Urinary Dimethylamine (DMA) and Its Precursor Asymmetric Dimethylarginine (ADMA) in Clinical Medicine, in the Context of Nitric Oxide (NO) and Beyond


Abstract
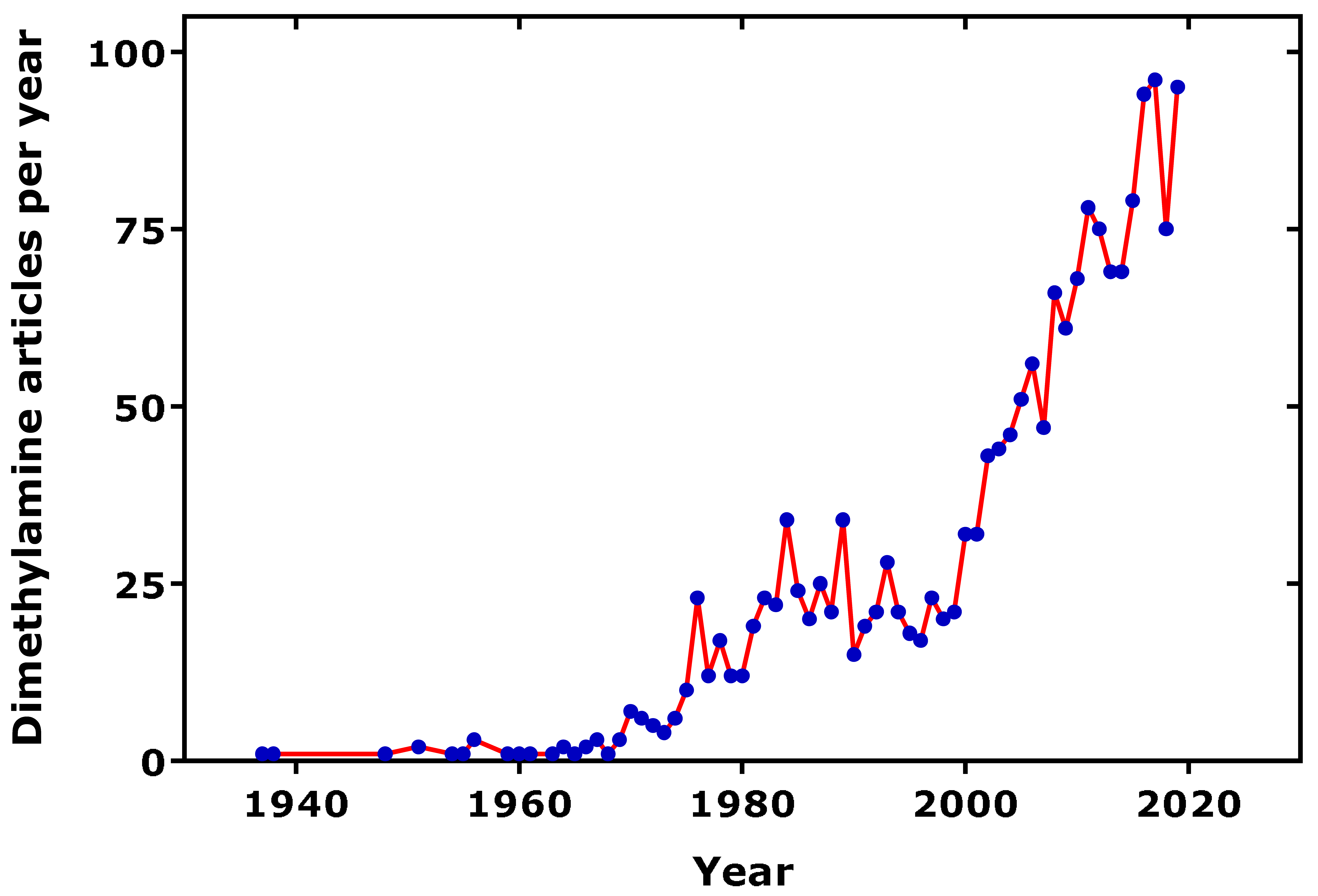
1. Introduction-A Brief Historical Retrospect and Aim of the Review
2. Post-translational Modification and Food: Two Major Origins of Urinary DMA
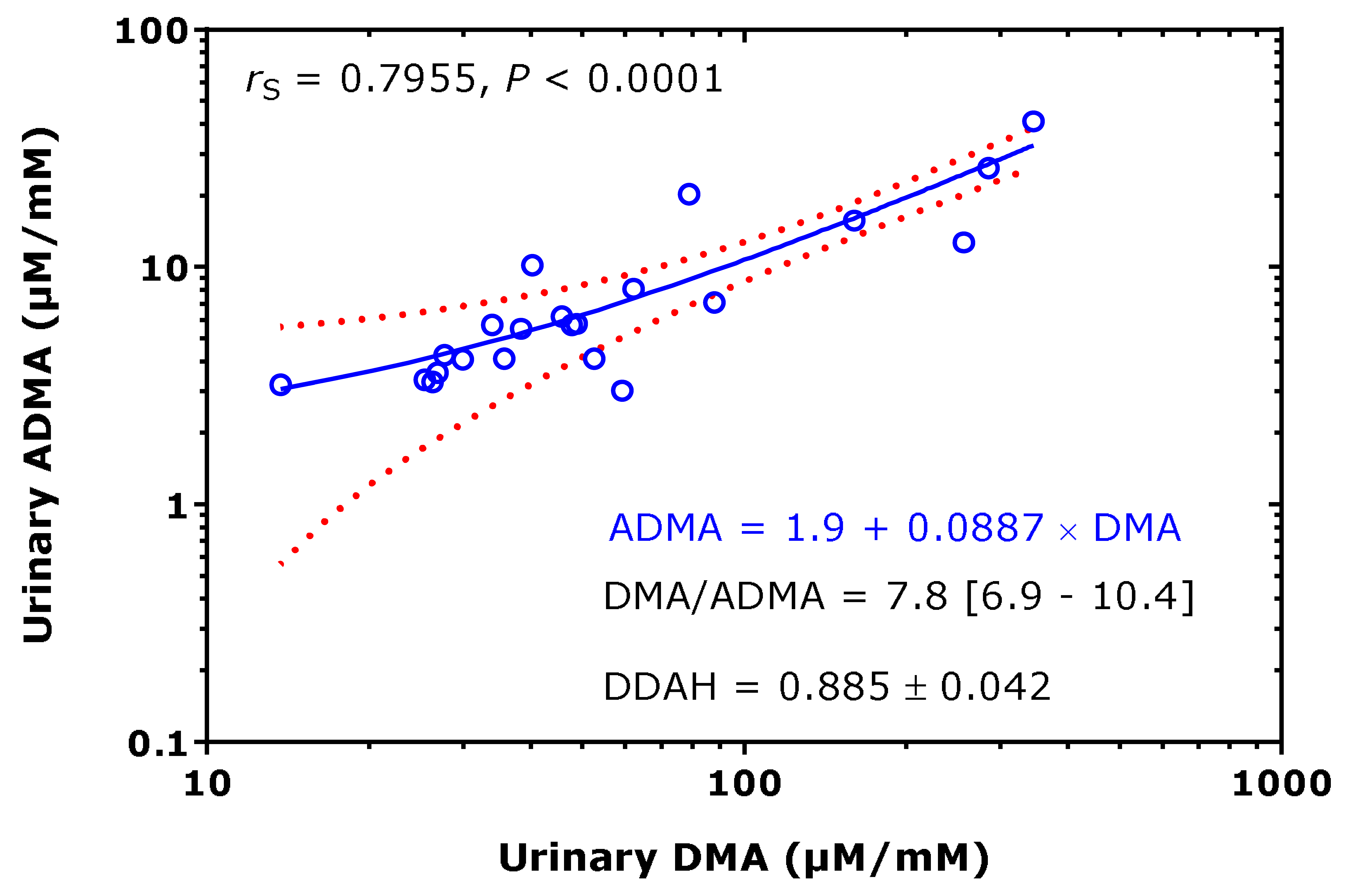
2.1. Asymmetric Protein-Arginine Dimethylation as the Major Contributor to Urinary DMA
2.2. Food as Considerable Contributor to Urinary DMA
3. Urinary DMA and ADMA in Children, Adolescents and Adults in Health and Disease
4. Statins, Metformin, L-Arginine, Catecholamines and the PRMT-DDAH-NOS Axis inAtherosclerosis and Beyond
4.1. Effect of Statins on ADMA and NO Biosynthesis
4.2. Effect of Metformin on ADMA Biosynthesis
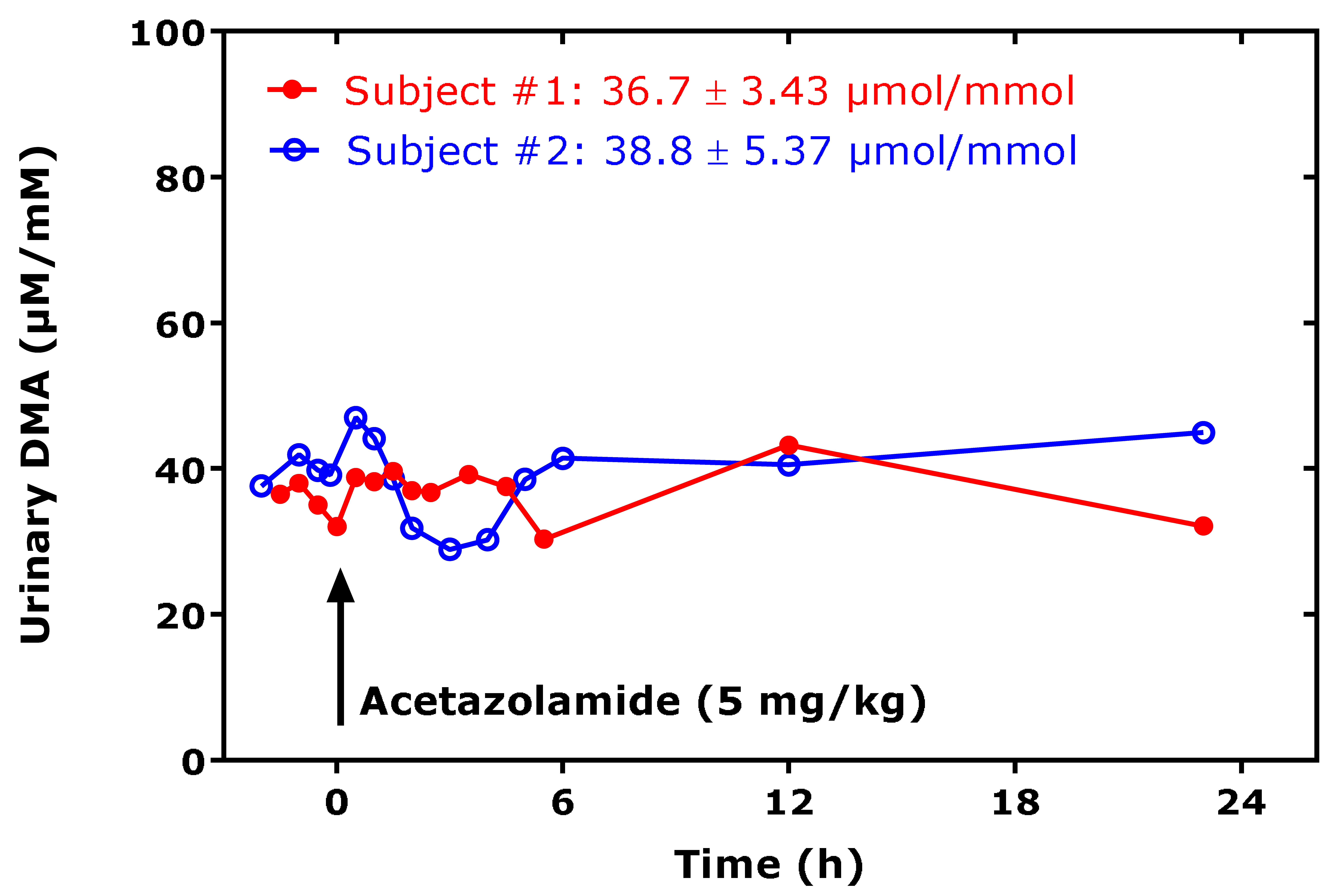
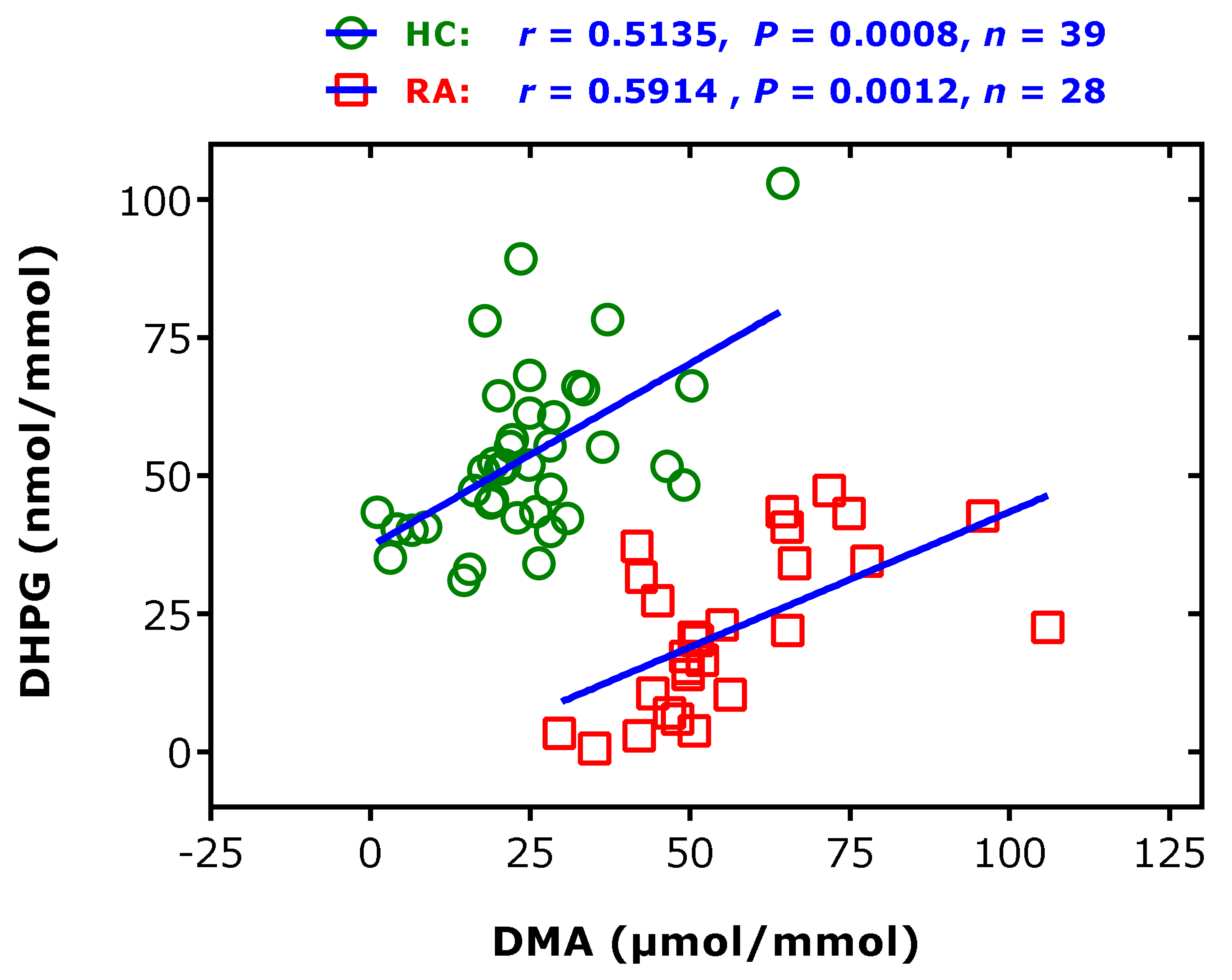
4.3. Inflammation, Catecholamines and DMA Excretion
5. DDAH Activity and Oxidative Stress
6. DDAH as a Pharmacological Target
7. Conclusions and Perspectives
Conflicts of Interest
References
- Löffler, H. Die flüchtigen amine des menschlichen harns. Hoppe Seylers Z. Physiol. Chem. 1935, 232, 259–262. [Google Scholar]
- Dowden, H.C. The determination of small amounts of dimethylamine in biological fluids. Biochem. J. 1938, 32, 455–459. [Google Scholar] [CrossRef]
- Magge, P.N.; Barnes, J.M. The experimental production of tumours in the rat by dimethylnitrosamine (N-nitroso dimethylamine). Acta Unio Int. Contra Cancrum. 1959, 15, 187–190. [Google Scholar]
- Mirvish, S.S. Kinetics of dimethylamine nitrosation in relation to nitrosamine carcinogenesis. J. Natl. Cancer Inst. 1970, 44, 633–639. [Google Scholar] [PubMed]
- Tannenbaum, S.R.; Wishnok, J.S.; Leaf, C.D. Inhibition of nitrosamine formation by ascorbic acid. Am. J. Clin. Nutr. 1991, 53, 247S–250S. [Google Scholar] [CrossRef] [PubMed]
- Blanc, R.S.; Richard, S. Arginine Methylation: The Coming of Age. Mol. Cell 2017, 65, 8–24. [Google Scholar] [CrossRef]
- Peng, C.; Wong, C.C. The story of protein arginine methylation: Characterization, regulation, and function. Expert Rev. Proteom. 2017, 14, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Tsikas, D. Does the inhibitory action of asymmetric dimethylarginine (ADMA) on the endothelial nitric oxide synthase activity explain its importance in the cardiovascular system? The ADMA paradox. J. Controv. Biomed. Res. 2017, 3, 16–22. [Google Scholar] [CrossRef][Green Version]
- Tsikas, D.; Bollenbach, A.; Hanff, E.; Kayacelebi, A.A. Asymmetric dimethylarginine (ADMA), symmetric dimethylarginine (SDMA) and homoarginine (hArg): The ADMA, SDMA and hArg paradoxes. Cardiovasc. Diabetol. 2018, 17, 1. [Google Scholar] [CrossRef]
- Tousoulis, D.; Kampoli, A.M.; Tentolouris, C.; Papageorgiou, N.; Stefanadis, C. The role of nitric oxide on endothelial function. Curr. Vasc. Pharmacol. 2012, 10, 4–18. [Google Scholar] [CrossRef]
- Tain, Y.L.; Hsu, C.N. Toxic Dimethylarginines: Asymmetric Dimethylarginine (ADMA) and Symmetric Dimethylarginine (SDMA). Toxins (Basel) 2017, 9, 92. [Google Scholar] [CrossRef] [PubMed]
- Busch, M.; Fleck, C.; Wolf, G.; Stein, G. Asymmetrical (ADMA) and symmetrical dimethylarginine (SDMA) as potential risk factors for cardiovascular and renal outcome in chronic kidney disease—Possible candidates for paradoxical epidemiology? Amino Acids 2006, 30, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, T.; Kimoto, M.; Sasaoka, K. Occurrence of a new enzyme catalyzing the direct conversion of NG,NG-dimethyl-l-arginine to L-citrulline in rats. Biochem. Biophys. Res. Commun. 1987, 148, 671–677. [Google Scholar] [CrossRef]
- Kimoto, M.; Tsuji, H.; Ogawa, T. N(G), N G)-Dimethyl-l-arginine, a dominant precursor of endogenous dimethylamine in rats. Amino Acids 1994, 6, 273–282. [Google Scholar] [CrossRef]
- Zhang, A.Q.; Mitchell, S.C.; Barrett, T.; Ayesh, R.; Smith, R.L. Fate of dimethylamine in man. Xenobiotica 1994, 24, 379–387. [Google Scholar] [CrossRef]
- Zhang, A.Q.; Mitchell, S.C.; Smith, R.L. Dimethylamine in human urine. Clin. Chim. Acta 1995, 233, 81–88. [Google Scholar] [CrossRef]
- Mitchell, S.C.; Zhang, A.Q.; Smith, R.L. Dimethylamine and diet. Food Chem. Toxicol. 2008, 46, 1734–17348. [Google Scholar] [CrossRef]
- Mitchell, S.C.; Zhang, A.Q.; Noblet, J.M.; Gillespie, S.; Jones, N.; Smith, R.L. Metabolic disposition of [14C]-trimethylamine N-oxide in rat: Variation with dose and route of administration. Xenobiotica 1997, 27, 1187–1197. [Google Scholar] [CrossRef]
- Kelly, R.H.; Yancey, P.H. High contents of trimethylamine oxide correlating with depth in deep-sea teleost fishes, skates, and decapod crustaceans. Biol. Bull. 1999, 196, 18–25. [Google Scholar] [CrossRef]
- Yancey, P.H.; Blake, W.R.; Conley, J. Unusual organic osmolytes in deep-sea animals: Adaptations to hydrostatic pressure and other perturbants. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 2002, 133, 667–676. [Google Scholar] [CrossRef]
- Samerotte, A.L.; Drazen, J.C.; Brand, G.L.; Seibel, B.A.; Yancey, P.H. Correlation of trimethylamine oxide and habitat depth within and among species of teleost fish: An analysis of causation. Physiol. Biochem. Zool. 2007, 80, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Laxson, C.J.; Condon, N.E.; Drazen, J.C.; Yancey, P.H. Decreasing urea:trimethylamine N-oxide ratios with depth in chondrichthyes: A physiological depth limit? Physiol. Biochem. Zool. 2011, 84, 494–505. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.; Thatje, S. Explaining bathymetric diversity patterns in marine benthic invertebrates and demersal fishes: Physiological contributions to adaptation of life at depth. Biol. Rev. Camb. Philos. Soc. 2014, 89, 406–426. [Google Scholar] [CrossRef] [PubMed]
- Yancey, P.H. Cellular responses in marine animals to hydrostatic pressure. J. Exp. Zool. A Ecol. Integr. Physiol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Asatoor, A.M.; Simenhoff, M.L. The origin of urinary dimethylamine. Biochim. Biophys. Act. 1965, 111, 384–392. [Google Scholar] [CrossRef]
- Zeisel, S.H.; DaCosta, K.A.; Fox, J.G. Endogenous formation of dimethylamine. Biochem. J. 1985, 232, 403–408. [Google Scholar] [CrossRef] [PubMed]
- Teerlink, T.; Hennekes, M.W.; Mulder, C.; Brulez, H.F. Determination of dimethylamine in biological samples by high-performance liquid chromatography. J. Chromatogr. B Biomed. Sci. Appl. 1997, 691, 269–276. [Google Scholar] [CrossRef]
- Tsikas, D.; Thum, T.; Becker, T.; Pham, V.V.; Chobanyan, K.; Mitschke, A.; Beckmann, B.; Gutzki, F.M.; Bauersachs, J.; Stichtenoth, D.O. Accurate quantification of dimethylamine (DMA) in human urine by gas chromatography-mass spectrometry as pentafluorobenzamide derivative: Evaluation of the relationship between DMA and its precursor asymmetric dimethylarginine (ADMA) in health and disease. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2007, 851, 229–239. [Google Scholar] [CrossRef]
- Tsikas, D.; Schubert, B.; Gutzki, F.M.; Sandmann, J.; Frölich, J.C. Quantitative determination of circulating and urinary asymmetric dimethylarginine (ADMA) in humans by gas chromatography-tandem mass spectrometry as methyl ester tri(N-pentafluoropropionyl) derivative. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2003, 798, 87–99. [Google Scholar] [CrossRef]
- Tsikas, D.; Wolf, A.; Mitschke, A.; Gutzki, F.M.; Will, W.; Bader, M. GC-MS determination of creatinine in human biological fluids as pentafluorobenzyl derivative in clinical studies and biomonitoring: Inter-laboratory comparison in urine with Jaffe, HPLC and enzymatic assays. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2010, 878, 2582–25929. [Google Scholar] [CrossRef]
- Hsu, C.N.; Chang-Chien, G.P.; Lin, S.; Hou, C.Y.; Ku, P.C.; Tain, Y.L. Association of trimethylamine, trimethylamine N-oxide, and dimethylamine with cardiovascular risk in children with chronic kidney disease. J. Clin. Med. 2020, 9, 336. [Google Scholar] [CrossRef] [PubMed]
- Lin, I.C.; Hsu, C.N.; Lo, M.H.; Chien, S.J.; Tain, Y.L. Low urinary citrulline/arginine ratio associated with blood pressure abnormalities and arterial stiffness in childhood chronic kidney disease. J. Am. Soc. Hypertens. 2016, 10, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Kuo, H.C.; Hsu, C.N.; Huang, C.F.; Lo, M.H.; Chien, S.J.; Tain, Y.L. Urinary arginine methylation index associated with ambulatory blood pressure abnormalities in children with chronic kidney disease. J. Am. Soc. Hypertens. 2012, 6, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Jansen, K.; Hanusch, B.; Pross, S.; Hanff, E.; Drabert, K.; Bollenbach, A.; Dugave, I.; Carmann, C.; Siefen, R.G.; Emons, B.; et al. Enhanced Nitric Oxide (NO) and Decreased ADMA Synthesis in Pediatric ADHD and Selective Potentiation of NO Synthesis by Methylphenidate. J. Clin. Med. 2020, 9, 175. [Google Scholar] [CrossRef] [PubMed]
- Bollenbach, A.; Schutte, A.E.; Kruger, R.; Tsikas, D. An Ethnic Comparison of Arginine Dimethylation and Cardiometabolic Factors in Healthy Black and White Youth: The ASOS and African-PREDICT Studies. J. Clin. Med. 2020, 9, 844. [Google Scholar] [CrossRef] [PubMed]
- Bollenbach, A.; Huneau, J.F.; Mariotti, F.; Tsikas, D. Asymmetric and Symmetric Protein Arginine Dimethylation: Concept and Postprandial Effects of High-Fat Protein Meals in Healthy Overweight Men. Nutrients 2019, 11, 1463. [Google Scholar] [CrossRef] [PubMed]
- Hanff, E.; Hafner, P.; Bollenbach, A.; Bonati, U.; Kayacelebi, A.A.; Fischer, D.; Tsikas, D. Effects of single and combined metformin and L-citrulline supplementation on L-arginine-related pathways in Becker muscular dystrophy patients: Possible biochemical and clinical implications. Amino Acids 2018, 50, 1391–1406. [Google Scholar] [CrossRef] [PubMed]
- Buck, A.; Kayacelebi, A.A.; Chobanyan-Jürgens, K.; Illsinger, S.; Bohnhorst, B.; Beckmann, B.; Hanff, E.; Das, A.M.; Tsikas, D.; Lücke, T. Comprehensive analysis of the L-arginine/L-homoarginine/nitric oxide pathway in preterm neonates: Potential roles for homoarginine and asymmetric dimethylarginine in foetal growth. Amino Acids 2017, 49, 783–794. [Google Scholar] [CrossRef]
- Schneider, J.Y.; Rothmann, S.; Schröder, F.; Langen, J.; Lücke, T.; Mariotti, F.; Huneau, J.F.; Frölich, J.C.; Tsikas, D. Effects of chronic oral L-arginine administration on the L-arginine/NO pathway in patients with peripheral arterial occlusive disease or coronary artery disease: L-Arginine prevents renal loss of nitrite, the major NO reservoir. Amino Acids 2015, 47, 1961–1974. [Google Scholar] [CrossRef]
- Hörster, I.; Weigt-Usinger, K.; Carmann, C.; Chobanyan-Jürgens, K.; Köhler, C.; Schara, U.; Kayacelebi, A.A.; Beckmann, B.; Tsikas, D.; Lücke, T. The L-arginine/NO pathway and homoarginine are altered in Duchenne muscular dystrophy and improved by glucocorticoids. Amino Acids 2015, 47, 1853–1863. [Google Scholar] [CrossRef] [PubMed]
- Carmann, C.; Lilienthal, E.; Weigt-Usinger, K.; Schmidt-Choudhury, A.; Hörster, I.; Kayacelebi, A.A.; Beckmann, B.; Chobanyan-Jürgens, K.; Tsikas, D.; Lücke, T. The L-arginine/NO pathway, homoarginine, and nitrite-dependent renal carbonic anhydrase activity in young people with type 1 diabetes mellitus. Amino Acids 2015, 47, 1865–1874. [Google Scholar] [CrossRef] [PubMed]
- Kanzelmeyer, N.K.; Pape, L.; Chobanyan-Jürgens, K.; Tsikas, D.; Hartmann, H.; Fuchs, A.J.; Vaske, B.; Das, A.M.; Haubitz, M.; Jordan, J.; et al. L-arginine/NO pathway is altered in children with haemolytic-uraemic syndrome (HUS). Oxid. Med. Cell. Longev. 2014, 2014, 203512. [Google Scholar] [CrossRef] [PubMed]
- Kanzelmeyer, N.; Tsikas, D.; Chobanyan-Jürgens, K.; Beckmann, B.; Vaske, B.; Illsinger, S.; Das, A.M.; Lücke, T. Asymmetric dimethylarginine in children with homocystinuria or phenylketonuria. Amino Acids 2012, 42, 1765–1772. [Google Scholar] [CrossRef] [PubMed]
- Chobanyan-Jürgens, K.; Fuchs, A.J.; Tsikas, D.; Kanzelmeyer, N.; Das, A.M.; Illsinger, S.; Vaske, B.; Jordan, J.; Lücke, T. Increased asymmetric dimethylarginine (ADMA) dimethylaminohydrolase (DDAH) activity in childhood hypercholesterolemia type II. Amino Acids 2012, 43, 805–811. [Google Scholar] [CrossRef] [PubMed]
- Wolf, C.; Lorenzen, J.M.; Stein, S.; Tsikas, D.; Störk, S.; Weidemann, F.; Ertl, G.; Anker, S.D.; Bauersachs, J.; Thum, T. Urinary asymmetric dimethylarginine (ADMA) is a predictor of mortality risk in patients with coronary artery disease. Int. J. Cardiol. 2012, 156, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Chobanyan-Jürgens, K.; Pham, V.V.; Stichtenoth, D.O.; Tsikas, D. Elevated dimethylarginine dimethylaminohydrolase (DDAH) activity in rheumatoid arthritis and spondyloarthritis. Nitric Oxide. 2011, 25, 436–438. [Google Scholar] [CrossRef] [PubMed]
- Becker , T.; Mevius, I.; de Vries, D.K.; Schaapherder, A.F.; zu Vilsendorf, A.M.; Klempnauer, J.; Frölich, J.C.; Tsikas, D. The L-arginine/NO pathway in end-stage liver disease and during orthotopic liver and kidney transplantation: Biological and analytical ramifications. Nitric Oxide. 2009, 20, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Lücke, T.; Kanzelmeyer, N.; Chobanyan, K.; Tsikas, D.; Franke, D.; Kemper, M.J.; Ehrich, J.H.; Das, A.M. Elevated asymmetric dimethylarginine (ADMA) and inverse correlation between circulating ADMA and glomerular filtration rate in children with sporadic focal segmental glomerulosclerosis (FSGS). Nephrol. Dial. Transplant. 2008, 23, 734–740. [Google Scholar] [CrossRef][Green Version]
- Lücke, T.; Tsikas, D.; Kanzelmeyer, N.; Vaske, B.; Das, A.M. Elevated plasma concentrations of the endogenous nitric oxide synthase inhibitor asymmetric dimethylarginine in citrullinemia. Metabolism 2006, 55, 1599–1603. [Google Scholar] [CrossRef]
- Lücke, T.; Tsikas, D.; Kanzelmeyer, N.K.; Boerkoel, C.F.; Clewing, J.M.; Vaske, B.; Ehrich, J.H.; Das, A.M. Vaso-occlusion in Schimke-immuno-osseous dysplasia: Is the NO pathway involved? Horm. Metab. Res. 2006, 38, 678–682. [Google Scholar] [CrossRef]
- Achan, V.; Broadhead, M.; Malaki, M.; Whitley, G.; Leiper, J.; MacAllister, R.; Vallance, P. Asymmetric dimethylarginine causes hypertension and cardiac dysfunction in humans and is actively metabolized by dimethylarginine dimethylaminohydrolase. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1455–1459. [Google Scholar] [CrossRef] [PubMed]
- Bassareo, P.P.; Fanos, V.; Puddu, M.; Flore, G.; Mercuro, G. Advanced Intrauterine Growth Restriction Is Associated with Reduced Excretion of Asymmetric Dimethylarginine. Early Hum. Dev. 2014, 90, 173–176. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.M.; Ding, Y.A.; Leu, H.B.; Yin, W.H.; Sheu, W.H.; Chu, K.M. Effect of rosuvastatin on plasma levels of asymmetric dimethylarginine in patients with hypercholesterolemia. Am. J. Cardiol. 2004, 94, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Thum, T.; Tsikas, D.; Stein, S.; Schultheiss, M.; Eigenthaler, M.; Anker, S.D.; Poole-Wilson, P.A.; Ertl, G.; Bauersachs, J. Suppression of endothelial progenitor cells in human coronary artery disease by the endogenous nitric oxide synthase inhibitor asymmetric dimethylarginine. J. Am. Coll. Cardiol. 2005, 46, 1693–1701. [Google Scholar] [CrossRef]
- Zhou, R.; Ma, P.; Xiong, A.; Xu, Y.; Wang, Y.; Xu, Q. Protective effects of low-dose rosuvastatin on isoproterenol-induced chronic heart failure in rats by regulation of DDAH-ADMA-NO pathway. Cardiovasc. Ther. 2017, 35. [Google Scholar] [CrossRef] [PubMed]
- Günes, D.N.; Kayacelebi, A.A.; Hanff, E.; Lundgren, J.; Redfors, B.; Tsikas, D. Metabolism and distribution of pharmacological homoarginine in plasma and main organs of the anesthetized rat. Amino Acids 2017, 49, 2033–2044. [Google Scholar] [CrossRef]
- Tsikas, D.; Kayacelebi, A.A. Do homoarginine and asymmetric dimethylarginine act antagonistically in the cardiovascular system? Circ. J. 2014, 8, 2094–2095. [Google Scholar] [CrossRef] [PubMed]
- Cortese, F.; Gesualdo, M.; Cortese, A.; Carbonara, S.; Devito, F.; Zito, A.; Ricci, G.; Scicchitano, P.; Ciccone, M.M. Rosuvastatin: Beyond the cholesterol-lowering effect. Pharmacol. Res. 2016, 107, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.H.; Neil, C.J.; Sverdlov, A.L.; Ngo, D.T.; Chan, W.P.; Heresztyn, T.; Chirkov, Y.Y.; Tsikas, D.; Frenneaux, M.P.; Horowitz, J.D. Enhanced NO signaling in patients with Takotsubo cardiomyopathy: Short-term pain, long-term gain? Cardiovasc. Drugs Ther. 2013, 27, 541–547. [Google Scholar] [CrossRef]
- Cheung, K.W.K.; Hsueh, C.H.; Zhao, P.; Meyer, T.W.; Zhang, L.; Huang, S.M.; Giacomini, K.M. The Effect of Uremic Solutes on the Organic Cation Transporter 2. J. Pharm. Sci. 2017, 106, 2551–2557. [Google Scholar] [CrossRef]
- Pham, V.V.; Stichtenoth, D.O.; Tsikas, D. Nitrite correlates with 3-nitrotyrosine but not with the F(2)-isoprostane 15(S)-8-iso-PGF(2α) in urine of rheumatic patients. Nitric Oxide 2009, 21, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Zoerner, A.A.; Heusser, K.; Gutzki, F.M.; Mitschke, A.; Tank, J.; Stichtenoth, D.O.; Jordan, J.; Tsikas, D. Unique pentafluorobenzylation and collision-induced dissociation for specific and accurate GC-MS/MS quantification of the catecholamine metabolite 3,4-dihydroxyphenylglycol (DHPG) in human urine. J. Chromatogr. B Analyt. Technol. Biomed. Life. Sci. 2011, 879, 1444–1456. [Google Scholar] [CrossRef] [PubMed]
- Kielbasa, W.; Pan, A.; Pereira, A. A pharmacokinetic/pharmacodynamic investigation: Assessment of edivoxetine and atomoxetine on systemic and central 3,4-dihydroxyphenylglycol, a biochemical marker for norepinephrine transporter inhibition. Eur. Neuropsychopharmacol. 2015, 25, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Bieck, P.R.; Leibowitz, M.; Lachno, D.R.; Ledent, E.; Padich, R.; Jhee, S. Dihydroxyphenylglycol as a Biomarker of Norepinephrine Transporter Inhibition by Atomoxetine: Human Model to Assess Central and Peripheral Effects of Dosing. J. Clin. Psychopharmacol. 2016, 36, 675–683. [Google Scholar] [CrossRef] [PubMed]
- Mallamaci, F.; Tripepi, G.; Maas, R.; Malatino, L.; Böger, R.; Zoccali, C. Analysis of the relationship between norepinephrine and asymmetric dimethyl arginine levels among patients with end-stage renal disease. J. Am. Soc. Nephrol. 2004, 15, 435–441. [Google Scholar] [CrossRef]
- Augustyniak, R.A.; Victor, R.G.; Morgan, D.A.; Zhang, W. L-NAME- and ADMA-induced sympathetic neural activation in conscious rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2006, 290, R726–R732. [Google Scholar] [CrossRef]
- Gamboa, A.; Shibao, C.; Diedrich, A.; Paranjape, S.Y.; Farley, G.; Christman, B.; Raj, S.R.; Robertson, D.; Biaggioni, I. Excessive nitric oxide function and blood pressure regulation in patients with autonomic failure. Hypertension 2008, 51, 1531–1536. [Google Scholar] [CrossRef]
- Gamboa, A.; Shibao, C.; Diedrich, A.; Choi, L.; Pohar, B.; Jordan, J.; Paranjape, S.; Farley, G.; Biaggioni, I. Contribution of endothelial nitric oxide to blood pressure in humans. Hypertension 2007, 49, 170–177. [Google Scholar] [CrossRef]
- Goldstein, D.S.; Polinsky, R.J.; Garty, M.; Robertson, D.; Brown, R.T.; Biaggioni, I.; Stull, R.; Kopin, I.J. Patterns of plasma levels of catechols in neurogenic orthostatic hypotension. Ann. Neurol. 1989, 26, 558–563. [Google Scholar] [CrossRef]
- Sharabi, Y.; Imrich, R.; Holmes, C.; Pechnik, S.; Goldstein, D.S. Generalized and neurotransmitter-selective noradrenergic denervation in Parkinson’s disease with orthostatic hypotension. Mov. Disord. 2008, 23, 1725–1732. [Google Scholar] [CrossRef]
- Leiper, J.; Murray-Rust, J.; McDonald, N.; Vallance, P. S-Nitrosylation of dimethylarginine dimethylaminohydrolase regulates enzyme activity: Further interactions between nitric oxide synthase and dimethylarginine dimethylaminohydrolase. Proc. Natl. Acad. Sci. USA 2002, 99, 13527–13532. [Google Scholar] [CrossRef] [PubMed]
- Grosse, G.M.; Schwedhelm, E.; Worthmann, H.; Choe, C. Arginine derivatives in cerebrovascular diseases: Mechanisms and clinical implications. Int. J. Mol. Sci. 2020, 21, 1798. [Google Scholar] [CrossRef] [PubMed]
- Chobanyan, K.; Thum, T.; Suchy, M.T.; Zhu, B.; Mitschke, A.; Gutzki, F.M.; Beckmann, B.; Stichtenoth, D.O.; Tsikas, D. GC-MS assay for hepatic DDAH activity in diabetic and non-diabetic rats by measuring dimethylamine (DMA) formed from asymmetric dimethylarginine (ADMA): Evaluation of the importance of S-nitrosothiols as inhibitors of DDAH activity in vitro and in vivo in humans. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2007, 858, 32–41. [Google Scholar] [PubMed]
- Schneider, J.Y.; Pham, V.V.; Frölich, J.C.; Tsikas, D. DDAH activity is not associated with oxidative stress in elderly patients with peripheral arterial occlusive disease. Exp. Gerontol. 2014, 55, 159–160. [Google Scholar] [CrossRef]
- Tsikas, D.; Rothmann, S.; Schneider, J.Y.; Suchy, M.T.; Trettin, A.; Modun, D.; Stuke, N.; Maassen, N.; Frölich, J.C. Development, validation and biomedical applications of stable-isotope dilution GC-MS and GC-MS/MS techniques for circulating malondialdehyde (MDA) after pentafluorobenzyl bromide derivatization: MDA as a biomarker of oxidative stress and its relation to 15(S)-8-iso-prostaglandin F2α and nitric oxide (NO). J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2016, 1019, 95–111. [Google Scholar]
- Hackman, A.; Abe, Y.; Insull, W.; Pownall, H.; Smith, L.; Dunn, K.; Gotto, A.M., Jr.; Ballantyne, C.M. Levels of soluble cell adhesion molecules in patients with dyslipidemia. Circulation 1996, 93, 1334–1338. [Google Scholar] [CrossRef]
- Leiper, J.; Nandi, M. The therapeutic potential of targeting endogenous inhibitors of nitric oxide synthesis. Nat. Rev. Drug Discov. 2011, 10, 277–291. [Google Scholar] [CrossRef]
- Lambden, S.; Tomlinson, J.; Piper, S.; Gordon, A.C.; Leiper, J. Evidence for a protective role for the rs805305 single nucleotide polymorphism of dimethylarginine dimethylaminohydrolase 2 (DDAH2) in septic shock through the regulation of DDAH activity. Crit. Care 2018, 22, 336. [Google Scholar] [CrossRef]
- Zelzer, S.; Enko, D.; Pilz, S.; Tomaschitz, A.; März, W.; Meinitzer, A. Cardiovascular Risk Patients: Cross-sectional Findings from the Ludwigshafen Risk and Cardiovascular Health (LURIC) Study. Myeloperoxidase, asymmetric dimethyl-arginine and the renin-angiotensin-aldosterone-system in cardiovascular risk patients: Cross-sectional findings from the Ludwigshafen Risk and Cardiovascular Health (LURIC) study. Clin. Biochem. 2017, 50, 739–745. [Google Scholar]
- Lin, C.; Li, H.; Liu, J.; Hu, Q.; Zhang, S.; Zhang, N.; Liu, L.; Dai, Y.; Cao, D.; Li, X.; et al. Arginine Hypomethylation-Mediated Proteasomal Degradation of Histone H4-an Early Biomarker of Cellular Senescence. Cell Death Differ. 2020. [Google Scholar] [CrossRef]
- Cuyàs, E.; Verdura, S.; Llorach-Pares, L.; Fernández-Arroyo, S.; Luciano-Mateo, F.; Cabré, N.; Stursa, J.; Werner, L.; Martin-Castillo, B.; Viollet, B.; et al. Metformin directly targets the H3K27me3 demethylase KDM6A/UTX. Aging Cell 2018, 17, e12772. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Condition | DMA (µM/mM) | ADMA (µM/mM) | DMA/ADMA | Relative DDAH Activity | Ref. |
|---|---|---|---|---|---|
| Pediatric CKD (n = 115) | stage G1: 0.57 stage G2–4: 0.51 | not measured | n.a. | [31] | |
| Mild-to-Moderate Pediatric CKD (n = 55) | G1: 3.28 G2–3a: 2.47 | G1: 16.9 G2–3a: 16.5 | G1: 0.19 G2–3a: 0.15 | [32] | |
| Pediatric CKD (n = 45) | G1: 28 G2–4: 26.8 | G1: 3.1 G2–4: 1.9 | G1: 9.03 G2–4: 14.1 | [33] | |
| Pediatric ADHD (n = 23) | 45.7 | 6.2 | 7.37 | 0.881 | [34] |
| Black Children (n = 41) White Children (n = 39) | 33.9 38.4 | 5.7 5.5 | 5.95 6.98 | 0.861 0.873 | [35] |
| Black Young Men (n = 292) White Young Men (n = 281) | 25.4 26.3 | 3.34 3.28 | 7.60 8.02 | 0.884 0.889 | [35] |
| Black Young Women (n = 309) White Young Women (n = 312) | 27.7 29.9 | 4.25 4.09 | 6.52 7.31 | 0.867 0.880 | [35] |
| Healthy Overweight Men (n = 11) | 26.9 | 3.59 | 7.49 | 0.882 | [36] |
| BMD, Group I (n = 10) BMD Group II (n = 8) | 48.8 53.0 | 5.75 8.05 | 8.49 6.58 | 0.895 0.953 | [37] |
| Preterm Neonates (n = 73 or 75) | 256 | 12.7 | 20.2 | 0.941 | [38] |
| PAOD, Group I (n = 20) PAOD, Group II (n = 20) | 59.2 69.3 | 3.02 2.89 | 19.6 24.0 | 0.951 0.897 | [39] |
| CAD, Group I (n = 29) CAD, Group II (n = 31) | 35.7 35.7 | 4.10 3.58 | 8.71 9.97 | 0.795 0.798 | [39] |
| DMD (n = 55) | 78.9 | 20.3 | 3.89 | [40] | |
| Pediatric T1DM ND (n = 10) Pediatric T1DM Treated (n = 92) | 40.3 30.5 | 10.2 5.32 | 3.95 5.74 | [41] | |
| Pediatric HUS (n = 4 or 5) | 13.7 | 3.3 | 4.15 | 0.811 | [42] |
| Pediatric Homocystinuria (n = 6) | 62.2 | 8.1 | 7.68 | 0.885 | [43] |
| Pediatric PKU (n = 52) | not measured | 6.8 | n.a. | [43] | |
| Pediatric HyperCh Type II (n = 64) Pediatric NormoCh (n = 54) | 88 56 | 7.1 7.2 | 12.4 7.78 | 0.925 | [44] |
| CAD (Stage 0/1/2/3) (n = 77) | 52.5/53/53/53 | 4.1/4.3/4/3 | 13.7/13.3/15/22 | 0.928 | [45] |
| Rheumatoid Arthritis RA (n = 10) Undifferentiated RA (n = 10) Spondyloarthritis (n = 5) Vasculitis (n = 3) Health (n = 10) | 74.3 38.4 82.5 46.9 10.1 | 2.82 3.29 2.77 3.86 1.35 | 26.3 11.7 29.8 12.2 7.48 | [46] | |
| End-Stage Liver Disease (n = 9) | 47.8 | 5.7 | 8.39 | 0.893 | [47] |
| Pediatric FSGS (n = 9) Non-FSGS (n = 11) | 345 130 | 41 5.7 | 8.41 22.8 | 0.894 | [48] |
| Pediatric Citrullinemia (n = 8) | 285 | 26.1 | 10.9 | 0.916 | [49] |
| Schimke Disease (n = 10) Healthy Controls (n = 10) | not measured not measured | 13.3 17.0 | n.a. n.a. | [50] | |
| Condition | DMA (µmol/d) | ADMA | DMA/ADMA | Ref. | |
| Healthy Males (n = 6) Placebo ADMA Intravenous ADMA (3 mg/kg) | 260 560 | not measured not measured | n.a. n.a. | [51] | |
| Healthy Females (n = 101) Healthy Males (n = 102) Healthy Humans | 300 470 160–1280 | not measured not measured not measured | n.a. n.a. n.a. | [16] | |
| Healthy Men (n = 4) | 340 | not measured | n.a. | [25] |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsikas, D. Urinary Dimethylamine (DMA) and Its Precursor Asymmetric Dimethylarginine (ADMA) in Clinical Medicine, in the Context of Nitric Oxide (NO) and Beyond. J. Clin. Med. 2020, 9, 1843. https://doi.org/10.3390/jcm9061843
Tsikas D. Urinary Dimethylamine (DMA) and Its Precursor Asymmetric Dimethylarginine (ADMA) in Clinical Medicine, in the Context of Nitric Oxide (NO) and Beyond. Journal of Clinical Medicine. 2020; 9(6):1843. https://doi.org/10.3390/jcm9061843
Chicago/Turabian StyleTsikas, Dimitrios. 2020. "Urinary Dimethylamine (DMA) and Its Precursor Asymmetric Dimethylarginine (ADMA) in Clinical Medicine, in the Context of Nitric Oxide (NO) and Beyond" Journal of Clinical Medicine 9, no. 6: 1843. https://doi.org/10.3390/jcm9061843
APA StyleTsikas, D. (2020). Urinary Dimethylamine (DMA) and Its Precursor Asymmetric Dimethylarginine (ADMA) in Clinical Medicine, in the Context of Nitric Oxide (NO) and Beyond. Journal of Clinical Medicine, 9(6), 1843. https://doi.org/10.3390/jcm9061843