLH1 | Deletion exons 14 and 15 b | p.V520Gfs*7 |
| HNPCC001 | Ams | MLH1 | c.1731 + 3A > T a | p.S556Rfs*14 |
| HNPCC073 | Ams | MLH1 | c.2041G > A a | p.A681T |
| HNPCC082 | Ams | MLH1 | c.2041G > A a | p.A681T |
| HNPCC086 | Beth | MLH1 | c.2041G > A a | p.A681T |
| HNPCC011 | Beth | MLH1 | c.2041G > A a | p.A681T |
| HNPCC057 | Ams | MLH1 | c.2041G > A a | p.A681T |
| HNPCC019 | Beth | MLH1 | c.2092_2093delTC a | p.S698Rfs*5 |
| HNPCC004 | Ams | MLH1 | Deletion exon 19 b | p.S702_X757del |
| HNPCC010 | Ams | MLH1 | Deletion exon 19 b | p.S702_X757del |
| HNPCC047 | Ams | MSH2 | Deletion exon 2 b | p.A72Ffs*9 |
| HNPCC100 | Ams | MSH2 | c.388_389delCA c | p.Q130Vfs*2 |
| HNPCC118 | Ams | MSH2 | c.942 + 3A > T c | p.V265_Q314del |
| HNPCC027 | Ams | MSH2 | c.1215C > A a | p.Y405* |
| HNPCC111 | Beth | MSH2 | c.1861C > T c | p.R621* |
| HNPCC031 | Ams | MSH2 | c.2038C > T a | p.R680* |
| HNPCC075 | Ams | MSH2 | c.2131C > T c | p.R711* |
| HNPCC012 | Ams | MSH2 | c.2185_2192del7insCCCT a | p.M729_E731delinsP729_*730 |
| HNPCC088 | Ams | EPCAM-MSH2 | Deletion EPCAM and exons 1–6 MSH2 c | p.? |
| HNPCC094 | Ams | EPCAM | Deletion exons 6–9 c | p.? |
| HNPCC106 | US | PMS2 | c.903G > T d | p.K301N (Skips exon 8) |
| HNPCC084 | Beth | PMS2 | c.2016delG c | p.M672Ifs*15 |
| HNPCC093 | Ams | PMS2 | Deletion exon 14 c | p.A759Gfs*8 |
| HNPCC116 | US | PMS2 | Deletion exon 14 c | p.A759Gfs*8 |
| Clinical Information | Group 1 | Group 2 |
|---|---|---|
| Gender: n (%) | ||
| Female | 109 (51) | 37 (59) |
| Male | 104 (49) | 27 (42) |
| Total | 213 | 64 |
| Neoplasms: n (%) | ||
| CRC | 170/140 subjects | 88/61 subjects |
| Extracolonic cancer | 138/109 subjects | 35/23 subjects |
| Total | 308 | 123 |
| Neoplasm by gender **: n (%) | ||
| Female | 173 | 73 |
| CRC | 72 (41.6) | 43 (58.9) |
| Extracolonic cancer | 101 (58.4) | 30 (41.1) |
| Male | 135 | 50 |
| CRC | 98 (72.6) | 45 (90) |
| Extracolonic cancer | 37 (27.4) | 5 (10) |
| Age (years) at diagnosis: mean and range | ||
| Any first cancer | 45.1 (1–89) | 40.8 (18–84) |
| Female | 45.9 (1–89) | 39.5 (18–84) |
| Male | 44.2 (10–84) | 42.5 (27–65) |
| First CRC | 43.4 (18–89) | 41.2 (18–65) |
| Female | 43.1 (18–89) | 39.6 (18–63) |
| Male | 42.5 (21–73) | 43.2 (27–65) |
| First extracolonic cancer | 51.0 (1–88) | 43.3 (26–84) |
| Female | 50.8 (1–88) | 48.1 (26–84) |
| Male | 51.5 (10–84) | 55.0 (40–66) |
| Group 1 | Group 2 | |||||||
|---|---|---|---|---|---|---|---|---|
| Neoplasm | path_MLH1 n = 195 | path_MSH2 n = 99 | path_PMS2 n = 14 | Total n = 308 | path_MLH1 n = 77 | path_MSH2 n = 41 | path_PMS2 n = 5 | Total n = 123 |
| Colorectal: n (%) | 116 (59.5) | 47 (47.5) | 7 (50) | 170 | 57 (74) | 27 (66) | 4 (80) | 88 |
| Extracolonic: n (%) | 79 (40.5) | 52 (52.5) | 7 (50) | 138 | 20 (26) | 14 (34) | 1 (20) | 35 |
| Gynecological cancer | ||||||||
| Uterus | 21 (10.8) | 11 (11.1) | 1 (7.1) | 33 | 4 (5.2) | 2 (4.9) | 0 | 6 |
| Ovary | 2 (1.0) | 5 (5.1) | 1 (7.1) | 8 | 1 (1.3) | 4 (9.8) | 0 | 5 |
| Upper gastrointestinal cancer | ||||||||
| Stomach | 10 (5.1) | 7 (7.1) | 0 | 17 | 1 (1.3) | 0 | 0 | 1 |
| Pancreas | 6 (3.0) | 1 (1.0) | 1 (7.1) | 8 | 2 (2.6) | 0 | 0 | 2 |
| Liver | 3 (1.5) | 0 | 0 | 3 | 0 | 0 | 0 | 0 |
| Gallbladder | 1 (0.5) | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
| Small Intestine | 0 | 1 (1.0) | 0 | 1 | 0 | 1 (2.4) | 0 | 1 |
| Genitourinary tract cancer | ||||||||
| Kidney | 2 (1.0) | 6 (6.1) | 1 (7.1) | 9 | 1 (1.3) | 1 (2.4)) | 0 | 2 |
| Bladder | 0 | 1(1.0) | 0 | 1 | 0 | 1(2.4) | 0 | 1 |
| Ureter | 3(1.5) | 0 | 0 | 3 | 0 | 0 | 0 | 0 |
| Other cancers | ||||||||
| Breast | 12 (6.2) | 7 (7.1) | 1 (7.1) | 20 | 4 (5.2) | 2 (4.9) | 0 | 6 |
| Skin | 7 (3.6) | 6 (6.1) | 0 | 13 | 5 (6.5) | 3 (7.3) | 0 | 8 |
| Brain | 2 (1.0) | 3 (3.0) | 0 | 5 | 0 | 0 | 0 | 0 |
| Lung | 2 (1.0) | 2 (2.0) | 0 | 4 | 1 (1.3) | 0 | 0 | 1 |
| Prostate | 5 (2.6) | 0 | 0 | 5 | 0 | 0 | 0 | 0 |
| Soft tissue | 1 (0.5) | 0 | 0 | 1 | 1(1.3) | 0 | 0 | 1 |
| Bone | 1 (0.5) | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
| Leukemia | 1 (0.5) | 0 | 1 (7.1) | 2 | 0 | 0 | 0 | 0 |
| Head and Neck | 0 | 1 (1.0) | 0 | 1 | 0 | 0 | 0 | 0 |
| Thyroid | 0 | 1 (1.0) | 0 | 1 | 0 | 0 | 0 | 0 |
| Cervix | 0 | 0 | 1 (7.1) | 1 | 0 | 0 | 1 (20) | 1 |
| Cancer Type | Age, Year | All % | CI95 | Gender | path_MLH1 | path_MSH2 | path_PMS2 ** | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Female | CI95 | Male | CI95 | Female | CI95 | Male | CI95 | Female | CI95 | Male | CI95 | Both | CI95 | ||||
| Colorectal (n = 61) | 50 | 59.5 | 46.4–69.4 | 59.5 | 49.1–72.2 | 60.1 | 38.9–74.0 | 63.5 | 39.1–78.2 | 67.9 | 39.7–83.0 | 53.5 | 9.8–76.0 | 68.0 | 9.2–88.7 | 20.5 | 0–42.3 |
| 60 | 75.7 | 62.3–84.4 | 76.5 | 56.2–87.4 | 74.6 | 52.9–86.3 | 80.5 | 52.1–92.0 | 78.6 | 50.1–90.8 | 82.6 | 10.4–96.6 | 84.0 | 9.3–97.2 | 36.4 | 0–63.1 | |
| 70 | 88.2 | 73.7–94.7 | 89.5 | 66.6–96.7 | 86.5 | 59.3–95.5 | 87.0 | 56.7–96.1 | 89.3 | 45.7–97.9 | 100.0 | - | 100.0 | - | 68.2 | 0–92.8 | |
| Extracolonic (n = 22) | 50 | 18.5 | 8.6–27.3 | 31.3 | 14.4–44.9 | 2.7 | 0–7.8 | 23.2 | 4.6–38.1 | 0 | - | 64.1 | 12.8–85.2 | 10.0 | 0–26.8 | 0.0 | - |
| 60 | 28.0 | 14.8–39.2 | 39.6 | 19.7–54.5 | 13.5 | 0–27.2 | 36.6 | 10.2–55.2 | 15.4 | 0–32.9 | 64.1 | 12.8–85.2 | 10.0 | 0–26.8 | 0.0 | - | |
| 70 | 50.0 | 27.9–65.4 | 71.7 | 35.0–87.7 | 24.3 | 0–44.7 | 60.4 | 15.7–81.4 | 15.4 | 0–32.9 | 100 | - | 55.0 | 0–88.9 | 33.3 | 0–70.0 | |
| Gynecological (n = 10) | 50 | 22.1 | 7.1–34.7 | 22.1 | 7.1–34.7 | - | - | 12.1 | 0–24.2 | - | - | 60.0 | 5.7–83.0 | - | - | 0.0 | - |
| 60 | 30.0 | 11.8–44.4 | 30.0 | 11.8–44.4 | - | - | 24.2 | 2.1–41.2 | - | - | 60.0 | 5.7–83.0 | - | - | 0.0 | - | |
| 70 | 30.0 | 11.8–44.4 | 30.0 | 11.8–44.4 | - | - | 24.2 | 2.1–41.2 | - | - | 60.0 | 5.7–83.0 | - | - | 0.0 | - | |
| Skin (n = 5) | 50 | 2.4 | 0–5.6 | 2.1 | 0–6.0 | 2.7 | 0–7.8 | 0.0 | - | 0.0 | - | 7.7 | 0–21.1 | 10.0 | 0–26.8 | 0.0 | - |
| 60 | 5.0 | 0–10.7 | 2.1 | 0–6.0 | 8.1 | 0–18.8 | 0.0 | - | 7.7 | 0–21.1 | 7.7 | 0–21.1 | 10.0 | 0–26.8 | 0.0 | - | |
| 70 | 14.5 | 0–27.0 | 11.0 | 0–26.5 | 18.3 | 0–37.2 | 12.5 | 0–32.7 | 7.7 | 0–21.1 | 7.7 | 0–21.1 | 55.0 | 0–88.9 | 0.0 | - | |
| Upper gastrointestinal tract (n = 3) | 50 | 1.7 | 0–5.0 | - | - | - | - | - | - | - | - | - | - | - | - | - | - |
| 60 | 4.3 | 0–10.1 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | |
| 70 | 4.3 | 0–10.1 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | |
| Breast (n = 3) | 50 | 5.2 | 0–12.1 | 5.2 | 0–12.1 | - | - | 3.4 | 0–9.9 | - | - | 12.5 | 0–32.7 | - | - | 0.0 | - |
| 60 | 9.0 | 0–18.5 | 9.0 | 0–18.5 | - | - | 9.5 | 0–21.6 | - | - | 12.5 | 0–32.7 | - | - | 0.0 | - | |
| 70 | 9.0 | 0–18.5 | 9.0 | 0–18.5 | - | - | 9.5 | 0–21.6 | - | 12.5 | 0–32.7 | - | - | 0.0 | - | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Álvarez, K.; Orellana, P.; De la Fuente, M.; Canales, T.; Pinto, E.; Heine, C.; Solar, B.; Hurtado, C.; Møller, P.; Kronberg, U.; et al. Spectrum and Frequency of Tumors, Cancer Risk and Survival in Chilean Families with Lynch Syndrome: Experience of the Implementation of a Registry. J. Clin. Med. 2020, 9, 1861. https://doi.org/10.3390/jcm9061861
Álvarez K, Orellana P, De la Fuente M, Canales T, Pinto E, Heine C, Solar B, Hurtado C, Møller P, Kronberg U, et al. Spectrum and Frequency of Tumors, Cancer Risk and Survival in Chilean Families with Lynch Syndrome: Experience of the Implementation of a Registry. Journal of Clinical Medicine. 2020; 9(6):1861. https://doi.org/10.3390/jcm9061861
Chicago/Turabian StyleÁlvarez, Karin, Paulina Orellana, Marjorie De la Fuente, Tamara Canales, Eliana Pinto, Claudio Heine, Benjamín Solar, Claudia Hurtado, Pål Møller, Udo Kronberg, and et al. 2020. "Spectrum and Frequency of Tumors, Cancer Risk and Survival in Chilean Families with Lynch Syndrome: Experience of the Implementation of a Registry" Journal of Clinical Medicine 9, no. 6: 1861. https://doi.org/10.3390/jcm9061861
APA StyleÁlvarez, K., Orellana, P., De la Fuente, M., Canales, T., Pinto, E., Heine, C., Solar, B., Hurtado, C., Møller, P., Kronberg, U., Zarate, A. J., Dominguez-Valentin, M., & López-Köstner, F. (2020). Spectrum and Frequency of Tumors, Cancer Risk and Survival in Chilean Families with Lynch Syndrome: Experience of the Implementation of a Registry. Journal of Clinical Medicine, 9(6), 1861. https://doi.org/10.3390/jcm9061861