Periodontitis: A Multifaceted Disease of Tooth-Supporting Tissues


{kind=link}
{kind=link}
Abstract
1. Introduction
2. Pathogenic Biofilms
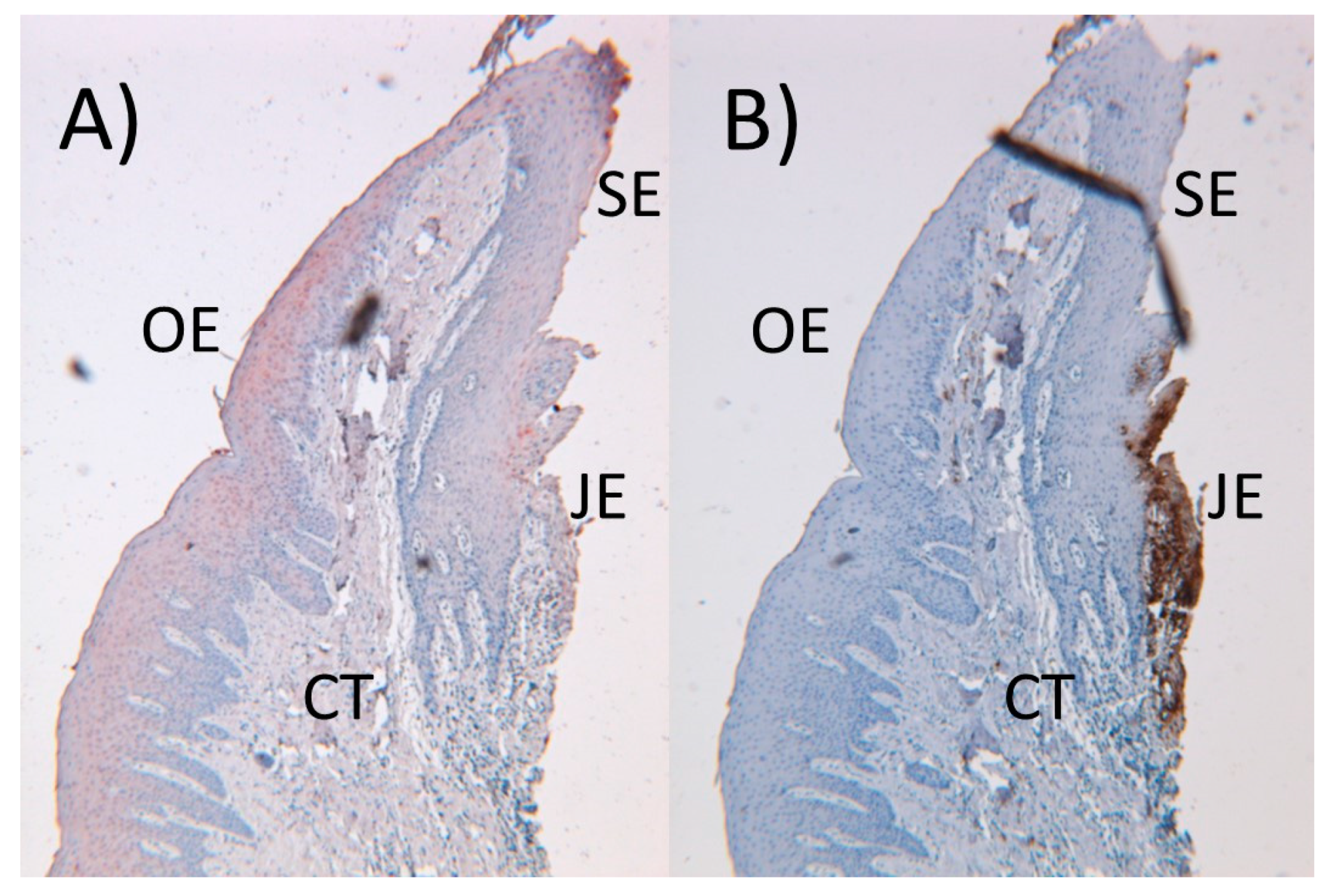
3. Immunologic Players of the Periodontium
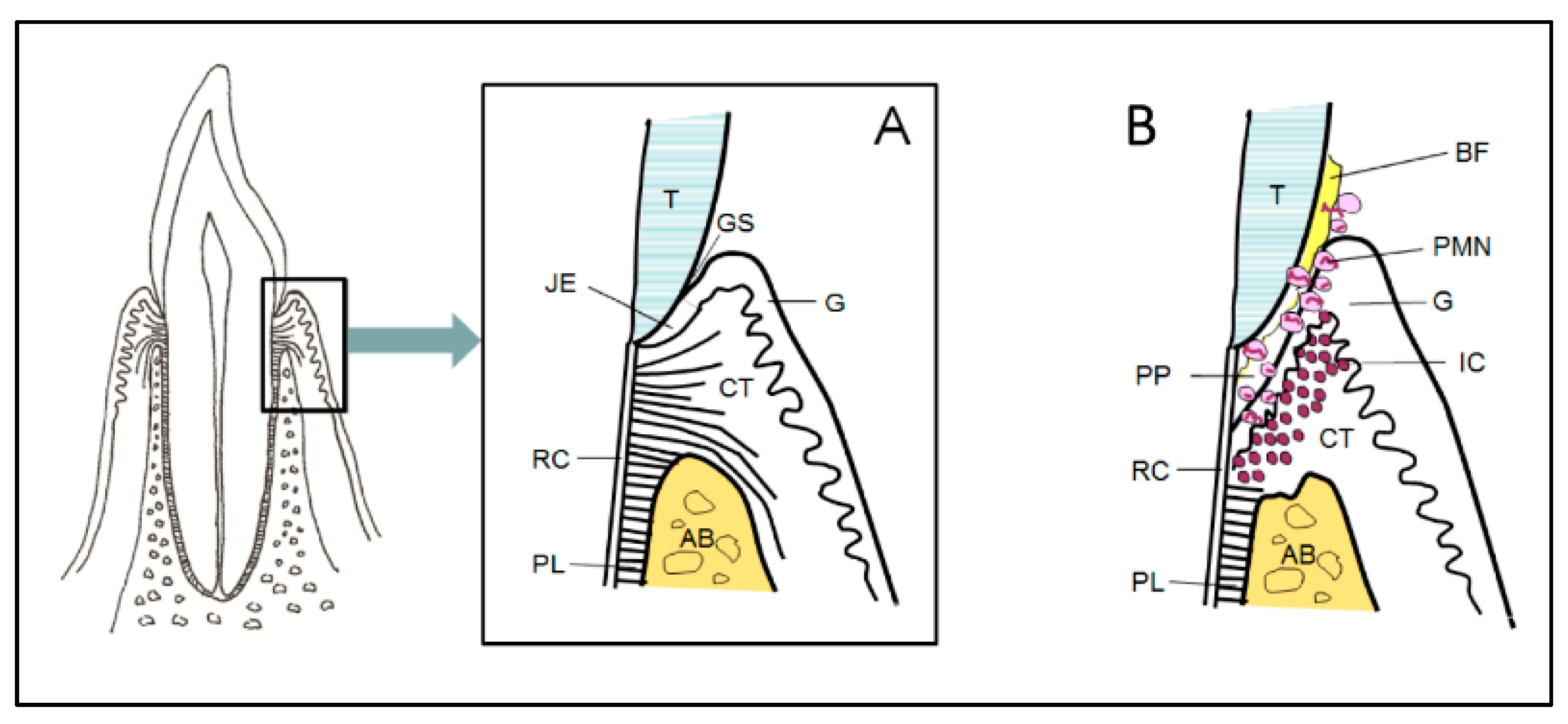
4. Inflammatory Process and Periodontal Tissue Destruction
5. Periodontal Therapy—Impact on Oral and General Health
6. Future Considerations
Author Contributions
Conflicts of Interest
References
- Eke, P.I.; Dye, B.A.; Wei, L.; Thornton-Evans, G.O.; Genco, R.J. Prevalence of periodontitis in adults in the United States: 2009 and 2010. J. Dent. Res. 2012, 91, 914–920. [Google Scholar] [CrossRef] [PubMed]
- Kassebaum, N.J.; Bernabé, E.; Dahiya, M.; Bhandari, B.; Murray, C.J.; Marcenes, W. Global burden of severe periodontitis in 1990–2010: A systematic review and meta-regression. J. Dent. Res. 2014, 93, 1045–1053. [Google Scholar] [CrossRef] [PubMed]
- Genco, R.J.; Borgnakke, W.S. Risk factors for periodontal disease. Periodontol. 2000 2013, 62, 59–94. [Google Scholar] [CrossRef] [PubMed]
- Lalla, E.; Papapanou, P.N. Diabetes mellitus and periodontitis: A tale of two common interrelated diseases. Nat. Rev. Endocrinol. 2011, 7, 738–748. [Google Scholar] [CrossRef] [PubMed]
- Lalla, E.; Cheng, B.; Lal, S.; Kaplan, S.; Softness, B.; Greenberg, E.; Goland, R.S.; Lamster, I.B. Diabetes mellitus promotes periodontal destruction in children. J. Clin. Periodontol. 2007, 34, 294–298. [Google Scholar] [CrossRef] [PubMed]
- Heikkinen, A.M.; Pajukanta, R.; Pitkäniemi, J.; Broms, U.; Sorsa, T.; Koskenvuo, M.; Meurman, J.H. The effect of smoking on periodontal health of 15- to 16-year-old adolescents. J. Periodontol. 2008, 79, 2042–2047. [Google Scholar] [CrossRef] [PubMed]
- Thomson, W.M.; Shearer, D.M.; Broadbent, J.M.; Foster Page, L.A.; Poulton, R. The natural history of periodontal attachment loss during the third and fourth decades of life. J. Clin. Periodontol. 2013, 40, 672–680. [Google Scholar] [CrossRef] [PubMed]
- Ylöstalo, P.; Sakki, T.; Laitinen, J.; Järvelin, M.R.; Knuuttila, M. The relation of tobacco smoking to tooth loss among young adults. Eur. J. Oral Sci. 2004, 112, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Kosaka, T.; Ono, T.; Yoshimuta, Y.; Kida, M.; Kikui, M.; Nokubi, T.; Maeda, Y.; Kokubo, Y.; Watanabe, M.; Miyamoto, Y. The effect of periodontal status and occlusal support on masticatory performance: The Suita study. J. Clin. Periodontol. 2014, 41, 497–503. [Google Scholar] [CrossRef]
- Tonetti, M.S.; Greenwell, H.; Kornman, K.S. Staging and grading of periodontitis: Framework and proposal of a new classification and case definition. J. Clin. Periodontol. 2018, 45 (Suppl. 20), S149–S161. [Google Scholar] [CrossRef]
- Darveau, R.P. Periodontitis: A polymicrobial disruption of host homeostasis. Nat. Rev. Microbiol. 2010, 8, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Sanz, M.; Beighton, D.; Curtis, M.A.; Cury, J.A.; Dige, I.; Dommisch, H.; Ellwood, R.; Giacaman, R.A.; Herrera, D.; Herzberg, M.C.; et al. Role of microbial biofilms in the maintenance of oral health and in the development of dental caries and periodontal diseases. Consensus report of group 1 of the joint EFP/ORCA workshop on the boundaries between caries and periodontal disease. J. Clin. Periodontol. 2017, 44 (Suppl. 18), 5–11. [Google Scholar] [CrossRef] [PubMed]
- Kolenbrander, P.E.; Palmer, R.J., Jr.; Periasamy, S.; Jakubovics, N.S. Oral multispecies biofilm development and the key role of cell-cell distance. Nat. Rev. Microbiol. 2010, 8, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Paster, B.J.; Boches, S.K.; Galvin, J.L.; Ericson, R.E.; Lau, C.N.; Levanos, V.A.; Sahasrabudhe, A.; Dewhirst, F.E. Bacterial diversity in human subgingival plaque. J. Bacteriol. 2001, 183, 3770–3783. [Google Scholar] [CrossRef]
- Könönen, E. Development of oral bacterial flora in young children. Ann. Med. 2000, 32, 107–112. [Google Scholar] [CrossRef]
- Haraldsson, G.; Holbrook, W.P.; Könönen, E. Clonal persistence of oral Fusobacterium nucleatum in infancy. J. Dent. Res. 2004, 83, 500–504. [Google Scholar] [CrossRef]
- Gursoy, U.K.; Pöllänen, M.; Könönen, E.; Uitto, V.J. Biofilm formation enhances the oxygen tolerance and invasiveness of Fusobacterium nucleatum in an oral mucosa culture model. J. Periodontol. 2010, 81, 1084–1091. [Google Scholar] [CrossRef]
- Mendes, R.T.; Nguyen, D.; Stephens, D.; Pamuk, F.; Fernandes, D.; Hasturk, H.; Van Dyke, T.E.; Kantarci, A. Hypoxia-induced endothelial cell responses - possible roles during periodontal disease. Clin. Exp. Dent. Res. 2018, 4, 241–248. [Google Scholar] [CrossRef]
- Socransky, S.S.; Haffajee, A.D.; Cugini, M.A.; Smith, C.; Kent, R.L., Jr. Microbial complexes in subgingival plaque. J. Clin. Periodontol. 1998, 25, 134–144. [Google Scholar] [CrossRef]
- Griffen, A.L.; Beall, C.J.; Campbell, J.H.; Firestone, N.D.; Kumar, P.S.; Yang, Z.K.; Podar, M.; Leys, E.J. Distinct and complex bacterial profiles in human periodontitis and health revealed by 16S pyrosequencing. ISME J. 2012, 6, 1176–1185. [Google Scholar] [CrossRef]
- Hajishengallis, G. The inflammophilic character of the periodontitis-associated microbiota. Mol. Oral Microbiol. 2014, 29, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Könönen, E.; Paju, S.; Pussinen, P.J.; Hyvönen, M.; Di Tella, P.; Suominen-Taipale, L.; Knuuttila, M. Population-based study of salivary carriage of periodontal pathogens in adults. J. Clin. Microbiol. 2007, 45, 2446–2451. [Google Scholar] [CrossRef]
- Könönen, E.; Müller, H.P. Microbiology of aggressive periodontitis. Periodontol. 2000 2014, 65, 46–78. [Google Scholar] [CrossRef] [PubMed]
- Curtis, M.A. Periodontal microbiology - the lid’s off the box again. J. Dent. Res. 2014, 93, 840–842. [Google Scholar] [CrossRef] [PubMed]
- Saglie, F.R.; Marfany, A.; Camargo, P. Intragingival occurrence of Actinobacillus actinomycetemcomitans and Bacteroides gingivalis in active destructive periodontal lesions. J. Periodontol. 1988, 59, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Gursoy, U.K.; Könönen, E.; Uitto, V.J. Intracellular replication of fusobacteria requires new actin filament formation of epithelial cells. APMIS 2008, 116, 1063–1070. [Google Scholar] [CrossRef] [PubMed]
- Baek, K.; Ji, S.; Choi, Y. Complex intratissue microbiota forms biofilms in periodontal lesions. J. Dent. Res. 2018, 97, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Hasegawa, M.; Inohara, N. The role of oral pathobionts in dysbiosis during periodontitis development. J. Dent. Res. 2014, 93, 539–546. [Google Scholar] [CrossRef] [PubMed]
- Herrero, E.R.; Fernandes, S.; Verspecht, T.; Ugarte-Berzal, E.; Boon, N.; Proost, P.; Bernaerts, K.; Quirynen, M.; Teughels, W. Dysbiotic biofilms deregulate the periodontal inflammatory response. J. Dent. Res. 2018, 97, 547–555. [Google Scholar] [CrossRef]
- Peyyala, R.; Kirakodu, S.S.; Novak, K.F.; Ebersole, J.L. Oral microbial biofilm stimulation of epithelial cell responses. Cytokine 2012, 58, 65–72. [Google Scholar] [CrossRef]
- Mason, M.R.; Preshaw, P.M.; Nagaraja, H.N.; Dabdoub, S.M.; Rahman, A.; Kumar, P.S. The subgingival microbiome of clinically healthy current and never smokers. ISME J. 2015, 9, 268–272. [Google Scholar] [CrossRef] [PubMed]
- Pussinen, P.J.; Könönen, E.; Paju, S.; Hyvärinen, K.; Gursoy, U.K.; Huumonen, S.; Knuuttila, M.; Suominen, A.L. Periodontal pathogen carriage, rather than periodontitis, determines the serum antibody levels. J. Clin. Periodontol. 2011, 38, 405–411. [Google Scholar] [CrossRef] [PubMed]
- Pussinen, P.J.; Tuomisto, K.; Jousilahti, P.; Havulinna, A.S.; Sundvall, J.; Salomaa, V. Endotoxemia, immune response to periodontal pathogens, and systemic inflammation associate with incident cardiovascular disease events. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1433–1439. [Google Scholar] [CrossRef] [PubMed]
- Damgaard, C.; Reinholdt, J.; Enevold, C.; Fiehn, N.E.; Nielsen, C.H.; Holmstrup, P. Immunoglobulin G antibodies against Porphyromonas gingivalis or Aggregatibacter actinomycetemcomitans in cardiovascular disease and periodontitis. J. Oral Microbiol. 2017, 9, 1374154. [Google Scholar] [CrossRef] [PubMed]
- Palm, F.; Lahdentausta, L.; Sorsa, T.; Tervahartiala, T.; Gokel, P.; Buggle, F.; Safer, A.; Becher, H.; Grau, A.J.; Pussinen, P. Biomarkers of periodontitis and inflammation in ischemic stroke: A case-control study. Innate Immun. 2014, 20, 511–518. [Google Scholar] [CrossRef]
- Demmer, R.T.; Jacobs, D.R., Jr.; Singh, R.; Zuk, A.; Rosenbaum, M.; Papapanou, P.N.; Desvarieux, M. Periodontal bacteria and prediabetes prevalence in ORIGINS: The oral infections, glucose intolerance, and insulin resistance study. J. Dent. Res. 2015, 94, 201–211. [Google Scholar] [CrossRef]
- Scher, J.U.; Ubeda, C.; Equinda, M.; Khanin, R.; Buischi, Y.; Viale, A.; Lipuma, L.; Attur, M.; Pillinger, M.H.; Weissmann, G.; et al. Periodontal disease and the oral microbiota in new-onset rheumatoid arthritis. Arthritis Rheum. 2012, 64, 3083–3094. [Google Scholar] [CrossRef]
- Eriksson, K.; Fei, G.; Lundmark, A.; Benchimol, D.; Lee, L.; Hu, Y.O.O.; Kats, A.; Saevarsdottir, S.; Catrina, A.I.; Klinge, B.; et al. Periodontal health and oral microbiota in patients with rheumatoid arthritis. J. Clin. Med. 2019, 8, 630. [Google Scholar] [CrossRef]
- Fan, X.; Alekseyenko, A.V.; Wu, J.; Peters, B.A.; Jacobs, E.J.; Gapstur, S.M.; Purdue, M.P.; Abnet, C.C.; Stolzenberg-Solomon, R.; Miller, G.; et al. Human oral microbiome and prospective risk for pancreatic cancer: A population-based nested case-control study. Gut 2018, 67, 120–127. [Google Scholar] [CrossRef]
- Han, Y.W. Fusobacterium nucleatum: A commensal-turned pathogen. Curr. Opin. Microbiol. 2015, 23, 141–147. [Google Scholar] [CrossRef]
- Abed, J.; Maalouf, N.; Parhi, L.; Chaushu, S.; Mandelboim, O.; Bachrach, G. Tumor targeting by Fusobacterium nucleatum: A pilot study and future perspectives. Front. Cell. Infect. Microbiol. 2017, 7, 295. [Google Scholar] [CrossRef] [PubMed]
- Gursoy, U.K.; Könönen, E.; Luukkonen, N.; Uitto, V.J. Human neutrophil defensins and their effect on epithelial cells. J. Periodontol. 2013, 84, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Benakanakere, M.; Kinane, D.F. Innate cellular responses to the periodontal biofilm. Front. Oral Biol. 2012, 15, 41–55. [Google Scholar] [PubMed]
- Cekici, A.; Kantarci, A.; Hasturk, H.; Van Dyke, T.E. Inflammatory and immune pathways in the pathogenesis of periodontal disease. Periodontol. 2000 2014, 64, 57–80. [Google Scholar] [CrossRef] [PubMed]
- Damgaard, C.; Holmstrup, P.; Van Dyke, T.E.; Nielsen, C.H. The complement system and its role in the pathogenesis of periodontitis: Current concepts. J. Periodontal Res. 2015, 50, 283–293. [Google Scholar] [CrossRef]
- Liu, J.; Du, X.; Chen, J.; Hu, L.; Chen, L. The induction expression of human β-defensins in gingival epithelial cells and fibroblasts. Arch. Oral Biol. 2013, 58, 1415–1421. [Google Scholar] [CrossRef]
- Kasnak, G.; Könönen, E.; Syrjänen, S.; Gürsoy, M.; Zeidán-Chuliá, F.; Firatli, E.; Gürsoy, U.K. NFE2L2/NRF2, OGG1, and cytokine responses of human gingival keratinocytes against oxidative insults of various origin. Mol. Cell. Biochem. 2019, 452, 63–70. [Google Scholar] [CrossRef]
- Song, B.; Zhang, Y.L.; Chen, L.J.; Zhou, T.; Huang, W.K.; Zhou, X.; Shao, L.Q. The role of Toll-like receptors in periodontitis. Oral Dis. 2017, 23, 168–180. [Google Scholar] [CrossRef]
- Elmanfi, S.; Zhou, J.; Sintim, H.O.; Könönen, E.; Gürsoy, M.; Gürsoy, U.K. Regulation of gingival epithelial cytokine response by bacterial cyclic dinucleotides. J. Oral Microbiol. 2018, 11, 1538927. [Google Scholar] [CrossRef]
- Fteita, D.; Könönen, E.; Gürsoy, M.; Ma, X.; Sintim, H.O.; Gürsoy, U.K. Quorum sensing molecules regulate epithelial cytokine response and biofilm-related virulence of three Prevotella species. Anaerobe 2018, 54, 128–135. [Google Scholar] [CrossRef]
- Hiroshima, Y.; Bando, M.; Kataoka, M.; Inagaki, Y.; Herzberg, M.C.; Ross, K.F.; Hosoi, K.; Nagata, T.; Kido, J. Regulation of antimicrobial peptide expression in human gingival keratinocytes by interleukin-1α. Arch. Oral Biol. 2011, 56, 761–767. [Google Scholar] [CrossRef] [PubMed]
- Jura, J.; Skalniak, L.; Koj, A. Monocyte chemotactic protein-1-induced protein-1 (MCPIP1) is a novel multifunctional modulator of inflammatory reactions. Biochim. Biophys. Acta 2012, 1823, 1905–1913. [Google Scholar] [CrossRef] [PubMed]
- Cavalla, F.; Hernández-Rios, P.; Sorsa, T.; Biguetti, C.; Hernández, M. Matrix metalloproteinases as regulators of periodontal inflammation. Int. J. Mol. Sci. 2017, 18, 440. [Google Scholar]
- Wilensky, A.; Segev, H.; Mizraji, G.; Shaul, Y.; Capucha, T.; Shacham, M.; Hovav, A.H. Dendritic cells and their role in periodontal disease. Oral Dis. 2014, 20, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Dong, G.; Guo, L.; Graves, D.T. The function of dendritic cells in modulating the host response. Mol. Oral Microbiol. 2018, 33, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.C.; Hughes, F.J.; Taams, L.S. The presence, function and regulation of IL-17 and Th17 cells in periodontitis. J. Clin. Periodontol. 2014, 41, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Alnaeeli, M.; Penninger, J.M.; Teng, Y.T. Immune interactions with CD4+ T cells promote the development of functional osteoclasts from murine CD11c+ dendritic cells. J. Immunol. 2006, 177, 3314–3326. [Google Scholar] [CrossRef] [PubMed]
- Parkos, C.A. Neutrophil-epithelial interactions: A double-edged sword. Am. J. Pathol. 2016, 186, 1404–1416. [Google Scholar] [CrossRef] [PubMed]
- Fine, N.; Hassanpour, S.; Borenstein, A.; Sima, C.; Oveisi, M.; Scholey, J.; Cherney, D.; Glogauer, M. Distinct oral neutrophil subsets define health and periodontal disease states. J. Dent. Res. 2016, 95, 931–938. [Google Scholar] [CrossRef] [PubMed]
- Hajishengallis, G.; Moutsopoulos, N.M.; Hajishengallis, E.; Chavakis, T. Immune and regulatory functions of neutrophils in inflammatory bone loss. Semin. Immunol. 2016, 28, 146–158. [Google Scholar] [CrossRef] [PubMed]
- Matthews, J.B.; Wright, H.J.; Roberts, A.; Ling-Mountford, N.; Cooper, P.R.; Chapple, I.L. Neutrophil hyper-responsiveness in periodontitis. J. Dent. Res. 2007, 86, 718–722. [Google Scholar] [CrossRef] [PubMed]
- Ling, M.R.; Chapple, I.L.; Matthews, J.B. Peripheral blood neutrophil cytokine hyper-reactivity in chronic periodontitis. Innate Immun. 2015, 21, 714–725. [Google Scholar] [CrossRef] [PubMed]
- Olsen, I.; Hajishengallis, G. Major neutrophil functions subverted by Porphyromonas gingivalis. J. Oral Microbiol. 2016, 8, 30936. [Google Scholar] [CrossRef] [PubMed]
- Sochalska, M.; Potempa, J. Manipulation of neutrophils by Porphyromonas gingivalis in the development of periodontitis. Front. Cell. Infect. Microbiol. 2017, 7, 197. [Google Scholar] [CrossRef] [PubMed]
- Davies, L.C.; Rosas, M.; Jenkins, S.J.; Liao, C.T.; Scurr, M.J.; Brombacher, F.; Fraser, D.J.; Allen, J.E.; Jones, S.A.; Taylor, P.R. Distinct bone marrow-derived and tissue-resident macrophage lineages proliferate at key stages during inflammation. Nat. Commun. 2013, 4, 1886. [Google Scholar] [CrossRef] [PubMed]
- Hajishengallis, G.; Sahingur, S.E. Novel inflammatory pathways in periodontitis. Adv. Dent. Res. 2014, 26, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Garlet, G.P.; Giannobile, W.V. Macrophages: The bridge between inflammation resolution and tissue repair? J. Dent. Res. 2018, 97, 1079–1081. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, G.; Shaik-Dasthagirisaheb, Y.B.; Huang, N.; Viglianti, G.A.; Henderson, A.J.; Kantarci, A.; Gibson, F.C. Immunologic environment influences macrophage response to Porphyromonas gingivalis. Mol. Oral Microbiol. 2017, 32, 250–261. [Google Scholar] [CrossRef] [PubMed]
- Dutzan, N.; Konkel, J.E.; Greenwell-Wild, T.; Moutsopoulos, N.M. Characterization of the human immune cell network at the gingival barrier. Mucosal Immunol. 2016, 9, 1163–1172. [Google Scholar] [CrossRef] [PubMed]
- Campbell, L.; Millhouse, E.; Malcolm, J.; Culshaw, S. T cells, teeth and tissue destruction - what do T cells do in periodontal disease? Mol. Oral Microbiol. 2016, 31, 445–456. [Google Scholar] [CrossRef] [PubMed]
- Loos, B.G.; Papantonopoulos, G.; Jepsen, S.; Laine, M.L. What is the contribution of genetics to periodontal risk? Dent. Clin. North Am. 2015, 59, 761–780. [Google Scholar] [CrossRef]
- Bosshardt, D.D.; Lang, N.P. The junctional epithelium: From health to disease. J Dent. Res. 2005, 84, 9–20. [Google Scholar] [CrossRef]
- Lang, N.P.; Schätzle, M.A.; Löe, H. Gingivitis as a risk factor in periodontal disease. J. Clin. Periodontol. 2009, 36 (Suppl. 10), 3–8. [Google Scholar] [CrossRef]
- Henderson, B.; Kaiser, F. Bacterial modulators of bone remodeling in the periodontal pocket. Periodontol. 2000 2018, 76, 97–108. [Google Scholar] [CrossRef]
- Sorsa, T.; Tjäderhane, L.; Konttinen, Y.T.; Lauhio, A.; Salo, T.; Lee, H.M.; Golub, L.M.; Brown, D.L.; Mäntylä, P. Matrix metalloproteinases: Contribution to pathogenesis, diagnosis and treatment of periodontal inflammation. Ann. Med. 2006, 38, 306–321. [Google Scholar] [CrossRef]
- Kurgan, S.; Kantarci, A. Molecular basis for immunohistochemical and inflammatory changes during progression of gingivitis to periodontitis. Periodontol. 2000 2018, 76, 51–67. [Google Scholar] [CrossRef]
- Belibasakis, G.N.; Bostanci, N. The RANKL-OPG system in clinical periodontology. J. Clin. Periodontol. 2012, 39, 239–248. [Google Scholar] [CrossRef]
- Gursoy, U.K.; Könönen, E.; Huumonen, S.; Tervahartiala, T.; Pussinen, P.J.; Suominen, A.L.; Sorsa, T. Salivary type I collagen degradation end-products and related matrix metalloproteinases in periodontitis. J. Clin. Periodontol. 2013, 40, 18–25. [Google Scholar] [CrossRef]
- Reynolds, M.A.; Kao, R.T.; Camargo, P.M.; Caton, J.G.; Clem, D.S.; Fiorellini, J.P.; Geisinger, M.L.; Mills, M.P.; Nares, S.; Nevins, M.L. Periodontal regeneration - intrabony defects: A consensus report from the AAP regeneration workshop. J. Periodontol. 2015, 86 (Suppl. 2), 105–107. [Google Scholar] [CrossRef]
- Bunaes, D.F.; Lie, S.A.; Enersen, M.; Aastrøm, A.N.; Mustafa, K.; Leknes, K.N. Site-specific treatment outcome in smokers following non-surgical and surgical periodontal therapy. J. Clin. Periodontol. 2015, 42, 933–942. [Google Scholar] [CrossRef]
- Ryder, M.I.; Couch, E.T.; Chaffee, B.W. Personalized periodontal treatment for the tobacco- and alcohol-using patient. Periodontol. 2000 2018, 78, 30–46. [Google Scholar] [CrossRef]
- Delima, S.L.; McBride, R.K.; Preshaw, P.M.; Heasman, P.A.; Kumar, P.S. Response of subgingival bacteria to smoking cessation. J. Clin. Microbiol. 2010, 48, 2344–2449. [Google Scholar] [CrossRef]
- Haffajee, A.D.; Teles, R.P.; Socransky, S.S. The effect of periodontal therapy on the composition of the subgingival microbiota. Periodontol 2006, 42, 219–258. [Google Scholar] [CrossRef]
- Axelsson, P.; Nyström, B.; Lindhe, J. The long-term effect of a plaque control program on tooth mortality, caries and periodontal disease in adults. Results after 30 years of maintenance. J. Clin. Periodontol. 2004, 31, 749–757. [Google Scholar] [CrossRef]
- Rosling, B.; Serino, G.; Hellström, M.K.; Socransky, S.S.; Lindhe, J. Longitudinal periodontal tissue alterations during supportive therapy. Findings from subjects with normal and high susceptibility to periodontal disease. J. Clin. Periodontol. 2001, 28, 241–249. [Google Scholar] [CrossRef]
- Müller, S.; Eickholz, P.; Reitmeir, P.; Eger, T. Long-term tooth loss in periodontally compromised but treated patients according to the type of prosthodontic treatment. A retrospective study. J. Oral Rehabil. 2013, 40, 358–367. [Google Scholar] [CrossRef]
- Paraskevas, S.; Huizinga, J.D.; Loos, B.G. A systematic review and meta-analyses on C-reactive protein in relation to periodontitis. J. Clin. Periodontol. 2008, 35, 277–290. [Google Scholar] [CrossRef]
- Tonetti, M.S.; D’Aiuto, F.; Nibali, L.; Donald, A.; Storry, C.; Parkar, M.; Suvan, J.; Hingorani, A.D.; Vallance, P.; Deanfield, J. Treatment of periodontitis and endothelial function. N. Engl. J. Med. 2007, 356, 911–920. [Google Scholar] [CrossRef]
- Teeuw, W.J.; Slot, D.E.; Susanto, H.; Gerdes, V.E.; Abbas, F.; D’Aiuto, F.; Kastelein, J.J.; Loos, B.G.J. Treatment of periodontitis improves the atherosclerotic profile: A systematic review and meta-analysis. J. Clin. Periodontol. 2014, 41, 70–79. [Google Scholar] [CrossRef]
- Wang, X.; Han, X.; Guo, X.; Luo, X.; Wang, D. The effect of periodontal treatment on hemoglobin A1c levels of diabetic patients: A systematic review and meta-analysis. PLoS ONE 2014, 9, e108412. [Google Scholar] [CrossRef]
- D’Aiuto, F.; Gkranias, N.; Bhowruth, D.; Khan, T.; Orlandi, M.; Suvan, J.; Masi, S.; Tsakos, G.; Hurel, S.; Hingorani, A.D.; et al. Systemic effects of periodontitis treatment in patients with type 2 diabetes: A 12 month, single-centre, investigator-masked, randomised trial. Lancet Diabetes Endocrinol. 2018, 6, 954–965. [Google Scholar]
- Morand, D.N.; Davideau, J.L.; Clauss, F.; Jessel, N.; Tenenbaum, H.; Huck, O. Cytokines during periodontal wound healing: Potential application for new therapeutic approach. Oral Dis. 2017, 23, 300–311. [Google Scholar] [CrossRef]
- Alvarez, C.; Rojas, C.; Rojas, L.; Cafferata, E.A.; Monasterio, G.; Vernal, R. Regulatory T lymphocytes in periodontitis: A translational view. Mediators Inflamm. 2018, 2018, 7806912. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Könönen, E.; Gursoy, M.; Gursoy, U.K. Periodontitis: A Multifaceted Disease of Tooth-Supporting Tissues. J. Clin. Med. 2019, 8, 1135. https://doi.org/10.3390/jcm8081135
Könönen E, Gursoy M, Gursoy UK. Periodontitis: A Multifaceted Disease of Tooth-Supporting Tissues. Journal of Clinical Medicine. 2019; 8(8):1135. https://doi.org/10.3390/jcm8081135
Chicago/Turabian StyleKönönen, Eija, Mervi Gursoy, and Ulvi Kahraman Gursoy. 2019. "Periodontitis: A Multifaceted Disease of Tooth-Supporting Tissues" Journal of Clinical Medicine 8, no. 8: 1135. https://doi.org/10.3390/jcm8081135
APA StyleKönönen, E., Gursoy, M., & Gursoy, U. K. (2019). Periodontitis: A Multifaceted Disease of Tooth-Supporting Tissues. Journal of Clinical Medicine, 8(8), 1135. https://doi.org/10.3390/jcm8081135