Is Hyperuricemia, an Early-Onset Metabolic Disorder, Causally Associated with Cardiovascular Disease Events in Han Chinese?

 , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Onset Sequence Study
2.3. UA Mendelian Randomization Study
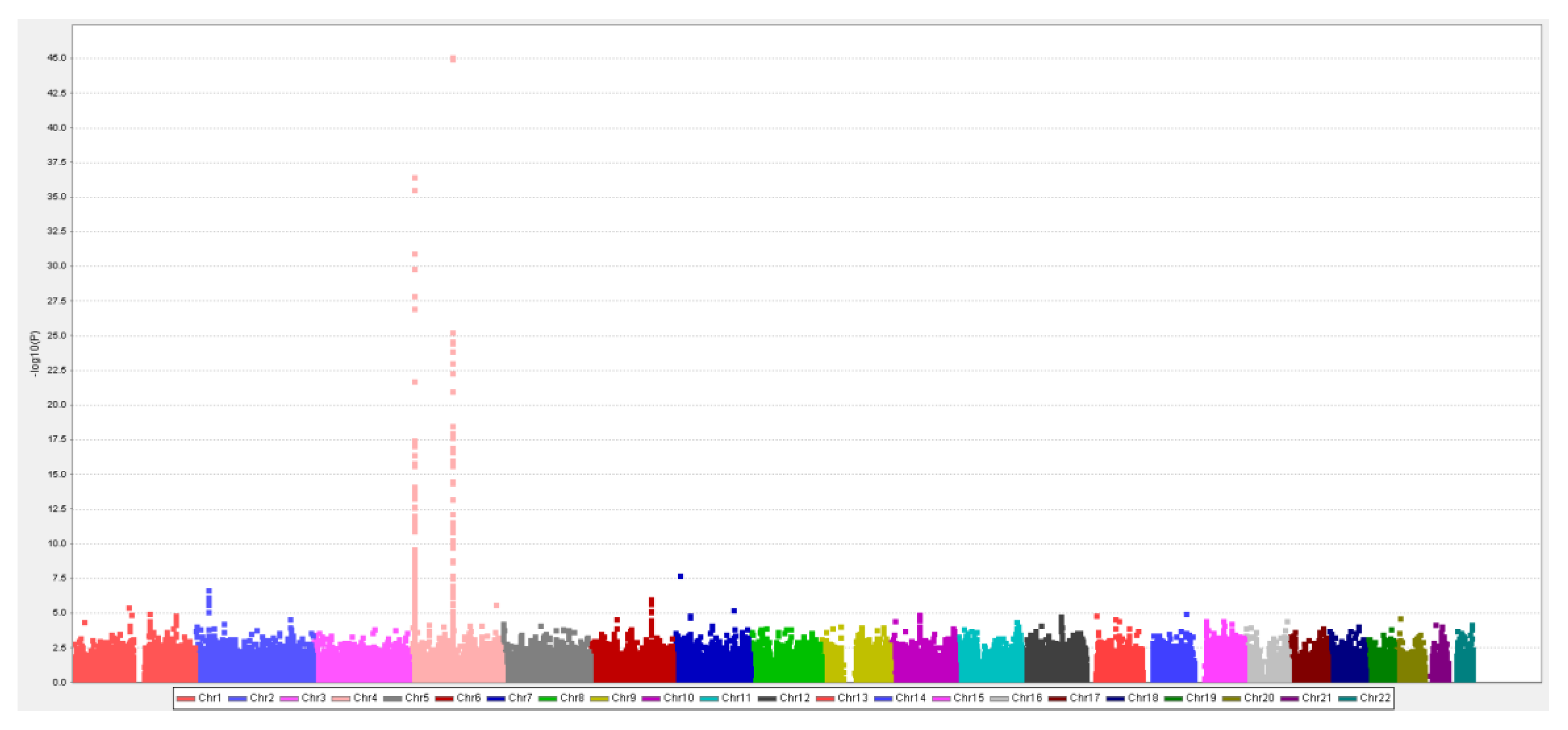
2.3.1. Han-Chinese SUA-SNP Selection with a Two-Stage GWAS
2.3.2. SUA-SNPs from Literature Review
2.3.3. Calculating the WGRS
2.3.4. Association between SUA and CVD Events
2.4. UAMR Study: Sensitivity Analyses
3. Results
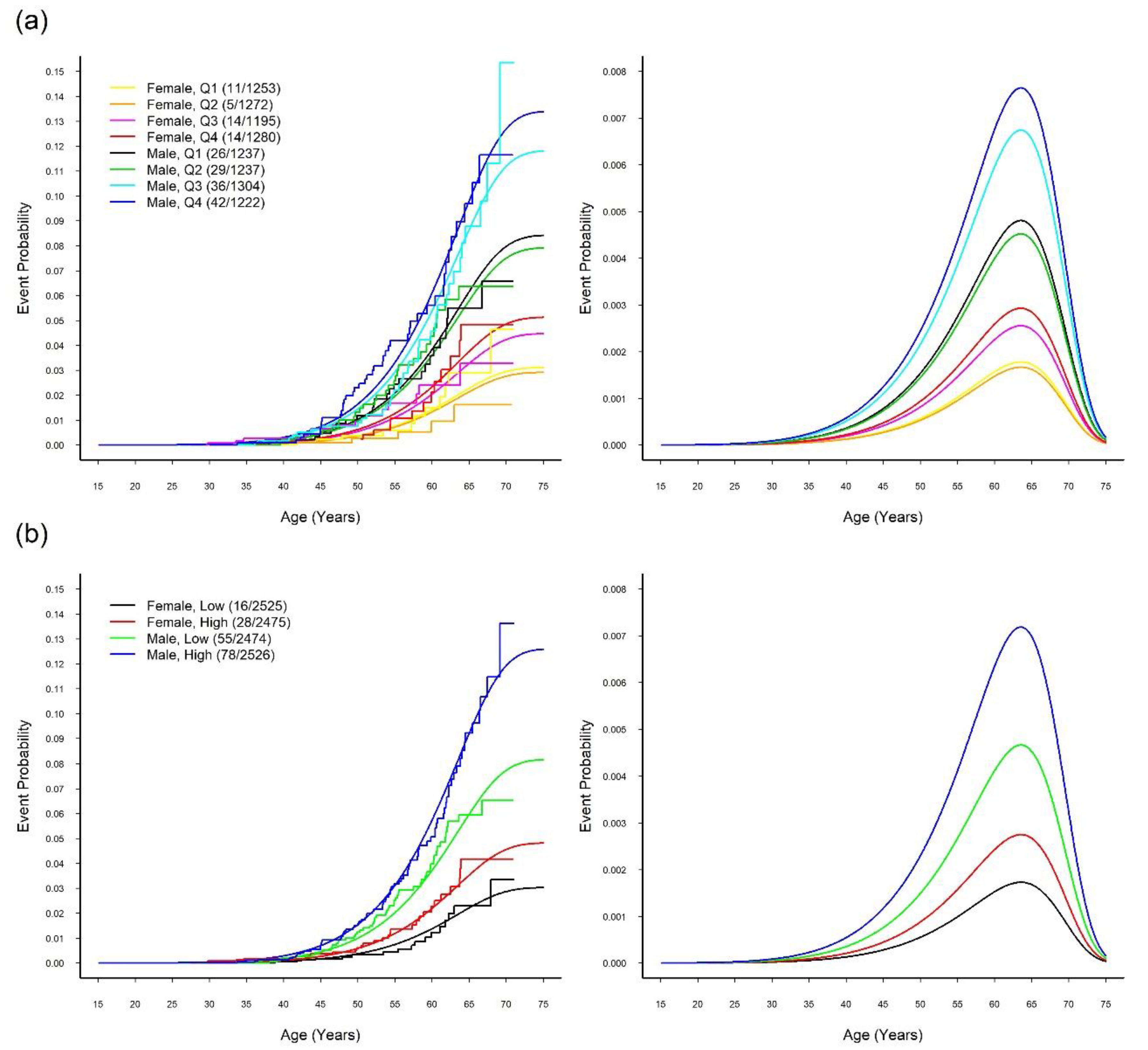
3.1. Onset Sequence Study
3.2. MR Study: SUA-SNP Discovery and Selection
3.3. MR Study: WGRS
3.4. MR Study: Relationship between SUA and CVD
4. Discussions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mozaffarian, D.; Benjamin, E.J.; Go, A.S.; Arnett, D.K.; Blaha, M.J.; Cushman, M.; De Ferranti, S.; Després, J.P.; Fullerton, H.J.; Howard, V.J.; et al. Heart disease and stroke statistics 2015 update: A report from the American Heart Association. Circulation 2015, 131, 434–441. [Google Scholar] [CrossRef]
- Keenan, T.; Zhao, W.; Rasheed, A.; Ho, W.K.; Malik, R.; Felix, J.F.; Young, R.; Shah, N.; Samuel, M.; Sheikh, N.; et al. Causal assessment of serum urate levels in cardiometabolic diseases through a Mendelian Randomization study. J. Am. Coll. Cardiol. 2016, 67, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Huang, L.; Song, M.; Song, Y. Baseline serum uric acid level as a predictor of cardiovascular disease related mortality and all-cause mortality: A meta-analysis of prospective studies. Atherosclerosis 2013, 231, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Feig, D.I.; Kang, D.H.; Johnson, R.J. Uric Acid and Cardiovascular Risk. N. Engl. J. Med. 2008, 359, 1811–1821. [Google Scholar] [CrossRef] [PubMed]
- Chuang, S.Y.; Chen, J.H.; Yeh, W.T.; Wu, C.C.; Pan, W.H. Hyperuricemia and increased risk of ischemic heart disease in a large Chinese cohort. Int. J. Cardiol. 2012, 154, 316–321. [Google Scholar] [CrossRef] [PubMed]
- Chu, N.F.; Wang, D.J.; Liou, S.H.; Shieh, S.M. Relationship between hyperuricemia and other cardiovascular disease risk factors among adult males in Taiwan. Eur. J. Epidemiol. 2000, 16, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Iso, H.; Murakami, Y.; Miura, K.; Nagai, M.; Sugiyama, D.; Ueshima, H.; Okamura, T. Serum Uric Acid and Mortality Form Cardiovascular Disease: EPOCH-JAPAN Study. J. Atheroscler. Thromb. 2016, 23, 692–703. [Google Scholar] [CrossRef]
- Pan, W.H.; Flegal, K.M.; Chang, H.Y.; Yeh, W.T.; Yeh, C.J.; Lee, W.C. Body mass index and obesity-related metabolic disorders in Taiwanese and US whites and blacks: Implications for definitions of overweight and obesity for Asians. Am. J. Clin. Nutr. 2004, 79, 31–39. [Google Scholar] [CrossRef]
- Nejatinamini, S.; Ataie-Jafari, A.; Qorbani, M.; Nikoohemat, S.; Kelishadi, R.; Asayesh, H. Association between serum uric acid level and metabolic syndrome components. J. Diabetes Metab. Disord. 2015, 14, 70. [Google Scholar] [CrossRef]
- Tsay, Y.C.; Chen, C.H.; Pan, W.H. Ages at Onset of 5 Cardiometabolic Diseases Adjusting for Nonsusceptibility: Implications for the Pathogenesis of Metabolic Syndrome. Am. J. Epidemiol. 2016, 184, 366–377. [Google Scholar] [CrossRef]
- Smith, G.D.; Ebrahim, S. ‘Mendelian randomization’: Can genetic epidemiology contribute to understanding environmental determinants of disease? Int. J. Epidemiol. 2003, 32, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Tyrrell, J.; Jones, S.; Beaumont, R.; Astley, C.M.; Lovell, R.; Yaghootkar, H.; Tuke, M.; Ruth, K.S.; Freathy, R.M.; Hirschhorn, J.N.; et al. Height, body mass index, and socioeconomic status: Mendelian randomisation study in UK Biobank. BMJ 2016, 352, i582. [Google Scholar] [CrossRef] [PubMed]
- Glymour, M.M. Alcohol and cardiovascular disease. BMJ 2014, 349, g4334. [Google Scholar] [CrossRef] [PubMed]
- Kleber, M.E.; Delgado, G.; Grammer, T.B.; Silbernagel, G.; Huang, J.; Krämer, B.K.; Ritz, E.; März, W. Uric Acid and Cardiovascular Events: A Mendelian Randomization Study. J. Am. Soc. Nephrol. 2015, 26, 2831–2838. [Google Scholar] [CrossRef] [PubMed]
- White, J.; Sofat, R.; Hemani, G.; Shah, T.; Engmann, J.; Dale, C.; Shah, S.; Kruger, F.A.; Giambartolomei, C.; Swerdlow, D.I.; et al. Plasma urate concentration and risk of coronary heart disease: A Mendelian randomisation analysis. Lancet Diabetes Endocrinol. 2016, 4, 327–336. [Google Scholar] [CrossRef]
- Stark, K.; Reinhard, W.; Grassl, M.; Erdmann, J.; Schunkert, H.; Illig, T.; Hengstenberg, C. Common Polymorphisms Influencing Serum Uric Acid Levels Contribute to Susceptibility to Gout, but Not to Coronary Artery Disease. PLoS ONE 2009, 4, e7729. [Google Scholar] [CrossRef] [PubMed]
- Palmer, T.M.; Nordestgaard, B.G.; Benn, M.; Tybjærg-Hansen, A.; Smith, G.D.; Lawlor, D.A.; Timpson, N.J. Association of plasma uric acid with ischaemic heart disease and blood pressure: Mendelian randomisation analysis of two large cohorts. BMJ 2013, 347, f4262. [Google Scholar] [CrossRef]
- Yang, B.; Mo, Z.; Wu, C.; Yang, H.; Yang, X.; He, Y.; Gui, L.; Zhou, L.; Guo, H.; Zhang, X.; et al. A genome-wide association study identifies common variants influencing serum uric acid concentrations in a Chinese population. BMC Med. Genomics 2014, 7, 10. [Google Scholar] [CrossRef]
- Chen, C.H.; Tsay, Y.C.; Wu, Y.C.; Horng, C.F. Logistic-AFT location-scale mixture regression models with nonsusceptibility for left-truncated and general interval-censored data. Stat. Med. 2013, 32, 4285–4305. [Google Scholar] [CrossRef]
- Chuang, S.Y.; Bai, C.H.; Chen, W.H.; Lien, L.M.; Pan, W.H. Fibrinogen Independently Predicts the Development of Ischemic Stroke in a Taiwanese Population: CVDFACTS Study. Stroke 2009, 40, 1578–1584. [Google Scholar] [CrossRef]
- Weng, L.C.; Yeh, W.T.; Bai, C.H.; Chen, H.J.; Chuang, S.Y.; Chang, H.Y.; Lin, B.F.; Chen, K.J.; Pan, W.H. Is Ischemic Stroke Risk Related to Folate Status or Other Nutrients Correlated With Folate Intake? Stroke 2008, 39, 3152–3158. [Google Scholar] [CrossRef]
- Frydman, H. A Note on Nonparametric Estimation of the Distribution Function from Interval-Censored and Truncated Observations. J. R. Stat. Soc. Ser. B 1994, 56, 71–74. [Google Scholar] [CrossRef]
- Turnbull, B.W. The Empirical Distribution Function with Arbitrarily Grouped, Censored and Truncated Data. J. R. Stat. Soc. Ser. B 1976, 38, 290–295. [Google Scholar] [CrossRef]
- Chen, C.H.; Yang, J.H.; Chiang, C.W.; Hsiung, C.N.; Wu, P.E.; Chang, L.C.; Chu, H.W.; Chang, J.; Song, I.W.; Yang, S.L.; et al. Population structure of Han Chinese in the modern Taiwanese population based on 10,000 participants in the Taiwan Biobank project. Hum. Mol. Genet. 2016, 25, 5321–5331. [Google Scholar] [CrossRef]
- Yang, H.C.; Lin, C.H.; Hsu, C.L.; Hung, S.I.; Wu, J.Y.; Pan, W.H.; Chen, Y.T.; Fann, C.S.J. A comparison of major histocompatibility complex SNPs in Han Chinese residing in Taiwan and Caucasians. J. Biomed. Sci. 2006, 13, 489–498. [Google Scholar] [CrossRef]
- Benjamini, Y.; Drai, D.; Elmer, G.; Kafkafi, N.; Golani, I. Controlling the false discovery rate in behavior genetics research. Behav. Brain Res. 2001, 125, 279–284. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; De Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Cheng, Y.C.; Hsiao, F.C.; Yeh, E.C.; Lin, W.J.; Tang, C.Y.L.; Tseng, H.C.; Wu, H.T.; Liu, C.K.; Chen, C.C.; Chen, Y.T.; et al. VarioWatch: Providing large-scale and comprehensive annotations on human genomic variants in the next generation sequencing era. Nucleic Acids Res. 2012, 40, W76–W81. [Google Scholar] [CrossRef]
- Barrett, J.C.; Fry, B.; Maller, J.; Daly, M.J. Haploview: Analysis and visualization of LD and haplotype maps. Bioinformatics 2005, 21, 263–265. [Google Scholar] [CrossRef]
- Mokry, L.E.; Ross, S.; Timpson, N.J.; Sawcer, S.; Smith, G.D.; Richards, J.B. Obesity and Multiple Sclerosis: A Mendelian Randomization Study. PLoS Med. 2016, 13, e1002053. [Google Scholar] [CrossRef]
- Bowden, J.; Smith, G.D.; Burgess, S. Mendelian randomization with invalid instruments: Effect estimation and bias detection through Egger regression. Int. J. Epidemiol. 2015, 44, 512–525. [Google Scholar] [CrossRef]
- Pierce, B.L.; Ahsan, H.; Vanderweele, T.J. Power and instrument strength requirements for Mendelian randomization studies using multiple genetic variants. Int. J. Epidemiol. 2011, 40, 740–752. [Google Scholar] [CrossRef]
- Chen, J.H.; Chuang, S.Y.; Chen, H.J.; Yeh, W.T.; Pan, W.H. Serum uric acid level as an independent risk factor for all-cause, cardiovascular, and ischemic stroke mortality: A chinese cohort study. Arthritis Rheum. 2009, 61, 225–232. [Google Scholar] [CrossRef]
- Chen, J.H.; Lan, J.L.; Cheng, C.F.; Liang, W.M.; Lin, H.Y.; Tsay, G.J.; Yeh, W.T.; Pan, W.H. Effect of Urate-Lowering Therapy on All-Cause and Cardiovascular Mortality in Hyperuricemic Patients without Gout: A Case-Matched Cohort Study. PLoS ONE 2015, 10, e0145193. [Google Scholar] [CrossRef]
- Culleton, B.F.; Larson, M.G.; Kannel, W.B.; Levy, D. Serum uric acid and risk for cardiovascular disease and death: The Framingham Heart Study. Ann. Intern. Med. 1999, 131, 7–13. [Google Scholar] [CrossRef]
- Moriarity, J.T.; Folsom, A.R.; Iribarren, C.; Nieto, F.J.; Rosamond, W.D. Serum uric acid and risk of coronary heart disease: Atherosclerosis Risk in Communities (ARIC) Study. Ann. Epidemiol. 2000, 10, 136–143. [Google Scholar] [CrossRef]
- International Diabetes Federation. 2015 Diabetes Atlas; International Diabetes Federation: Brussels, Belgium, 2015. [Google Scholar]
- Chuang, S.Y.; Lee, S.C.; Hsieh, Y.T.; Pan, W.H. Trends in hyperuricemia and gout prevalence: Nutrition and Health Survey in Taiwan from 1993–1996 to 2005–2008. Asia Pac. J. Clin. Nutr. 2011, 20, 301–308. [Google Scholar]
- Annemans, L.; Spaepen, E.; Gaskin, M.; Bonnemaire, M.; Malier, V.; Gilbert, T.; Nuki, G. Gout in the UK and Germany: Prevalence, comorbidities and management in general practice 2000–2005. Ann. Rheum. Dis. 2008, 67, 960–966. [Google Scholar] [CrossRef]
- Feig, D.I.; Soletsky, B.; Johnson, R.J. Effect of Allopurinol on Blood Pressure of Adolescents with Newly Diagnosed Essential Hypertension: A Randomized Trial. JAMA 2008, 300, 924–932. [Google Scholar] [CrossRef]
- Robertson, A.J.; Struthers, A.D. A Randomized Controlled Trial of Allopurinol in Patients with Peripheral Arterial Disease. Can. J. Cardiol. 2016, 32, 190–196. [Google Scholar] [CrossRef]
- Gavin, A.D.; Struthers, A.D. Allopurinol reduces B-type natriuretic peptide concentrations and haemoglobin but does not alter exercise capacity in chronic heart failure. Heart 2005, 91, 749–753. [Google Scholar] [CrossRef]
- Mulay, S.R.; Evan, A.; Anders, H.J. Molecular mechanisms of crystal-related kidney inflammation and injury. Implications for cholesterol embolism, crystalline nephropathies and kidney stone disease. Nephrol. Dial. Transp. 2014, 29, 507–514. [Google Scholar] [CrossRef]
- Duncan, D.J.; Yang, Z.; Hopkins, P.M.; Steele, D.S.; Harrison, S.M. TNF-alpha and IL-1beta increase Ca2+ leak from the sarcoplasmic reticulum and susceptibility to arrhythmia in rat ventricular myocytes. Cell Calcium 2010, 47, 378–386. [Google Scholar] [CrossRef]
- Park, J.H.; Jin, Y.M.; Hwang, S.; Cho, D.H.; Kang, D.H.; Jo, I. Uric acid attenuates nitric oxide production by decreasing the interaction between endothelial nitric oxide synthase and calmodulin in human umbilical vein endothelial cells: A mechanism for uric acid-induced cardiovascular disease development. Nitric Oxide 2013, 32, 36–42. [Google Scholar] [CrossRef]
- Sautin, Y.Y.; Nakagawa, T.; Zharikov, S.; Johnson, R.J. Adverse effects of the classic antioxidant uric acid in adipocytes: NADPH oxidase-mediated oxidative/nitrosative stress. Am. J. Physiol. Cell Physiol. 2007, 293, C584–C596. [Google Scholar] [CrossRef]
- Johnson, R.J.; Bakris, G.L.; Borghi, C.; Chonchol, M.B.; Feldman, D.; Lanaspa, M.A.; Merriman, T.R.; Moe, O.W.; Mount, D.B.; Lozada, L.G.S.; et al. Hyperuricemia, Acute and Chronic Kidney Disease, Hypertension, and Cardiovascular Disease: Report of a Scientific Workshop Organized by the National Kidney Foundation. Am. J. Kidney Dis. 2018, 71, 851–865. [Google Scholar] [CrossRef]
- Sirker, A.; Zhang, M.; Shah, A.M. NADPH oxidases in cardiovascular disease: Insights from in vivo models and clinical studies. Basic Res. Cardiol. 2011, 106, 735–747. [Google Scholar] [CrossRef]
- Targher, G.; Byrne, C.D.; Lonardo, A.; Zoppini, G.; Barbui, C. Non-alcoholic fatty liver disease and risk of incident cardiovascular disease: A meta-analysis. J. Hepatol. 2016, 65, 589–600. [Google Scholar] [CrossRef]
- Lonardo, A.; Nascimbeni, F.; Mantovani, A.; Targher, G. Hypertension, diabetes, atherosclerosis and NASH: Cause or consequence? J. Hepatol. 2018, 68, 335–352. [Google Scholar] [CrossRef]
- Suzuki, A.; Angulo, P.; Lymp, J.; St Sauver, J.; Muto, A.; Okada, T.; Lindor, K. Chronological development of elevated aminotransferases in a nonalcoholic population. Hepatology 2005, 41, 64–71. [Google Scholar] [CrossRef]
- Vanni, E.; Bugianesi, E.; Kotronen, A.; De Minicis, S.; Yki-Jarvinen, H.; Svegliati-Baroni, G. From the metabolic syndrome to NAFLD or vice versa? Dig Liver Dis. 2010, 42, 320–330. [Google Scholar] [CrossRef]
- Yki-Järvinen, H. Non-alcoholic fatty liver disease as a cause and a consequence of metabolic syndrome. Lancet Diabetes Endocrinol. 2014, 2, 901–910. [Google Scholar] [CrossRef]
- Lonardo, A.; Ballestri, S.; Marchesini, G.; Angulo, P.; Loria, P. Nonalcoholic fatty liver disease: A precursor of the metabolic syndrome. Dig. Liver Dis. 2015, 47, 181–190. [Google Scholar] [CrossRef]
- Ballestri, S.; Zona, S.; Targher, G.; Romagnoli, D.; Baldelli, E.; Nascimbeni, F.; Roverato, A.; Guaraldi, G.; Lonardo, A. Nonalcoholic fatty liver disease is associated with an almost twofold increased risk of incident type 2 diabetes and metabolic syndrome. Evidence from a systematic review and meta-analysis. J. Gastroenterol. Hepatol. 2016, 31, 936–944. [Google Scholar] [CrossRef]
- Lonardo, A.; Nascimbeni, F.; Ballestri, S.; Fairweather, D.; Win, S.; Than, T.A.; Abdelmalek, M.F.; Suzuki, A. Sex Differences in NAFLD: State of the Art and Identification of Research Gaps. Hepatology 2019. [Google Scholar] [CrossRef]
- Yang, J.D.; Abdelmalek, M.F.; Guy, C.D.; Gill, R.M.; Lavine, J.E.; Yates, K.; Klair, J.; Terrault, N.A.; Clark, J.M.; Unalp-Arida, A.; et al. Patient Sex, Reproductive Status, and Synthetic Hormone Use Associate with Histologic Severity of Nonalcoholic Steatohepatitis. Clin. Gastroenterol. Hepatol. 2017, 15, 127–131. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| 1st Stage (N = 7000) | 2nd Stage (N = 3000) | Combined (N = 10,000) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Chr | SNP | BP | Gene | Alleles | Beta | P-Value | FDR | Beta | P-Value | Beta | P-Value | F | R2 |
| 4 | rs4148155 | 89054667 | ABCG2 | A/G | 0.31 | 9.35 × 10−46 | 2.86 × 10−40 | 0.32 | 5.60 × 10−22 | 0.31 | 4.34 × 10−66 | 37.7 | 0.022 |
| 4 | rs3733588 | 9997303 | SLC2A9 | A/G | −0.26 | 2.91 × 10−37 | 5.93 × 10−32 | −0.21 | 2.80 × 10−11 | −0.24 | 1.73 × 10−46 | 25.9 | 0.015 |
| 4 | rs2725211 | 88970375 | PKD2 | C/T | 0.25 | 1.86 × 10−25 | 1.14 × 10−20 | 0.30 | 1.50 × 10−17 | 0.27 | 3.18 × 10−41 | 24.2 | 0.014 |
| 4 | rs17013282 | 88765873 | MEPE | G/A | 0.19 | 1.03 × 10−11 | 1.15 × 10−7 | 0.21 | 1.38 × 10−6 | 0.20 | 4.95 × 10−17 | 11.1 | 0.007 |
| 4 | rs17013187 | 88733531 | IBSP | C/T | 0.16 | 1.49 × 10−9 | 1.35 × 10−5 | 0.14 | 6.63 × 10−4 | 0.15 | 2.85 × 10−12 | 8.8 | 0.005 |
| 4 | rs3756224 | 10105739 | WDR1 | T/C | 0.11 | 1.05 × 10−7 | 6.67 × 10−4 | 0.10 | 1.73 × 10−3 | 0.11 | 9.45 × 10−10 | 7.9 | 0.005 |
| 2 | rs1260326 | 27730940 | GCKR | C/T | 0.10 | 7.90 × 10−7 | 4.38 × 10−3 | 0.11 | 6.55 × 10−4 | 0.10 | 2.33 × 10−9 | 8.5 | 0.005 |
| 1 | rs4072037 | 155162067 | MUC1 | T/C | 0.11 | 8.18 × 10−6 | 3.60 × 10−2 | 0.11 | 4.56 × 10−3 | 0.11 | 1.43 × 10−7 | 6.8 | 0.004 |
| Characteristic | WGRS | P-Value † | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Q1 (N = 2490) | Q2 (N = 2509) | Q3 (N = 2499) | Q4 (N = 2502) | ||||||
| Mean | (SD) | Mean | (SD) | Mean | (SD) | Mean | (SD) | ||
| SUA (mg/dL) | 5.4 | (1.3) | 5.6 | (1.5) | 5.8 | (1.5) | 6.0 | (1.6) | <0.0001 * |
| Sex (Men%) | 49.7% | 49.3% | 52.2% | 48.8% | 0.080 | ||||
| Age (yr) | 49.1 | (11.4) | 48.9 | (11.0) | 48.6 | (11.0) | 48.8 | (11.1) | 0.140 |
| BMI (kg/m2) | 24.2 | (3.6) | 24.3 | (3.6) | 24.3 | (3.6) | 24.3 | (3.6) | 0.570 |
| FG (mg/dL) | 96.6 | (21.6) | 96.0 | (20.7) | 96.4 | (19.1) | 97.4 | (25.0) | 0.136 |
| T-CHO (mg/dL) | 193.4 | (35.9) | 191.8 | (35.1) | 191.9 | (34.1) | 194.1 | (36.6) | 0.060 |
| TG (mg/dL) | 118.0 | (104.0) | 113.8 | (83.0) | 117.9 | (81.9) | 124.7 | (118.3) | 0.002 * |
| HDL-C (mg/dL) | 53.7 | (13.4) | 53.3 | (13.2) | 52.8 | (12.8) | 52.8 | (13.1) | 0.050 |
| LDL-C (mg/dL) | 120.3 | (31.8) | 120.4 | (32.2) | 120.4 | (31.1) | 121.4 | (31.1) | 0.520 |
| eGFR (mL/min/1.73m2) | 103.4 | (24.4) | 102.7 | (24.6) | 102.2 | (24.7) | 102.9 | (25.1) | 0.41 |
| SGOT (U/L) | 24.6 | (16.6) | 24.6 | (12.5) | 24.7 | (13.1) | 24.2 | (10.4) | 0.582 |
| SGPT (U/L) | 24.5 | (18.8) | 25.2 | (21.6) | 25.8 | (23.5) | 24.8 | (19.4) | 0.110 |
| SBP (mmHg) | 115.7 | (16.9) | 116.0 | (17.1) | 116.7 | (17.3) | 116.9 | (18.0) | 0.060 |
| DBP (mmHg) | 71.6 | (10.8) | 72.0 | (10.9) | 72.7 | (11.3) | 72.8 | (11.3) | 0.010 * |
| Characteristic | WGRS | P-Value † | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Q1 (N = 2490) | Q2 (N = 2509) | Q3 (N = 2499) | Q4 (N = 2502) | ||||||
| N | % | N | % | N | % | N | % | ||
| Sex | 0.08 | ||||||||
| Male | 1237 | 49.7 | 1237 | 49.3 | 1304 | 52.2 | 1222 | 48.8 | |
| Female | 1253 | 50.3 | 1272 | 50.7 | 1195 | 47.8 | 1280 | 51.2 | |
| Drinking habit | 0.38 | ||||||||
| Yes | 207 | 8.3 | 183 | 7.3 | 204 | 8.2 | 204 | 8.2 | |
| No | 2194 | 88.1 | 2239 | 89.2 | 2215 | 88.6 | 2232 | 89.2 | |
| Quit | 89 | 3.57 | 87 | 3.5 | 80 | 3.2 | 66 | 2.6 | |
| Smoking habit | 0.16 | ||||||||
| Yes | 263 | 10.6 | 289 | 11.5 | 312 | 12.5 | 295 | 11.8 | |
| Few | 214 | 8.6 | 206 | 8.2 | 207 | 8.3 | 213 | 8.5 | |
| No | 1748 | 70.2 | 1723 | 68.7 | 1656 | 66.3 | 1703 | 68.1 | |
| Quit | 265 | 10.6 | 291 | 11.6 | 324 | 13.0 | 291 | 11.6 | |
| Education | 0.071 | ||||||||
| Elementary School | 177 | 7.1 | 172 | 6.9 | 163 | 6.5 | 199 | 8.0 | |
| Junior-high/Senior-high | 1063 | 42.7 | 1045 | 41.7 | 984 | 39.4 | 1027 | 41.1 | |
| BS/MS/PhD | 1248 | 50.2 | 1289 | 51.4 | 1351 | 54.1 | 1276 | 51.0 | |
| Marriage | 0.66 | ||||||||
| Single | 300 | 12.1 | 279 | 11.1 | 298 | 11.9 | 274 | 11.0 | |
| Married | 1936 | 77.8 | 1989 | 79.4 | 1961 | 78.5 | 1963 | 78.6 | |
| Divorced/Widowed | 253 | 10.2 | 238 | 9.5 | 239 | 9.6 | 262 | 10.5 | |
| Regular Exercise | 0.94 | ||||||||
| No | 1453 | 58.4 | 1473 | 58.7 | 1472 | 58.9 | 1481 | 59.2 | |
| Yes | 1021 | 40.8 | 1027 | 41.1 | 1036 | 41.3 | 1037 | 41.7 | |
| Predictors | Logistic | Location | Scale | Shape | AIC | ||||
|---|---|---|---|---|---|---|---|---|---|
| OR (95% C) | P-Value | EST (95% CI) | P-Value | EST (95% CI) | P-Value | EST (95% CI) | P-Value | ||
| (a)continuousWGRS | |||||||||
| Intercept | 1 | Referent | 4.16 a (4.11, 4.21) | <0.001 | −2.37 a (−3.00, −1.73) | <0.001 | 1.42 a (0.35, 2.49) | 0.009 | 2594.09 |
| Male | 2.87 a (2.01, 4.10) | <0.001 | |||||||
| WGRS | 1.41 c (1.04, 1.91) | 0.029 | |||||||
| (b)four-group WGRS | |||||||||
| Intercept | 1 | Referent | 4.16 a (4.11, 4.21) | <0.001 | −2.37 a (−3.00, −1.73) | <0.001 | 1.42 b (0.35, 2.50) | 0.009 | 2593.36 |
| Male | 2.86 a (2.00, 4.10) | <0.001 | |||||||
| Q2 | 0.94 (0.57, 1.53) | 0.790 | |||||||
| Q3 | 1.46 (0.93, 2.29) | 0.103 | |||||||
| Q4 | 1.68 c (1.08, 2.62) | 0.022 | |||||||
| (c)two-group WGRS | |||||||||
| Intercept | 1 | Referent | 4.16 a (4.11, 4.21) | <0.001 | −2.37 a (−3.00, −1.73) | <0.001 | 1.42 b (0.35, 2.50) | 0.009 | 2589.89 |
| Male | 2.85 a (1.99, 4.07) | <0.001 | |||||||
| High | 1.62 b (1.17, 2.23) | 0.003 | |||||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiang, K.-M.; Tsay, Y.-C.; Vincent Ng, T.-C.; Yang, H.-C.; Huang, Y.-T.; Chen, C.-H.; Pan, W.-H. Is Hyperuricemia, an Early-Onset Metabolic Disorder, Causally Associated with Cardiovascular Disease Events in Han Chinese? J. Clin. Med. 2019, 8, 1202. https://doi.org/10.3390/jcm8081202
Chiang K-M, Tsay Y-C, Vincent Ng T-C, Yang H-C, Huang Y-T, Chen C-H, Pan W-H. Is Hyperuricemia, an Early-Onset Metabolic Disorder, Causally Associated with Cardiovascular Disease Events in Han Chinese? Journal of Clinical Medicine. 2019; 8(8):1202. https://doi.org/10.3390/jcm8081202
Chicago/Turabian StyleChiang, Kuang-Mao, Yuh-Chyuan Tsay, Ta-Chou Vincent Ng, Hsin-Chou Yang, Yen-Tsung Huang, Chen-Hsin Chen, and Wen-Harn Pan. 2019. "Is Hyperuricemia, an Early-Onset Metabolic Disorder, Causally Associated with Cardiovascular Disease Events in Han Chinese?" Journal of Clinical Medicine 8, no. 8: 1202. https://doi.org/10.3390/jcm8081202
APA StyleChiang, K.-M., Tsay, Y.-C., Vincent Ng, T.-C., Yang, H.-C., Huang, Y.-T., Chen, C.-H., & Pan, W.-H. (2019). Is Hyperuricemia, an Early-Onset Metabolic Disorder, Causally Associated with Cardiovascular Disease Events in Han Chinese? Journal of Clinical Medicine, 8(8), 1202. https://doi.org/10.3390/jcm8081202