Impact of Oxidative Stress and Protein S-Glutathionylation in Aortic Valve Sclerosis Patients with Overt Atherosclerosis

 ,
,
Abstract
1. Introduction
2. Patients and Methods
2.1. Patient Population
2.2. Echocardiographic Evaluation
2.3. Blood Sampling and Biochemical Measurements
2.4. Glutathione Measurements
2.5. Asymmetric Dimethylarginine Measurements
2.6. Dot-Blot, Western Blot and Immunohistochemistry
2.7. In Vitro Model of Protein S-Glutathionylation
2.8. Immunoprecipitation
2.9. Reverse Transcription and Real-Time PCR
2.10. Statistical Analysis
3. Results
3.1. Patient Characteristics
3.2. Oxidative Stress and Endothelial Dysfunction
3.3. Systemic Inflammation Status
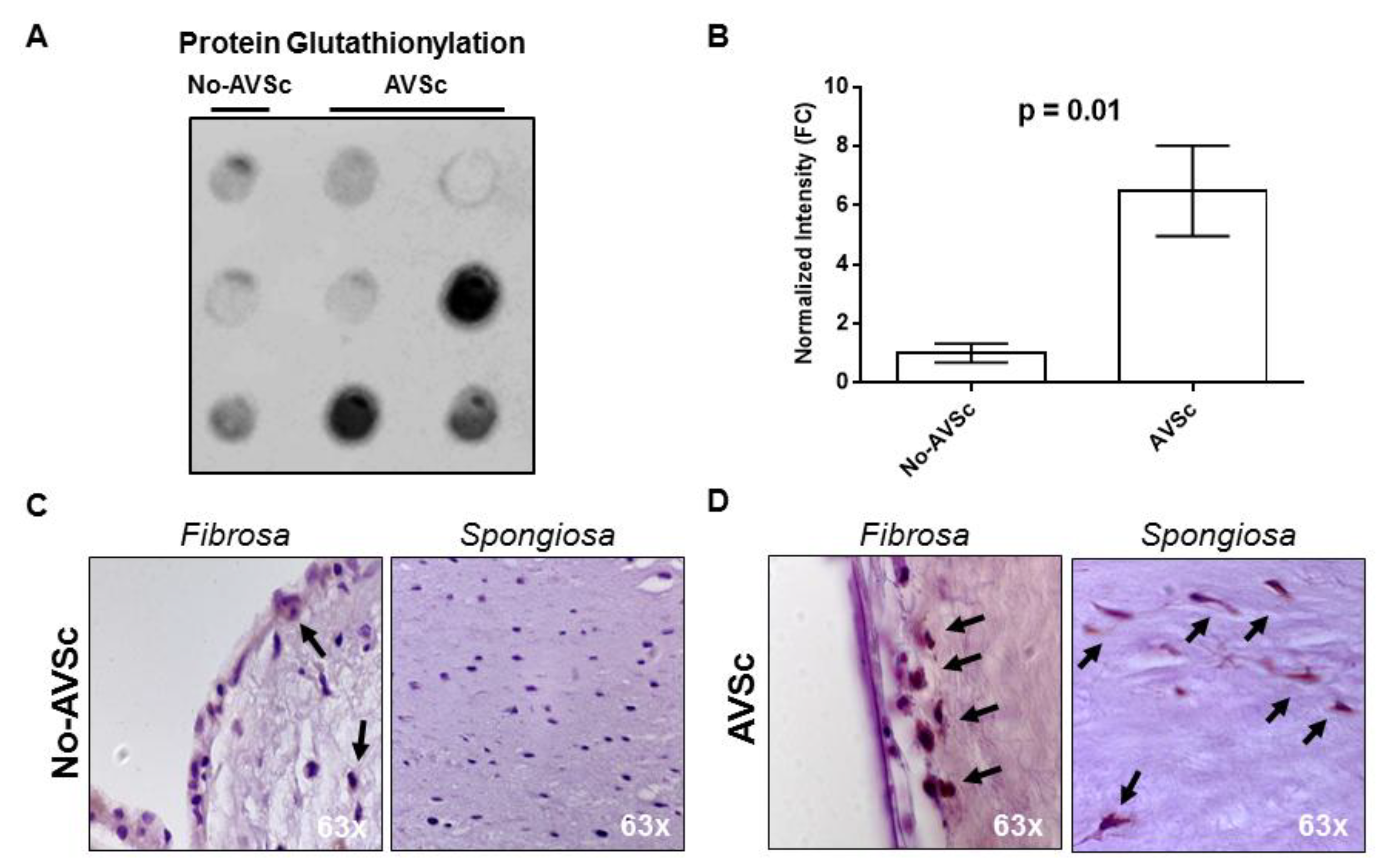
3.4. Aortic Valve Protein S-Glutathionylation
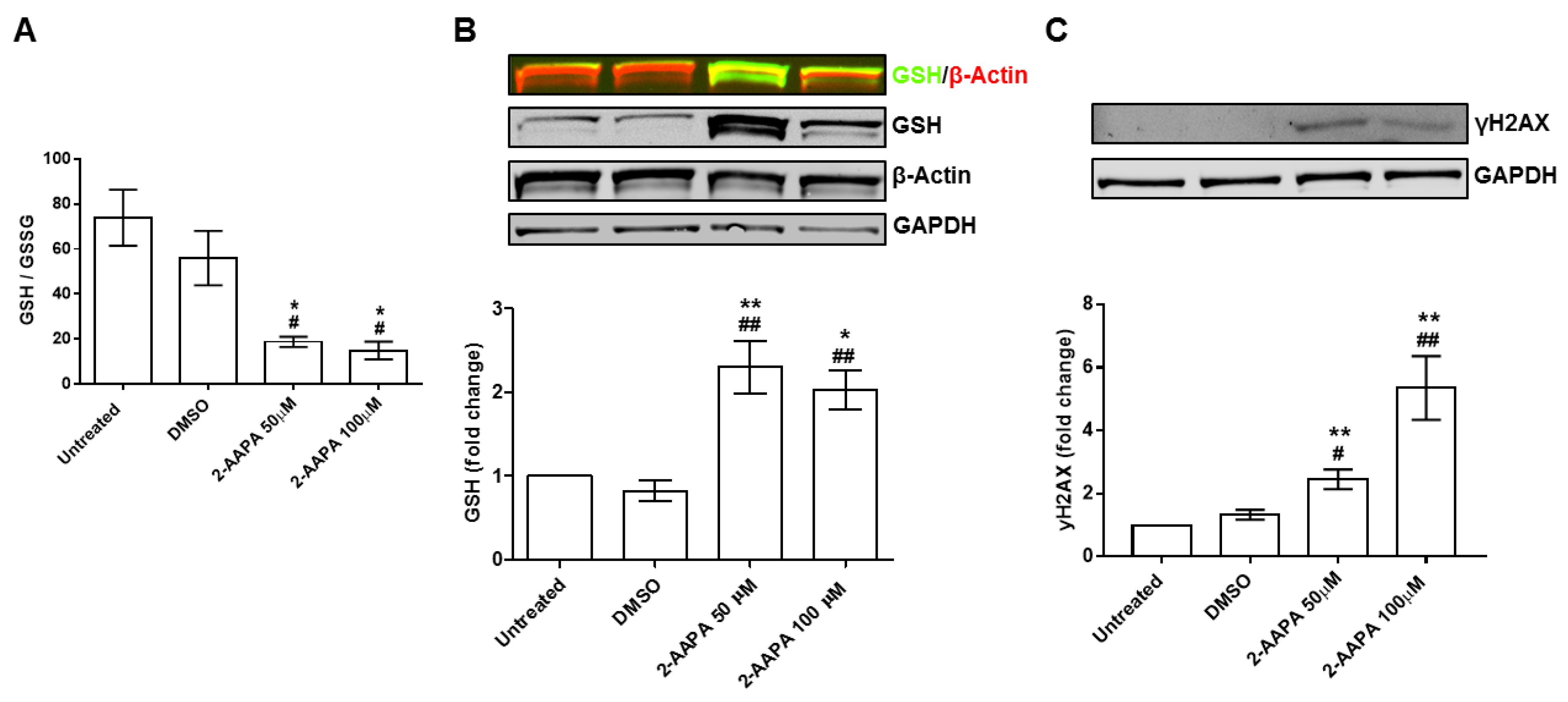
3.5. In Vitro Model of Protein S-Glutathionylation
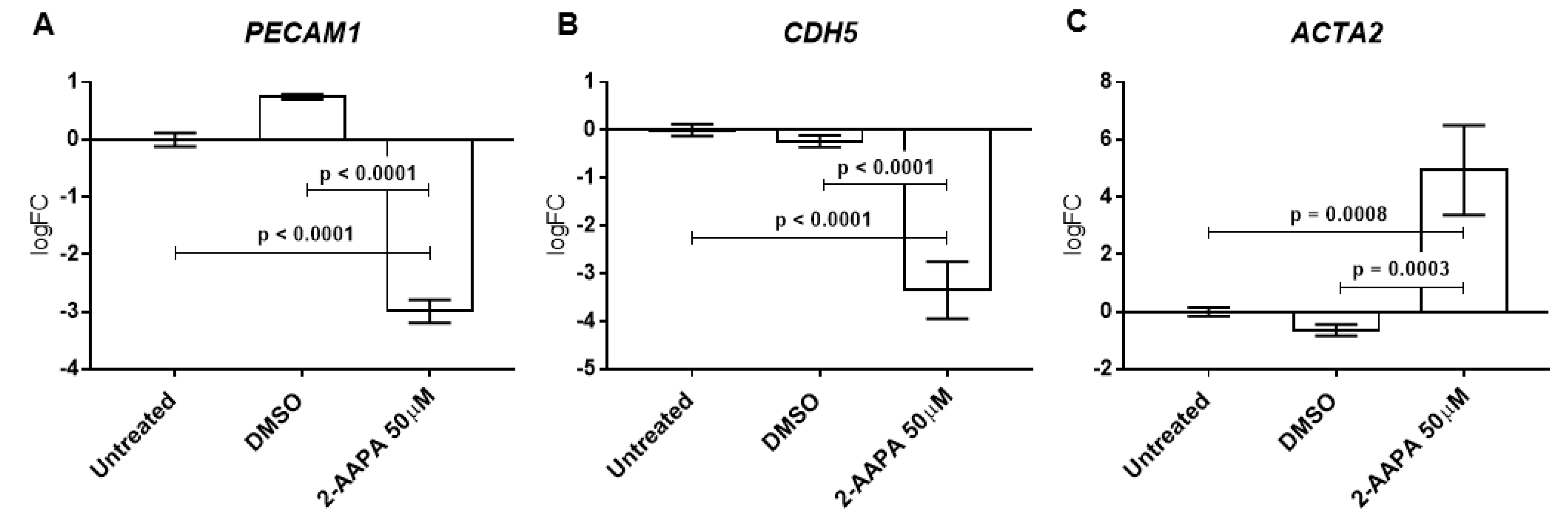
3.6. Protein S-Glutathionylation and Endothelial-to-Mesenchymal Transition
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rajamannan, N.M.; Evans, F.J.; Aikawa, E.; Grande-Allen, K.J.; Demer, L.L.; Heistad, D.D.; Simmons, C.A.; Masters, K.S.; Mathieu, P.; O’Brien, K.D.; et al. Calcific aortic valve disease: Not simply a degenerative process: A review and agenda for research from the National Heart and Lung and Blood Institute Aortic stenosis working group. Executive summary: Calcific aortic valve disease-2011 update. Circulation 2011, 124, 1783–1791. [Google Scholar] [CrossRef] [PubMed]
- Freeman, R.V.; Otto, C.M. Spectrum of calcific aortic valve disease: Pathogenesis, disease progression and treatment strategies. Circulation 2005, 111, 3316–3326. [Google Scholar] [CrossRef]
- Faggiano, P.; Antonini-Canterin, F.; Erlicher, A.; Romeo, C.; Cervesato, E.; Pavan, D.; Piazza, R.; Huang, G.; Nicolosi, G.L. Progression of Aortic valve sclerosis to Aortic stenosis. Am. J. Cardiol. 2003, 91, 99–101. [Google Scholar] [CrossRef]
- Coffey, S.; Cox, B.; Williams, M.J. The prevalence, incidence, progression and risks of Aortic valve sclerosis: A systematic review and meta-analysis. J. Am. Coll. Cardiol. 2014, 63, 2852–2861. [Google Scholar] [CrossRef]
- Beckmann, E.; Grau, J.B.; Sainger, R.; Poggio, P.; Ferrari, G. Insights into the use of biomarkers in calcific aortic valve disease. J. Heart Valve Dis. 2010, 19, 441–452. [Google Scholar]
- Di Minno, M.N.D.; Di Minno, A.; Ambrosino, P.; Songia, P.; Pepi, M.; Tremoli, E.; Poggio, P. Cardiovascular morbidity and mortality in patients with aortic valve sclerosis: A systematic review and meta-analysis. Int. J. Cardiol. 2018, 260, 138–144. [Google Scholar] [CrossRef]
- Di Minno, M.N.; Di Minno, A.; Songia, P.; Ambrosino, P.; Gripari, P.; Ravani, A.; Pepi, M.; Rubba, P.O.; Medda, E.; Tremoli, E.; et al. Markers of subclinical atherosclerosis in patients with aortic valve sclerosis: A meta-analysis of literature studies. Int. J. Cardiol. 2016, 223, 364–370. [Google Scholar] [CrossRef] [PubMed]
- Espinosa-Diez, C.; Miguel, V.; Vallejo, S.; Sanchez, F.J.; Sandoval, E.; Blanco, E.; Cannata, P.; Peiro, C.; Sanchez-Ferrer, C.F.; Lamas, S. Role of glutathione biosynthesis in endothelial dysfunction and fibrosis. Redox Biol. 2018, 14, 88–99. [Google Scholar] [CrossRef]
- Higashi, Y.; Maruhashi, T.; Noma, K.; Kihara, Y. Oxidative stress and endothelial dysfunction: Clinical evidence and therapeutic implications. Trends Cardiovasc. Med. 2014, 24, 165–169. [Google Scholar] [CrossRef] [PubMed]
- Asensi, M.; Sastre, J.; Pallardo, F.V.; Lloret, A.; Lehner, M.; Garcia-de-la Asuncion, J.; Vina, J. Ratio of reduced to oxidized glutathione as indicator of oxidative stress status and DNA damage. Methods Enzymol. 1999, 299, 267–276. [Google Scholar]
- Leopold, J.A.; Loscalzo, J. Oxidative enzymopathies and vascular disease. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1332–1340. [Google Scholar] [CrossRef]
- Emdin, M.; Pompella, A.; Paolicchi, A. Gamma-glutamyltransferase, atherosclerosis and cardiovascular disease: Triggering oxidative stress within the plaque. Circulation 2005, 112, 2078–2080. [Google Scholar] [CrossRef]
- Musthafa, Q.A.; Abdul Shukor, M.F.; Ismail, N.A.S.; Mohd Ghazi, A.; Mohd Ali, R.; IF, M.N.; Dimon, M.Z.; Wan Ngah, W.Z. Oxidative status and reduced glutathione levels in premature coronary artery disease and coronary artery disease. Free Radic. Res. 2017, 51, 787–798. [Google Scholar] [CrossRef]
- Widder, J.D.; Guzik, T.J.; Mueller, C.F.; Clempus, R.E.; Schmidt, H.H.; Dikalov, S.I.; Griendling, K.K.; Jones, D.P.; Harrison, D.G. Role of the multidrug resistance protein-1 in hypertension and vascular dysfunction caused by angiotensin II. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 762–768. [Google Scholar] [CrossRef]
- Xiong, Y.; Uys, J.D.; Tew, K.D.; Townsend, D.M. S-glutathionylation: From molecular mechanisms to health outcomes. Antioxid. Redox Signal. 2011, 15, 233–270. [Google Scholar] [CrossRef]
- Popov, D. Protein S-glutathionylation: From current basics to targeted modifications. Arch. Physiol. Biochem. 2014, 120, 123–130. [Google Scholar] [CrossRef]
- Ghezzi, P. Protein glutathionylation in health and disease. Biochim. Biophys. Acta 2013, 1830, 3165–3172. [Google Scholar] [CrossRef]
- Pastore, A.; Piemonte, F. Protein glutathionylation in cardiovascular diseases. Int. J. Mol. Sci. 2013, 14, 20845–20876. [Google Scholar] [CrossRef]
- Otto, C.M.; Lind, B.K.; Kitzman, D.W.; Gersh, B.J.; Siscovick, D.S. Association of aortic-valve sclerosis with cardiovascular mortality and morbidity in the elderly. N. Engl. J. Med. 1999, 341, 142–147. [Google Scholar] [CrossRef]
- Myasoedova, V.A.; Ravani, A.L.; Frigerio, B.; Valerio, V.; Moschetta, D.; Songia, P.; Poggio, P. Novel pharmacological targets for calcific aortic valve disease: Prevention and treatments. Pharmacol. Res. 2018, 136, 74–82. [Google Scholar] [CrossRef]
- Kim, D.B.; Jung, H.O.; Jeon, D.S.; Park, C.S.; Jang, S.W.; Park, H.J.; Kim, P.J.; Baek, S.H.; Seung, K.B.; Rho, T.H.; et al. Aortic valve sclerosis on echocardiography is a good predictor of coronary artery disease in patients with an inconclusive treadmill exercise test. Korean Circ. J. 2009, 39, 275–279. [Google Scholar] [CrossRef]
- Sui, S.J.; Ren, M.Y.; Xu, F.Y.; Zhang, Y. A high association of aortic valve sclerosis detected by transthoracic echocardiography with coronary arteriosclerosis. Cardiology 2007, 108, 322–330. [Google Scholar] [CrossRef]
- Roy, G.C.; Rahman, F.; Hoque, M.H.; Habib, M.A.; Banerjee, S.K.; Siddique, M.A.; Barua, U.K.; Hossain, A.S.; Bhuiyan, G.R.; Haider, M.S. Aortic valve sclerosis is an indicator of coronary artery diseases. Mymensingh Med. J. MMJ 2012, 21, 226–232. [Google Scholar]
- Shah, S.J.; Ristow, B.; Ali, S.; Na, B.Y.; Schiller, N.B.; Whooley, M.A. Acute myocardial infarction in patients with versus without aortic valve sclerosis and effect of statin therapy (from the Heart and Soul Study). Am. J. Cardiol. 2007, 99, 1128–1133. [Google Scholar] [CrossRef][Green Version]
- Owens, D.S.; Budoff, M.J.; Katz, R.; Takasu, J.; Shavelle, D.M.; Carr, J.J.; Heckbert, S.R.; Otto, C.M.; Probstfield, J.L.; Kronmal, R.A.; et al. Aortic valve calcium independently predicts coronary and cardiovascular events in a primary prevention population. JACC. Cardiovasc. Imaging 2012, 5, 619–625. [Google Scholar] [CrossRef]
- Chandra, H.R.; Goldstein, J.A.; Choudhary, N.; O’Neill, C.S.; George, P.B.; Gangasani, S.R.; Cronin, L.; Marcovitz, P.A.; Hauser, A.M.; O’Neill, W.W. Adverse outcome in Aortic sclerosis is associated with coronary artery disease and inflammation. J. Am. Coll. Cardiol. 2004, 43, 169–175. [Google Scholar] [CrossRef]
- Mazzone, A.; Venneri, L.; Berti, S. Aortic valve stenosis and coronary artery disease: Pathophysiological and clinical links. J. Cardiovasc. Med. (Hagerstown) 2007, 8, 983–989. [Google Scholar] [CrossRef]
- Soydinc, S.; Davutoglu, V.; Dundar, A.; Aksoy, M. Relationship between Aortic valve sclerosis and the extent of coronary artery disease in patients undergoing diagnostic coronary angiography. Cardiology 2006, 106, 277–282. [Google Scholar] [CrossRef]
- Milin, A.C.; Vorobiof, G.; Aksoy, O.; Ardehali, R. Insights into Aortic sclerosis and its relationship with coronary artery disease. J. Am. Heart Assoc. 2014, 3, e001111. [Google Scholar] [CrossRef]
- Poggianti, E.; Venneri, L.; Chubuchny, V.; Jambrik, Z.; Baroncini, L.A.; Picano, E. Aortic valve sclerosis is associated with systemic endothelial dysfunction. J. Am. Coll. Cardiol. 2003, 41, 136–141. [Google Scholar] [CrossRef]
- Erdogan, T.; Cetin, M.; Kocaman, S.A.; Durakoglugil, M.E.; Ergul, E.; Canga, A. Aortic valve sclerosis is a high predictive marker of systemic endothelial dysfunction in hypertensive patients. Herz 2013, 38, 915–921. [Google Scholar] [CrossRef] [PubMed]
- Sverdlov, A.L.; Ngo, D.T.; Chan, W.P.; Chirkov, Y.Y.; Gersh, B.J.; McNeil, J.J.; Horowitz, J.D. Determinants of aortic sclerosis progression: Implications regarding impairment of nitric oxide signalling and potential therapeutics. Eur. Heart J. 2012, 33, 2419–2425. [Google Scholar] [CrossRef]
- Cavalca, V.; Tremoli, E.; Porro, B.; Veglia, F.; Myasoedova, V.; Squellerio, I.; Manzone, D.; Zanobini, M.; Trezzi, M.; Di Minno, M.N.; et al. Oxidative stress and nitric oxide pathway in adult patients who are candidates for cardiac surgery: Patterns and differences. Interac. Cardiovasc. Thorac. Surg. 2013, 17, 923–930. [Google Scholar] [CrossRef]
- Farrar, E.J.; Huntley, G.D.; Butcher, J. Endothelial-derived oxidative stress drives myofibroblastic activation and calcification of the aortic valve. PLoS ONE 2015, 10, e0123257. [Google Scholar] [CrossRef]
- Ngo, D.T.; Heresztyn, T.; Mishra, K.; Marwick, T.H.; Horowitz, J.D. Aortic stenosis is associated with elevated plasma levels of asymmetric dimethylarginine (ADMA). Nitric Oxide Biol. Chem. 2007, 16, 197–201. [Google Scholar] [CrossRef]
- Miller, J.D.; Chu, Y.; Brooks, R.M.; Richenbacher, W.E.; Pena-Silva, R.; Heistad, D.D. Dysregulation of antioxidant mechanisms contributes to increased oxidative stress in calcific Aortic valvular stenosis in humans. J. Am. Coll. Cardiol. 2008, 52, 843–850. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.A.; Wang, T.Y.; Varadharaj, S.; Reyes, L.A.; Hemann, C.; Talukder, M.A.; Chen, Y.R.; Druhan, L.J.; Zweier, J.L. S-glutathionylation uncouples eNOS and regulates its cellular and vascular function. Nature 2010, 468, 1115–1118. [Google Scholar] [CrossRef] [PubMed]
- Hjortnaes, J.; Shapero, K.; Goettsch, C.; Hutcheson, J.D.; Keegan, J.; Kluin, J.; Mayer, J.E.; Bischoff, J.; Aikawa, E. Valvular interstitial cells suppress calcification of valvular endothelial cells. Atherosclerosis 2015, 242, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Richards, J.; El-Hamamsy, I.; Chen, S.; Sarang, Z.; Sarathchandra, P.; Yacoub, M.H.; Chester, A.H.; Butcher, J.T. Side-specific endothelial-dependent regulation of aortic valve calcification: Interplay of hemodynamics and nitric oxide signaling. Am. J. Pathol. 2013, 182, 1922–1931. [Google Scholar] [CrossRef] [PubMed]
- Dahal, S.; Huang, P.; Murray, B.T.; Mahler, G.J. Endothelial to mesenchymal transformation is induced by altered extracellular matrix in aortic valve endothelial cells. J. Biomed. Mater. Res. Part A 2017, 105, 2729–2741. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Duffhues, G.; Garcia de Vinuesa, A.; Ten Dijke, P. Endothelial-to-mesenchymal transition in cardiovascular diseases: Developmental signaling pathways gone awry. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 2018, 247, 492–508. [Google Scholar] [CrossRef]
- Souilhol, C.; Harmsen, M.C.; Evans, P.C.; Krenning, G. Endothelial-mesenchymal transition in atherosclerosis. Cardiovasc. Res. 2018, 114, 565–577. [Google Scholar] [CrossRef]
- Zhao, Y.; Seefeldt, T.; Chen, W.; Carlson, L.; Stoebner, A.; Hanson, S.; Foll, R.; Matthees, D.P.; Palakurthi, S.; Guan, X. Increase in thiol oxidative stress via glutathione reductase inhibition as a novel approach to enhance cancer sensitivity to X-ray irradiation. Free Radic. Biol. Med. 2009, 47, 176–183. [Google Scholar] [CrossRef]
- de Souza, L.F.; Schmitz, A.E.; da Silva, L.C.S.; de Oliveira, K.A.; Nedel, C.B.; Tasca, C.I.; de Bem, A.F.; Farina, M.; Dafre, A.L. Inhibition of reductase systems by 2-AAPA modulates peroxiredoxin oxidation and mitochondrial function in A172 glioblastoma cells. Toxicol. In Vitro Int. J. Publ. Assoc. BIBRA 2017, 42, 273–280. [Google Scholar] [CrossRef]
- Seefeldt, T.; Zhao, Y.; Chen, W.; Raza, A.S.; Carlson, L.; Herman, J.; Stoebner, A.; Hanson, S.; Foll, R.; Guan, X. Characterization of a novel dithiocarbamate glutathione reductase inhibitor and its use as a tool to modulate intracellular glutathione. J. Biol. Chem. 2009, 284, 2729–2737. [Google Scholar] [CrossRef] [PubMed]
- Lindman, B.R.; Clavel, M.A.; Mathieu, P.; Iung, B.; Lancellotti, P.; Otto, C.M.; Pibarot, P. Calcific aortic stenosis. Nat. Rev. Dis. Primers 2016, 2, 16006. [Google Scholar] [CrossRef] [PubMed]
- Mazzone, A.; Epistolato, M.C.; De Caterina, R.; Storti, S.; Vittorini, S.; Sbrana, S.; Gianetti, J.; Bevilacqua, S.; Glauber, M.; Biagini, A.; et al. Neoangiogenesis, T-lymphocyte infiltration and heat shock protein-60 are biological hallmarks of an immunomediated inflammatory process in end-stage calcified aortic valve stenosis. J. Am. Coll. Cardiol. 2004, 43, 1670–1676. [Google Scholar] [CrossRef] [PubMed]
- Gunduz, H.; Akdemir, R.; Binak, E.; Tamer, A.; Keser, N.; Uyan, C. Can serum lipid and CRP levels predict the “severity” of Aortic valve stenosis? Acta Cardiol. 2003, 58, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Galante, A.; Pietroiusti, A.; Vellini, M.; Piccolo, P.; Possati, G.; De Bonis, M.; Grillo, R.L.; Fontana, C.; Favalli, C. C-reactive protein is increased in patients with degenerative aortic valvular stenosis. J. Am. Coll. Cardiol. 2001, 38, 1078–1082. [Google Scholar] [CrossRef]
- Skowasch, D.; Schrempf, S.; Preusse, C.J.; Likungu, J.A.; Welz, A.; Luderitz, B.; Bauriedel, G. Tissue resident C reactive protein in degenerative aortic valves: Correlation with serum C reactive protein concentrations and modification by statins. Heart 2006, 92, 495–498. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, P.L.; Mazzone, A.M. C-reactive protein in aortic valve disease. Cardiovascular ultrasound 2006, 4, 37. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jeevanantham, V.; Singh, N.; Izuora, K.; D’Souza, J.P.; Hsi, D.H. Correlation of high sensitivity C-reactive protein and calcific aortic valve disease. Mayo Clin. Proc. 2007, 82, 171–174. [Google Scholar] [CrossRef]
- Novaro, G.M.; Katz, R.; Aviles, R.J.; Gottdiener, J.S.; Cushman, M.; Psaty, B.M.; Otto, C.M.; Griffin, B.P. Clinical factors but not C-reactive protein, predict progression of calcific aortic-valve disease: The Cardiovascular Health Study. J. Am. Coll. Cardiol. 2007, 50, 1992–1998. [Google Scholar] [CrossRef] [PubMed]
- Sverdlov, A.L.; Ngo, D.T.; Horowitz, J.D. Pathogenesis of Aortic sclerosis: Association with low BMI, tissue nitric oxide resistance but not systemic inflammatory activation. Am. J. Cardiovasc. Dis. 2012, 2, 43–49. [Google Scholar] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | CABG (n = 29) | CABG + AVSc (n = 29) | p Value |
|---|---|---|---|
| Age, years | 62.2 ± 6.2 | 65.2 ± 8.4 | 0.133 |
| Male sex, n (%) | 29 (100) | 29 (100) | 1.000 |
| Diabetes, n (%) | 7 (24) | 5 (17) | 0.525 |
| Hypertension, n (%) | 17 (59) | 22 (76) | 0.168 |
| Dyslipidemia, n (%) | 22 (76) | 19 (65.5) | 0.396 |
| Current Smoking, n (%) | 3 (10) | 7 (24) | 0.171 |
| Ex-Smokers, n (%) | 15 (52) | 13 (45) | 0.607 |
| Body mass index, kg/m2 | 26.7 ± 2.9 | 27.8 ± 3.6 | 0.156 |
| Creatinine, mg/dL | 0.91 ± 0.12 | 0.94 ± 0.17 | 0.411 |
| C-reactive protein, mg/L | 2.61 ± 2.56 | 2.73 ± 2.14 | 0.853 |
| New York Heart Association (NYHA) class | |||
| I | 10 (34) | 11 (38) | 1.000 |
| II | 16 (56) | 12 (41) | 0.593 |
| III | 3 (10) | 6 (21) | 0.470 |
| IV | - | - | - |
| 3-Vessels coronary disease, n (%) | 20 (69) | 19 (65.5) | 0.784 |
| Logistic EuroSCORE | 1.93 ± 1.79 | 2.68 ± 2.14 | 0.160 |
| Echocardiography | |||
| Left ventricle ejection fraction, n (%) | 61.3 ± 10.1 | 57.9 ± 10.1 | 0.210 |
| LV hypertrophy index, mm | 0.35 ± 0.13 | 0.41 ± 0.12 | 0.134 |
| Max. aortic velocity, m/s | 0.99 ± 0.54 | 1.23 ± 0.59 | 0.100 |
| Max. aortic gradient, mmHg | 5.14 ± 3.16 | 7.55 ± 6.79 | 0.090 |
| Therapies | |||
| Antiplatelets, n (%) | 21 (72) | 18 (62) | 0.410 |
| Angiotensin receptor blockers, n (%) | 5 (17) | 6 (21) | 0.743 |
| Converting enzyme inhibitors, n (%) | 8 (28) | 11 (38) | 0.410 |
| Calcium channel blockers, n (%) | 9 (31) | 9 (31) | 1.000 |
| Beta-blockers, n (%) | 19 (65.5) | 19 (65.5) | 1.000 |
| Nitrates, n (%) | 6 (21) | 10 (34.5) | 0.248 |
| Statins, n (%) | 18 (62) | 19 (65.5) | 0.789 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valerio, V.; Myasoedova, V.A.; Moschetta, D.; Porro, B.; Perrucci, G.L.; Cavalca, V.; Cavallotti, L.; Songia, P.; Poggio, P. Impact of Oxidative Stress and Protein S-Glutathionylation in Aortic Valve Sclerosis Patients with Overt Atherosclerosis. J. Clin. Med. 2019, 8, 552. https://doi.org/10.3390/jcm8040552
Valerio V, Myasoedova VA, Moschetta D, Porro B, Perrucci GL, Cavalca V, Cavallotti L, Songia P, Poggio P. Impact of Oxidative Stress and Protein S-Glutathionylation in Aortic Valve Sclerosis Patients with Overt Atherosclerosis. Journal of Clinical Medicine. 2019; 8(4):552. https://doi.org/10.3390/jcm8040552
Chicago/Turabian StyleValerio, Vincenza, Veronika A. Myasoedova, Donato Moschetta, Benedetta Porro, Gianluca L. Perrucci, Viviana Cavalca, Laura Cavallotti, Paola Songia, and Paolo Poggio. 2019. "Impact of Oxidative Stress and Protein S-Glutathionylation in Aortic Valve Sclerosis Patients with Overt Atherosclerosis" Journal of Clinical Medicine 8, no. 4: 552. https://doi.org/10.3390/jcm8040552
APA StyleValerio, V., Myasoedova, V. A., Moschetta, D., Porro, B., Perrucci, G. L., Cavalca, V., Cavallotti, L., Songia, P., & Poggio, P. (2019). Impact of Oxidative Stress and Protein S-Glutathionylation in Aortic Valve Sclerosis Patients with Overt Atherosclerosis. Journal of Clinical Medicine, 8(4), 552. https://doi.org/10.3390/jcm8040552