Curcumin and o-Vanillin Exhibit Evidence of Senolytic Activity in Human IVD Cells In Vitro



Abstract
1. Introduction
2. Experimental Section
2.1. Study Approval and Tissue Collection
2.2. In Vitro Disc Cell Cultures
2.3. SA-β-Gal Staining
2.4. Immunochemistry
2.5. Metabolic Activity
2.6. Caspase 3/7 Activity Assay
2.7. RT-qPCR
2.8. ELISA
2.9. DMMB
2.10. Western Blot Analysis
2.11. Statistical Analysis
3. Results
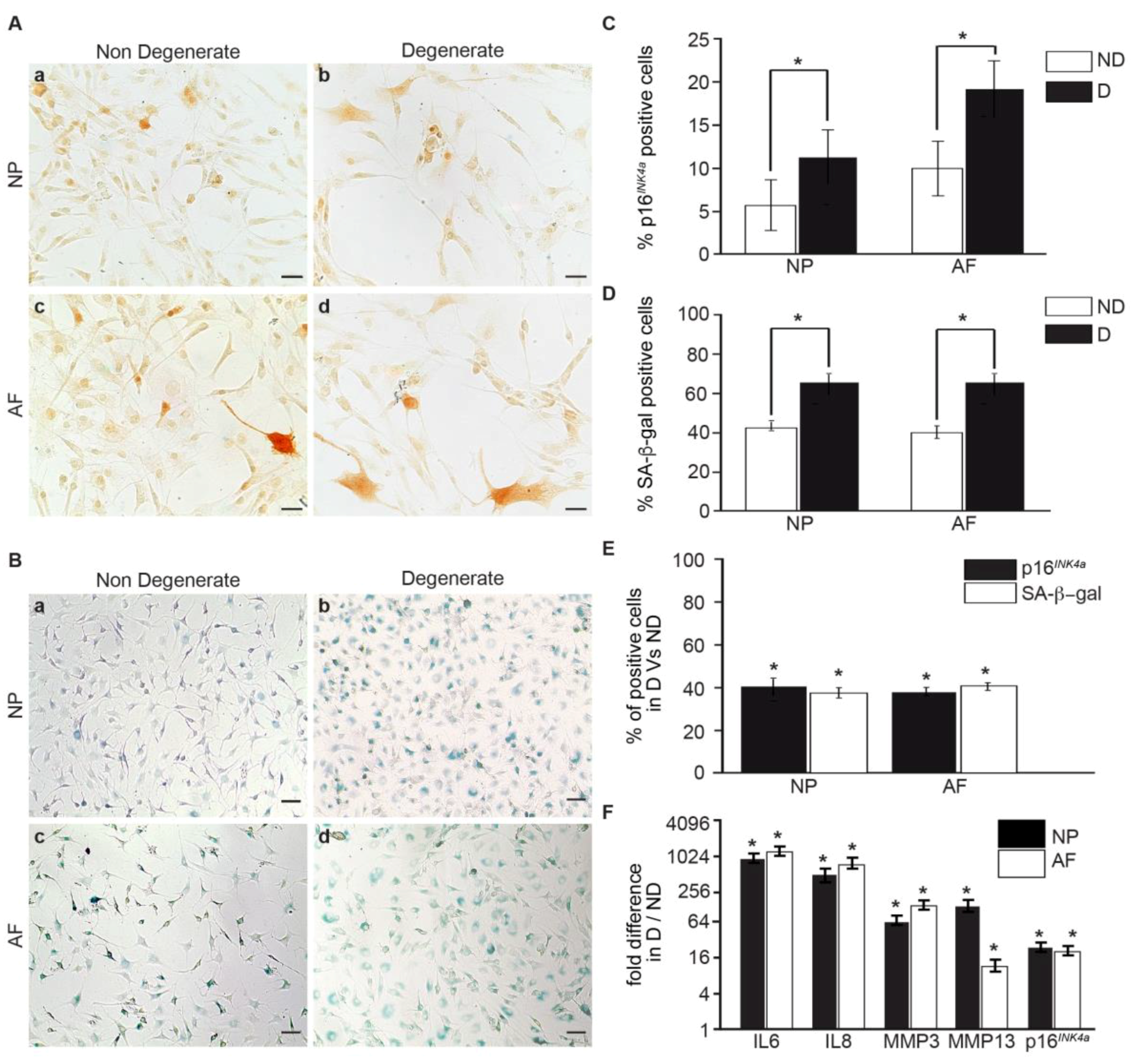
3.1. Senescent Human IVD Cells Increase with the Degree of Degeneration
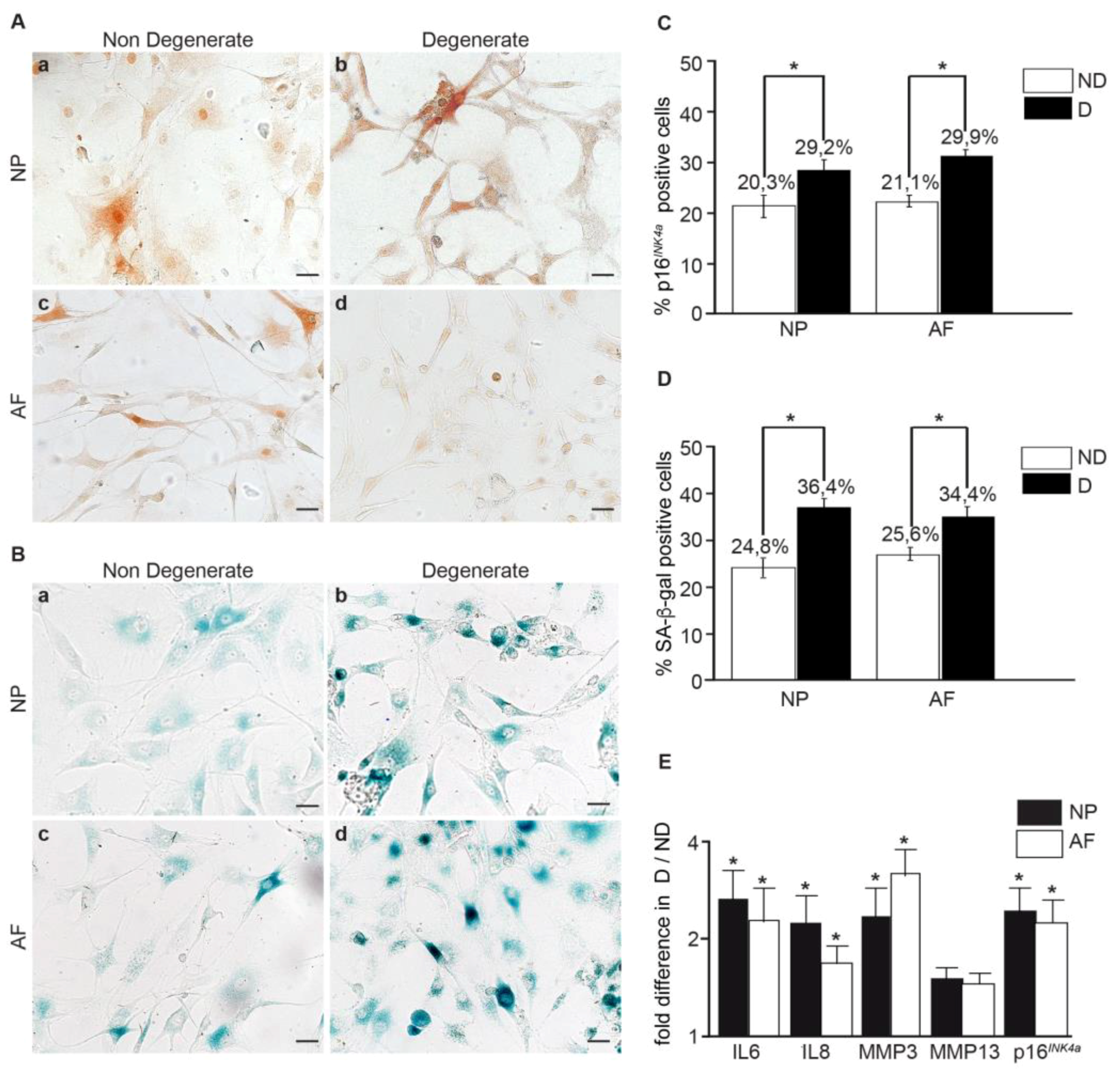
3.2. Within the Same Individual, Degenerate Discs Have Higher Numbers of Senescent Cells than Non-Mildly-Degenerate Tissues
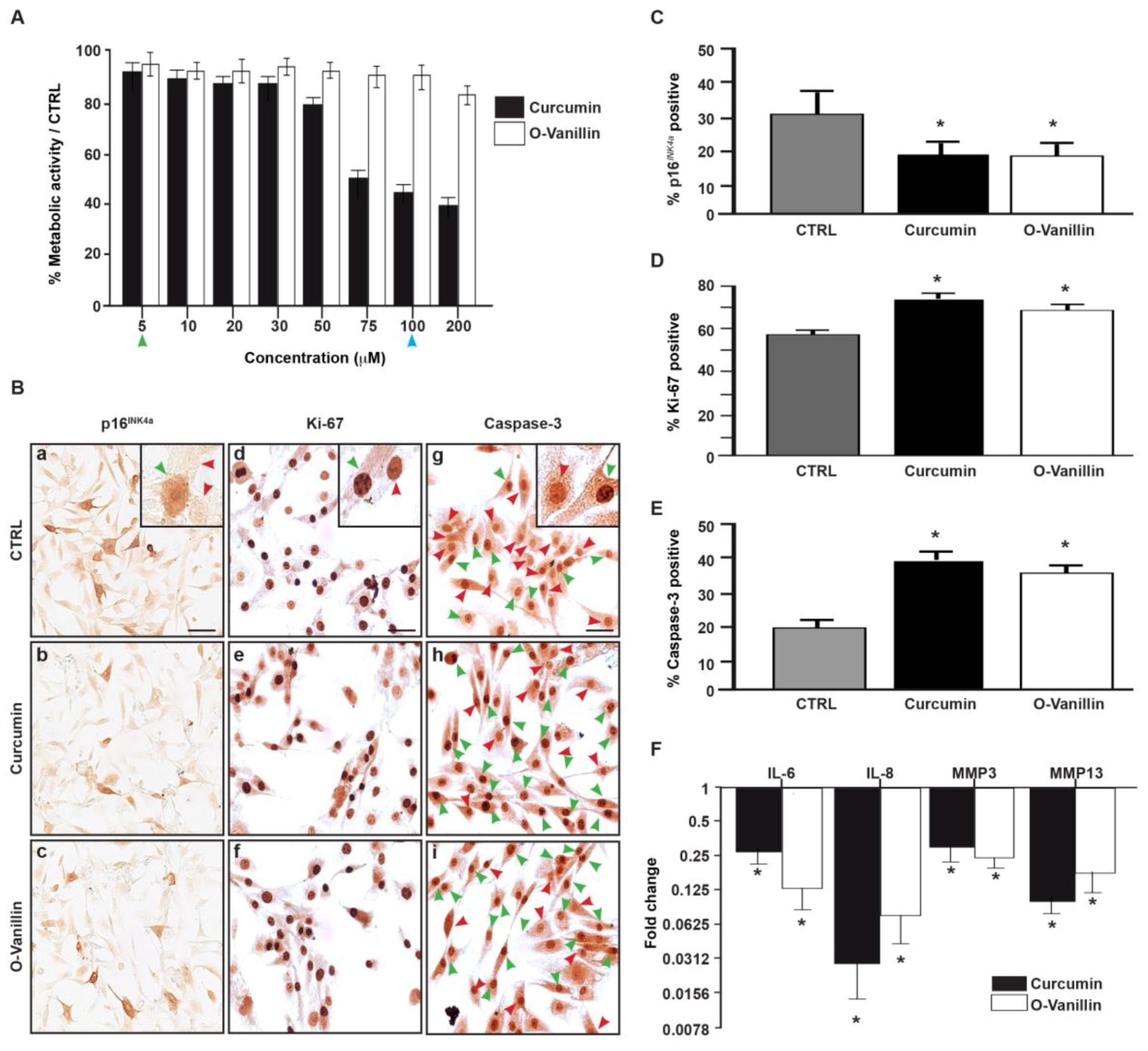
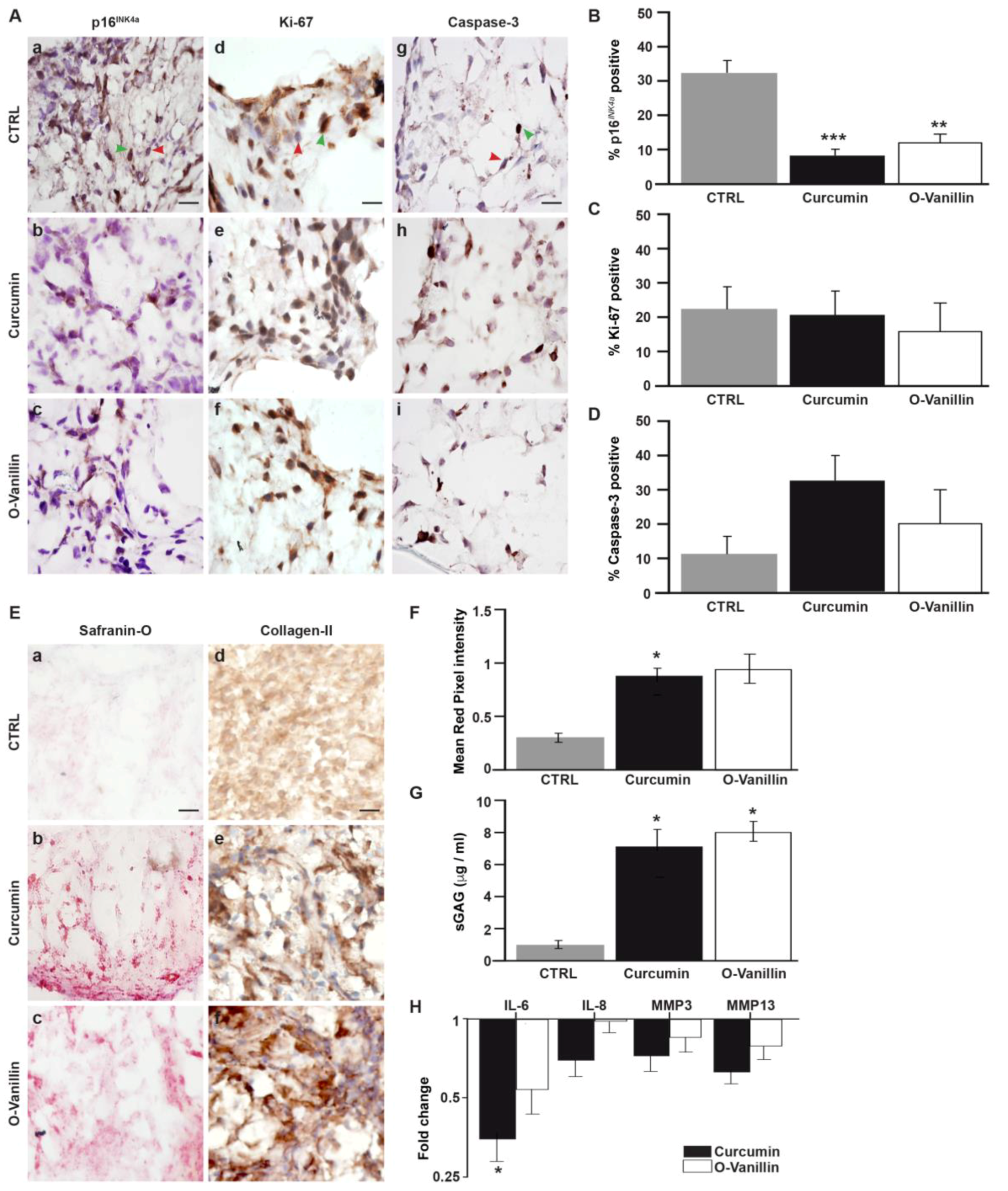
3.3. The Number of Senescent Cells and the SASP Factor Expression Are Reduced, While Proliferation and Apoptosis Are Increased, Following Curcumin and o-Vanillin Treatment of Degenerate IVD Cells Cultured in a Monolayer
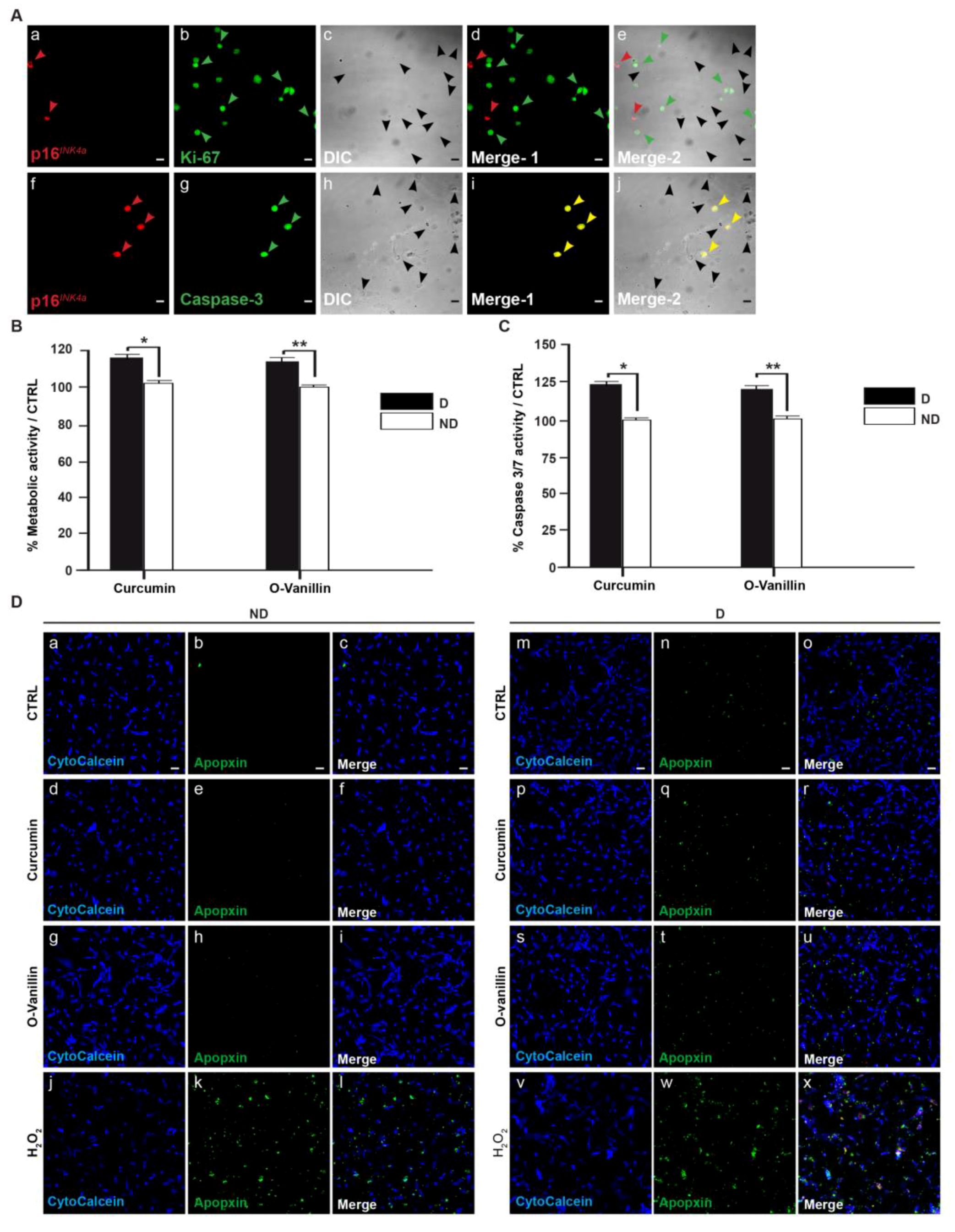
3.4. Increased Proliferation and Beneficial Apoptosis in Degenerate NP Cells Following Treatment Support the Senolytic Activity of Curcumin and o-Vanillin
3.4.1. Curcumin and o-Vanillin Reduced Senescent Cells in Pellet Culture
3.4.2. Curcumin and o-Vanillin Positively Affect Matrix Synthesis
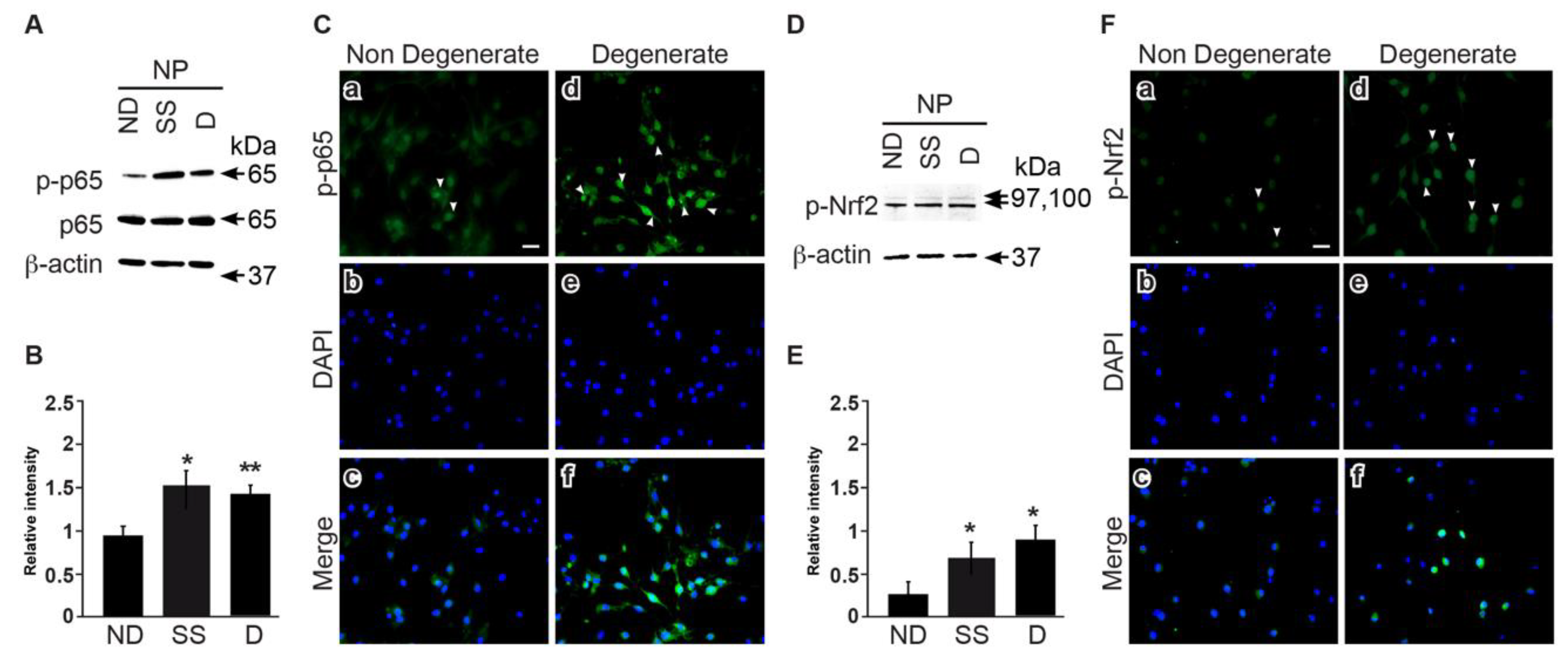
3.5. NFkB and Nrf2 Expression Is Higher in Degenerate Discs Compared to Non-Mildly-Degenerate Discs
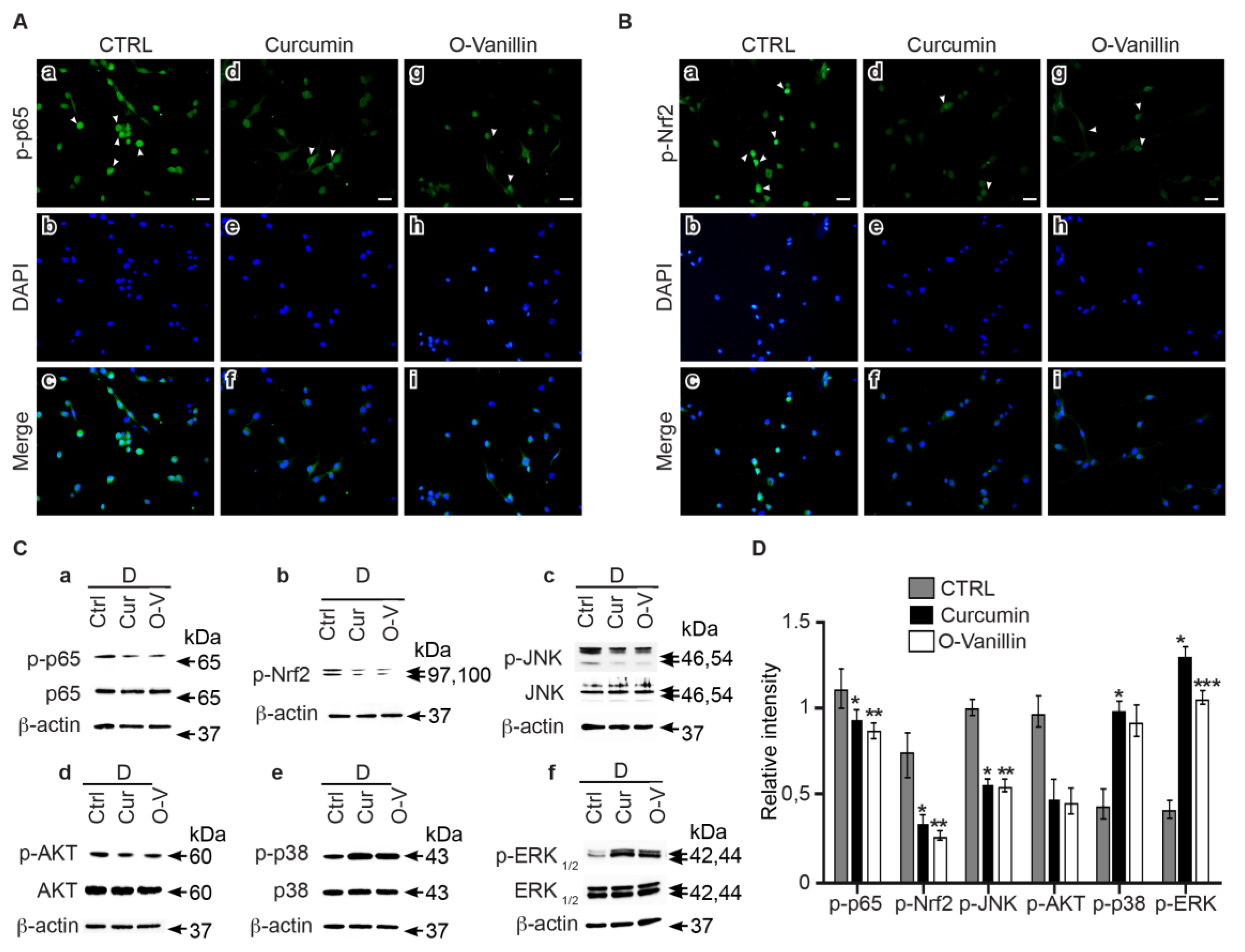
3.6. Curcumin and o-Vanillin Modulate Inflammatory Signaling Pathways
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Antoniou, J.; Steffen, T.; Nelson, F.; Winterbottom, N.; Hollander, A.P.; Poole, R.A.; Aebi, M.; Alini, M. The human lumbar intervertebral disc: Evidence for changes in the biosynthesis and denaturation of the extracellular matrix with growth, maturation, ageing, and degeneration. J. Clin. Investig. 1996, 98, 996–1003. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Coppe, J.P.; Lam, E.W. Cellular senescence: The sought or the unwanted? Trends Mol. Med. 2018, 24, 871–885. [Google Scholar] [CrossRef] [PubMed]
- Toussaint, O.; Medrano, E.E.; von Zglinicki, T. Cellular and molecular mechanisms of stress-induced premature senescence (SIPS) of human diploid fibroblasts and melanocytes. Exp. Gerontol. 2000, 35, 927–945. [Google Scholar] [CrossRef]
- De Keizer, P.L. The fountain of youth by targeting senescent cells? Trends Mol. Med. 2017, 23, 6–17. [Google Scholar] [CrossRef] [PubMed]
- Hou, A.; Chen, P.; Tang, H.; Meng, H.; Cheng, X.; Wang, Y.; Zhang, Y.; Peng, J. Cellular senescence in osteoarthritis and anti-aging strategies. Mech. Ageing Dev. 2018, 175, 83–87. [Google Scholar] [CrossRef]
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Janich, P.; Morton, J.P.; Athineos, D.; Kang, T.W.; Lasitschka, F.; Andrulis, M.; et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol. 2013, 15, 978–990. [Google Scholar] [CrossRef]
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of cellular senescence. Trends Cell Biol. 2018, 28, 436–453. [Google Scholar] [CrossRef] [PubMed]
- Tilstra, J.S.; Robinson, A.R.; Wang, J.; Gregg, S.Q.; Clauson, C.L.; Reay, D.P.; Nasto, L.A.; St Croix, C.M.; Usas, A.; Vo, N.; et al. NF-kappaB inhibition delays DNA damage-induced senescence and aging in mice. J. Clin. Investig. 2012, 122, 2601–2612. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Tchkonia, T.; Ding, H.; Ogrodnik, M.; Lubbers, E.R.; Pirtskhalava, T.; White, T.A.; Johnson, K.O.; Stout, M.B.; Mezera, V.; et al. JAK inhibition alleviates the cellular senescence-associated secretory phenotype and frailty in old age. Proc. Natl. Acad. Sci. USA 2015, 112, E6301–E6310. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Doornebal, E.J.; Pirtskhalava, T.; Giorgadze, N.; Wentworth, M.; Fuhrmann-Stroissnigg, H.; Niedernhofer, L.J.; Robbins, P.D.; Tchkonia, T.; Kirkland, J.L. New agents that target senescent cells: The flavone, fisetin, and the BCL-X-L inhibitors, A1331852 and A1155463. Aging 2017, 9, 955–963. [Google Scholar] [CrossRef]
- Kim, E.C.; Kim, J.R. Senotherapeutics: Emerging strategy for healthy aging and age-related disease. BMB Rep. 2019, 52, 47–55. [Google Scholar] [CrossRef]
- Mantzorou, M.; Pavlidou, E.; Vasios, G.; Tsagalioti, E.; Giaginis, C. Effects of Curcumin consumption on human chronic diseases: A narrative review of the most recent clinical data. Phytother. Res. 2018, 32, 957–975. [Google Scholar] [CrossRef]
- Ravindran, J.; Prasad, S.; Aggarwal, B.B. Curcumin and cancer cells: How many ways can curry kill tumor cells selectively? AAPS J. 2009, 11, 495–510. [Google Scholar] [CrossRef]
- Oliveira, A.; Sousa, E.; Sousa, E.; Vasconcelos, M.; Pinto, M.; Pinto, M. Curcumin: A natural lead for potential new drug candidates. Curr. Med. Chem. 2015, 22, 4196–4232. [Google Scholar] [CrossRef]
- Kumar, S.S.; Priyadarsini, K.I.; Sainis, K.B. Free radical scavenging activity of vanillin and O-Vanillin using 1,1-diphenyl-2-picrylhydrazyl (DPPH) radical. Redox Rep. 2002, 7, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Ji, H.F. Low stability remedies the low bioavailability of Curcumin. Trends Mol. Med. 2012, 18, 363–364. [Google Scholar] [CrossRef]
- Heger, M.; van Golen, R.F.; Broekgaarden, M.; Michel, M.C. The molecular basis for the pharmacokinetics and pharmacodynamics of Curcumin and its metabolites in relation to cancer. Pharmacol. Rev. 2014, 66, 222–307. [Google Scholar] [CrossRef] [PubMed]
- Tai, A.; Sawano, T.; Yazama, F.; Ito, H. Evaluation of antioxidant activity of vanillin by using multiple antioxidant assays. Biochim. Biophys. Acta Gen. Subj. 2011, 1810, 170–177. [Google Scholar] [CrossRef]
- Gawri, R.; Rosenzweig, D.H.; Krock, E.; Ouellet, J.A.; Stone, L.S.; Quinn, T.M.; Haglund, L. High mechanical strain of primary intervertebral disc cells promotes secretion of inflammatory factors associated with disc degeneration and pain. Arthritis Res. Ther. 2014, 16, R21. [Google Scholar] [CrossRef]
- Liu, H.; Shi, J.; Wilkerson, M.; Huang, Y.; Meschter, S.; Dupree, W.; Lin, F. Immunohistochemical detection of p16INK4a in liquid-based cytology specimens on cell block sections. Cancer 2007, 111, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Shidham, V.B.; Mehrotra, R.; Varsegi, G.; D’Amore, K.L.; Hunt, B.; Narayan, R. p16 (INK4a) immunocytochemistry on cell blocks as an adjunct to cervical cytology: Potential reflex testing on specially prepared cell blocks from residual liquid-based cytology specimens. Cytojournal 2011, 8. [Google Scholar] [CrossRef]
- Lee, S. Method for Quantitative Analysis of Glycosaminoglycans and Type II Collagen in Chondrocyte-Seeded Articular Cartilage Scaffolds with Varied Cross-Linking Density. Ph.D. Thesis, Massachusetts Institue of Technology, Cambridge, MA, USA, 2005. [Google Scholar]
- Page, B.; Page, M.; Noel, C. A new fluorometric assay for cytotoxicity measurements in-vitro. Int. J. Oncol. 1993, 3, 473–476. [Google Scholar] [CrossRef]
- Krock, E.; Rosenzweig, D.H.; Chabot-Dore, A.J.; Jarzem, P.; Weber, M.H.; Ouellet, J.A.; Stone, L.S.; Haglund, L. Painful, degenerating intervertebral discs up-regulate neurite sprouting and CGRP through nociceptive factors. J. Cell. Mol. Med. 2014, 18, 1213–1225. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(T) (-Delta Delta C) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Jeon, O.H.; Kim, C.; Laberge, R.M.; Demaria, M.; Rathod, S.; Vasserot, A.P.; Chung, J.W.; Kim, D.H.; Poon, Y.; David, N.; et al. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat. Med. 2017, 23, 775–781. [Google Scholar] [CrossRef]
- Mort, J.S.; Roughley, P.J. Measurement of glycosaminoglycan release from cartilage explants. Methods Mol. Med. 2007, 135, 201–209. [Google Scholar] [PubMed]
- Wang, Z.; Wei, D.; Xiao, H. Methods of cellular senescence induction using oxidative stress. Methods Mol. Biol. 2013, 1048, 135–144. [Google Scholar] [CrossRef]
- Wilke, H.J.; Rohlmann, F.; Neidlinger-Wilke, C.; Werner, K.; Claes, L.; Kettler, A. Validity and interobserver agreement of a new radiographic grading system for intervertebral disc degeneration: Part I. Lumbar spine. Eur. Spine J. 2006, 15, 720–730. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.P.; Pearce, R.H.; Schechter, M.T.; Adams, M.E.; Tsang, I.K.; Bishop, P.B. Preliminary evaluation of a scheme for grading the gross morphology of the human intervertebral disc. Spine 1990, 15, 411–415. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, A.; Roth, K.A.; Sayers, R.O.; Shindler, K.S.; Wong, A.M.; Fritz, L.C.; Tomaselli, K.J. In situ immunodetection of activated caspase-3 in apoptotic neurons in the developing nervous system. Cell Death Differ. 1998, 5, 1004–1016. [Google Scholar] [CrossRef]
- Park, J.B.; Byun, C.H.; Park, E.Y. Rat notochordal cells undergo premature stress-induced senescence by high glucose. Asian Spine J. 2015, 9, 495–502. [Google Scholar] [CrossRef]
- Yang, N.C.; Hu, M.L. The limitations and validities of senescence associated-beta-galactosidase activity as an aging marker for human foreskin fibroblast Hs68 cells. Exp. Gerontol. 2005, 40, 813–819. [Google Scholar] [CrossRef] [PubMed]
- Feng, C.; Liu, H.; Yang, M.; Zhang, Y.; Huang, B.; Zhou, Y. Disc cell senescence in intervertebral disc degeneration: Causes and molecular pathways. Cell Cycle 2016, 15, 1674–1684. [Google Scholar] [CrossRef] [PubMed]
- Klawitter, M.; Quero, L.; Klasen, J.; Gloess, A.N.; Klopprogge, B.; Hausmann, O.; Boos, N.; Wuertz, K. Curcuma DMSO extracts and Curcumin exhibit an anti-inflammatory and anti-catabolic effect on human intervertebral disc cells, possibly by influencing TLR2 expression and JNK activity. J. Inflamm. 2012, 9, 29. [Google Scholar] [CrossRef]
- Oliveira, C.B.; Meurer, Y.S.; Oliveira, M.G.; Medeiros, W.M.; Silva, F.O.; Brito, A.C.; Pontes Dde, L.; Andrade-Neto, V.F. Comparative study on the antioxidant and anti-Toxoplasma activities of vanillin and its resorcinarene derivative. Molecules 2014, 19, 5898–5912. [Google Scholar] [CrossRef] [PubMed]
- Shankar, S.; Srivastava, R.K. Bax and Bak genes are essential for maximum apoptotic response by Curcumin, a polyphenolic compound and cancer chemopreventive agent derived from turmeric, Curcuma longa. Carcinogenesis 2007, 28, 1277–1286. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, B.A.; Oegema, T.R., Jr. Initial characterization of the metabolism of intervertebral disc cells encapsulated in microspheres. J. Orthop. Res. 1992, 10, 677–690. [Google Scholar] [CrossRef]
- Zhou, H.; Beevers, C.S.; Huang, S. The targets of Curcumin. Curr. Drug Targets 2011, 12, 332–347. [Google Scholar] [CrossRef] [PubMed]
- Kaspar, J.W.; Niture, S.K.; Jaiswal, A.K. Nrf2: INrf2 (Keap1) signaling in oxidative stress. Free Radic. Biol. Med. 2009, 47, 1304–1309. [Google Scholar] [CrossRef]
- McMahon, M.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Keap1-dependent proteasomal degradation of transcription factor Nrf2 contributes to the negative regulation of antioxidant response element-driven gene expression. J. Biol. Chem. 2003, 278, 21592–21600. [Google Scholar] [CrossRef]
- Balogun, E.; Hoque, M.; Gong, P.; Killeen, E.; Green, C.J.; Foresti, R.; Alam, J.; Motterlini, R. Curcumin activates the haem oxygenase-1 gene via regulation of Nrf2 and the antioxidant-responsive element. Biochem. J. 2003, 371, 887–895. [Google Scholar] [CrossRef] [PubMed]
- Buhrmann, C.; Mobasheri, A.; Busch, F.; Aldinger, C.; Stahlmann, R.; Montaseri, A.; Shakibaei, M. Curcumin modulates nuclear factor kappaB (NF-kappaB)-mediated inflammation in human tenocytes in vitro: Role of the phosphatidylinositol 3-kinase/Akt pathway. J. Biol. Chem. 2011, 286, 28556–28566. [Google Scholar] [CrossRef]
- Wuertz, K.; Vo, N.; Kletsas, D.; Boos, N. Inflammatory and catabolic signalling in intervertebral discs: The roles of NF-kappaB and MAP kinases. Eur. Cell. Mater. 2012, 23, 103–119, discussion 119–120. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Nikulenkov, F.; Zawacka-Pankau, J.; Li, H.; Gabdoulline, R.; Xu, J.; Eriksson, S.; Hedstrom, E.; Issaeva, N.; Kel, A.; et al. ROS-dependent activation of JNK converts p53 into an efficient inhibitor of oncogenes leading to robust apoptosis. Cell Death. Differ. 2014, 21, 612. [Google Scholar] [CrossRef]
- Senthil, K.K.J.; Gokila, V.M.; Mau, J.L.; Lin, C.C.; Chu, F.H.; Wei, C.C.; Liao, V.H.C.; Wang, S.Y. A steroid like phytochemical Antcin M is an anti-aging reagent that eliminates hyperglycemia-accelerated premature senescence in dermal fibroblasts by direct activation of Nrf2 and SIRT-1. Oncotarget 2016, 7, 62836–62861. [Google Scholar] [CrossRef]
- Ouyang, Z.H.; Wang, W.J.; Yan, Y.G.; Wang, B.; Lv, G.H. The PI3K/Akt pathway: A critical player in intervertebral disc degeneration. Oncotarget 2017, 8, 57870–57881. [Google Scholar] [CrossRef]
- Liu, G.; Cao, P.; Chen, H.; Yuan, W.; Wang, J.; Tang, X. MiR-27a regulates apoptosis in nucleus pulposus cells by targeting PI3K. PLoS ONE 2013, 8, e75251. [Google Scholar] [CrossRef] [PubMed]
- Das, R.; Mahabeleshwar, G.; Kundu, G.C. Osteopontin stimulates cell motility and nuclear factor kappaB-mediated secretion of urokinase type plasminogen activator through phosphatidylinositol 3-kinase/Akt signaling pathways in breast cancer cells. J. Biol. Chem. 2003, 278, 31. [Google Scholar] [CrossRef]
- Pratsinis, H.; Kletsas, D. PDGF, bFGF and IGF-I stimulate the proliferation of intervertebral disc cells in vitro via the activation of the ERK and Akt signaling pathways. Eur. Spine J. 2007, 16, 1858–1866. [Google Scholar] [CrossRef]
- Iwasa, H.; Han, J.; Ishikawa, F. Mitogen-activated protein kinase p38 defines the common senescence-signalling pathway. Genes Cells 2003, 8, 131–144. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Donor | Age | Sex | Cause of Death | ICC | IHC | RT-qPCR | WB | ELISA | DMMB |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 84 | F | Motor vehicle accident | - | - | ||||
| 2 | 15 | M | Motor vehicle accident | - | - | ||||
| 3 | 49 | M | Unknown | - | NP | - | NP | NP | |
| 4 | 42 | M | Unknown | - | - | ||||
| 5 | 28 | M | Motor vehicle accident | - | - | ||||
| 6 | 50 | M | Anoxia | - | - | - | |||
| 7 | 68 | M | Cerebral hemorrhage | - | NP | ||||
| 8 | 41 | M | Cerebral anoxia | NP | |||||
| 9 | 44 | M | Anoxia | - | NP | NP | NP | NP | |
| 10 | 53 | F | Anoxia Carbon Monoxide | - | NP | - | NP | NP | |
| 11 | 27 | M | Trauma | - | |||||
| 12 | 51 | M | Unknown | - | NP | NP | NP | NP | |
| 13 | 66 | F | Head trauma | ||||||
| 14 | 60 | F | Hemorrhage/ischemic | - | - | - | |||
| 15 | 34 | M | Unknown | - | |||||
| 16 | 55 | F | Anoxia | - | |||||
| 17 | 49 | F | Arachnoid hemorrhage | - | |||||
| 18 | 53 | M | Subarachnoid hemorrhage | - | |||||
| 19 | 53 | M | cerebral aneurysm rupture | - | NP | - | NP | NP | NP |
| 20 | 62 | M | Anoxia | - | |||||
| 21 | 76 | F | Anoxia | NP | NP | - | NP | NP | NP |
| 22 | 52 | M | Cerebral vascular accident | NP | - | NP | |||
| 23 | 17 | M | Brain death | NP | - | NP | NP | ||
| 24 | 67 | M | Hemorrhagic stroke | NP | |||||
| 25 | 73 | F | Cerebrovascular accident | NP | |||||
| 26 | 68 | F | Cerebral hemorrhage | NP | |||||
| 27 | 39 | M | Gunshot to the neck | ||||||
| 28 | 35 | F | Surgical sample | - | - | NP | |||
| 29 | 30 | F | Surgical sample | - | - | NP | |||
| 30 | 32 | F | Surgical sample | - | - | NP | |||
| 31 | 45 | M | Surgical sample | - | |||||
| 32 | 27 | M | Surgical sample | - | |||||
| 33 | 38 | F | Surgical sample | - | - | ||||
| 34 | 36 | F | Surgical sample | - | - | ||||
| 35 | 47 | F | Surgical sample | - | - |
| Target | Forward Primer Sequence | Reverse Primer Sequence | Ref |
|---|---|---|---|
| p16INK4a | 5′- CTGCCCAACGCACCGAATA-3′ | 5′-GCTGCCCATCATCATGACCT-3′ | [26] |
| IL6 | 5′-TGAACCTTCCAAAGATGGCTG-3′ | 5′-CAAACTCCAAAAGACCAGTGATG-3′ | [26] |
| IL8 | 5′-TCCTGATTTCTGCAGCTCTG-3′ | 5′-GTCTTTATGCACTGACATCTAAGTTC-3′ | [26] |
| MMP3 | 5′-AATGGCATTCAGTCCCTCTATG-3′ | 5′-GACAGGTTCCGTGGGTAC-3′ | [26] |
| MMP13 | 5′-GATGACGATGTACAAGGGATCC-3′ | 5′-AGGGTCACATTTGTCTGGC-3′ | [26] |
| GAPDH | 5′-TCCCTGAGCTGAACGGGAAG-3′ | 5′-GGAGGAGTGGGTGTCGCTGT-3′ | [24] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cherif, H.; Bisson, D.G.; Jarzem, P.; Weber, M.; Ouellet, J.A.; Haglund, L. Curcumin and o-Vanillin Exhibit Evidence of Senolytic Activity in Human IVD Cells In Vitro. J. Clin. Med. 2019, 8, 433. https://doi.org/10.3390/jcm8040433
Cherif H, Bisson DG, Jarzem P, Weber M, Ouellet JA, Haglund L. Curcumin and o-Vanillin Exhibit Evidence of Senolytic Activity in Human IVD Cells In Vitro. Journal of Clinical Medicine. 2019; 8(4):433. https://doi.org/10.3390/jcm8040433
Chicago/Turabian StyleCherif, Hosni, Daniel G. Bisson, Peter Jarzem, Michael Weber, Jean A. Ouellet, and Lisbet Haglund. 2019. "Curcumin and o-Vanillin Exhibit Evidence of Senolytic Activity in Human IVD Cells In Vitro" Journal of Clinical Medicine 8, no. 4: 433. https://doi.org/10.3390/jcm8040433
APA StyleCherif, H., Bisson, D. G., Jarzem, P., Weber, M., Ouellet, J. A., & Haglund, L. (2019). Curcumin and o-Vanillin Exhibit Evidence of Senolytic Activity in Human IVD Cells In Vitro. Journal of Clinical Medicine, 8(4), 433. https://doi.org/10.3390/jcm8040433