
The Changing Landscape of Pulmonary Arterial Hypertension in the Adult with Congenital Heart Disease

Abstract
:1. Introduction
2. Epidemiology and Genetics
3. Classification of PAH
4. Clinical Evaluation of PAH-CHD
5. General Management
5.1. Expert Centers
5.2. Supportive Care
6. Advanced PAH-Specific Therapies
6.1. Endothelin Pathway
6.2. Nitric Oxide Pathway
6.3. Prostacyclin Pathway
6.4. Combination Therapy
7. Interventional Approaches
7.1. In Case of Moderately Elevated PVR
7.2. Decision to Intervene: To Close or Not to Close
7.3. Reversibility
7.4. (Lost-to-)Follow-Up after Repair
8. Future Implications
8.1. Risk Stratification
8.2. Challenging Patient Groups: Down Syndrome and Fontan
8.3. New Candidate Therapies in PAH
9. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lowe, B.S.; Therrien, J.; Ionescu-Ittu, R.; Pilote, L.; Martucci, G.; Marelli, A.J. Diagnosis of pulmonary hypertension in the congenital heart disease adult population impact on outcomes. J. Am. Coll. Cardiol. 2011, 58, 538–546. [Google Scholar] [CrossRef] [PubMed]
- Marelli, A.J.; Mackie, A.S.; Ionescu-Ittu, R.; Rahme, E.; Pilote, L. Congenital heart disease in the general population: Changing prevalence and age distribution. Circulation 2007, 115, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Moons, P.; Bovijn, L.; Budts, W.; Belmans, A.; Gewillig, M. Temporal trends in survival to adulthood among patients born with congenital heart disease from 1970 to 1992 in Belgium. Circulation 2010, 122, 2264–2272. [Google Scholar] [CrossRef] [PubMed]
- Van der Linde, D.; Konings, E.E.; Slager, M.A.; Witsenburg, M.; Helbing, W.A.; Takkenberg, J.J.; Roos-Hesselink, J.W. Birth prevalence of congenital heart disease worldwide: A systematic review and meta-analysis. J. Am. Coll. Cardiol. 2011, 58, 2241–2247. [Google Scholar] [CrossRef] [PubMed]
- Duffels, M.G.; Engelfriet, P.M.; Berger, R.M.; van Loon, R.L.; Hoendermis, E.; Vriend, J.W.; van der Velde, E.T.; Bresser, P.; Mulder, B.J. Pulmonary arterial hypertension in congenital heart disease: An epidemiologic perspective from a Dutch registry. Int. J. Cardiol. 2007, 120, 198–204. [Google Scholar] [CrossRef] [PubMed]
- Engelfriet, P.M.; Duffels, M.G.; Moller, T.; Boersma, E.; Tijssen, J.G.; Thaulow, E.; Gatzoulis, M.A.; Mulder, B.J. Pulmonary arterial hypertension in adults born with a heart septal defect: The Euro Heart Survey on adult congenital heart disease. Heart 2007, 93, 682–687. [Google Scholar] [CrossRef] [PubMed]
- Van Riel, A.C.; Schuuring, M.J.; van Hessen, I.D.; Zwinderman, A.H.; Cozijnsen, L.; Reichert, C.L.; Hoorntje, J.C.; Wagenaar, L.J.; Post, M.C.; van Dijk, A.P.J.; et al. Contemporary prevalence of pulmonary arterial hypertension in adult congenital heart disease following the updated clinical classification. Int. J. Cardiol. 2014, 174, 299–305. [Google Scholar] [CrossRef] [PubMed]
- Van de Bruaene, A.; Delcroix, M.; Pasquet, A.; De Backer, J.; De Pauw, M.; Naeije, R.; Vachiery, J.L.; Paelinck, B.; Morissens, M.; Budts, W. The Belgian Eisenmenger syndrome registry: Implications for treatment strategies? Acta Cardiol. 2009, 64, 447–453. [Google Scholar] [CrossRef] [PubMed]
- Dimopoulos, K.; Wort, S.J.; Gatzoulis, M.A. Pulmonary hypertension related to congenital heart disease: A call for action. Eur. Heart J. 2014, 35, 691–700. [Google Scholar] [CrossRef] [PubMed]
- Korten, M.A.; Helm, P.C.; Abdul-Khaliq, H.; Baumgartner, H.; Kececioglu, D.; Schlensak, C.; Bauer, U.M.; Diller, G.P.; Competence Network for Congenital Heart Defects Investigators. Eisenmenger syndrome and long-term survival in patients with Down syndrome and congenital heart disease. Heart 2016, 102, 1552–1557. [Google Scholar] [CrossRef] [PubMed]
- Roberts, K.E.; McElroy, J.J.; Wong, W.P.; Yen, E.; Widlitz, A.; Barst, R.J.; Knowles, J.A.; Morse, J.H. BMPR2 mutations in pulmonary arterial hypertension with congenital heart disease. Eur. Respir. J. 2004, 24, 371–374. [Google Scholar] [CrossRef] [PubMed]
- Opotowsky, A.R. Clinical Evaluation and Management of Pulmonary Hypertension in the Adult with Congenital Heart Disease. Circulation 2015, 131, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Galie, N.; Humbert, M.; Vachiery, J.L.; Gibbs, S.; Lang, I.; Torbicki, A.; Simonneau, G.; Peacock, A.; Vonk Noordegraaf, A.; Beghetti, M.; et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur. Heart J. 2016, 37, 67–119. [Google Scholar] [PubMed]
- Simonneau, G.; Gatzoulis, M.A.; Adatia, I.; Celermajer, D.; Denton, C.; Ghofrani, A.; Gomez Sanchez, M.A.; Krishna Kumar, R.; Landzberg, M.; Machado, R.F.; et al. Updated clinical classification of pulmonary hypertension. J. Am. Coll. Cardiol. 2013, 62, D34–D41. [Google Scholar] [CrossRef] [PubMed]
- Baumgartner, H.; Bonhoeffer, P.; De Groot, N.M.; de Haan, F.; Deanfield, J.E.; Galie, N.; Gatzoulis, M.A.; Gohlke-Baerwolf, C.; Kaemmerer, H.; Kilner, P.; et al. ESC Guidelines for the management of grown-up congenital heart disease (new version 2010). Eur. Heart J. 2010, 31, 2915–2957. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, V.V.; Presberg, K.W.; Doyle, R.L.; Abman, S.H.; McCrory, D.C.; Fortin, T.; Ahearn, G. Prognosis of pulmonary arterial hypertension: ACCP evidence-based clinical practice guidelines. Chest 2004, 126, 78s–92s. [Google Scholar] [CrossRef] [PubMed]
- Daliento, L.; Somerville, J.; Presbitero, P.; Menti, L.; Brach-Prever, S.; Rizzoli, G.; Stone, S. Eisenmenger syndrome. Factors relating to deterioration and death. Eur. Heart J. 1998, 19, 1845–1855. [Google Scholar] [CrossRef] [PubMed]
- Diller, G.P.; Dimopoulos, K.; Broberg, C.S.; Kaya, M.G.; Naghotra, U.S.; Uebing, A.; Harries, C.; Goktekin, O.; Gibbs, J.S.; Gatzoulis, M.A. Presentation, survival prospects, and predictors of death in Eisenmenger syndrome: A combined retrospective and case-control study. Eur. Heart J. 2006, 27, 1737–1742. [Google Scholar] [CrossRef] [PubMed]
- Kempny, A.; Hjortshoj, C.S.; Gu, H.; Li, W.; Opotowsky, A.R.; Landzberg, M.; Jensen, A.S.; Sondergaard, L.; Estensen, M.E.; Thilen, U.; et al. Predictors of Death in Contemporary Adult Patients with Eisenmenger Syndrome: A Multicentre Study. Circulation 2016. [Google Scholar] [CrossRef] [PubMed]
- Manes, A.; Palazzini, M.; Leci, E.; Bacchi Reggiani, M.L.; Branzi, A.; Galie, N. Current era survival of patients with pulmonary arterial hypertension associated with congenital heart disease: A comparison between clinical subgroups. Eur. Heart J. 2014, 35, 716–724. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, W.E. The remarkable right ventricle of patients with Eisenmenger syndrome. Coron. Artery Dis. 2005, 16, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Diller, G.P.; Dimopoulos, K.; Kafka, H.; Ho, S.Y.; Gatzoulis, M.A. Model of chronic adaptation: Right ventricular function in Eisenmenger syndrome. Eur. Heart J. Suppl. 2007, 9, H54–H60. [Google Scholar] [CrossRef]
- Diller, G.P.; Dimopoulos, K.; Okonko, D.; Li, W.; Babu-Narayan, S.V.; Broberg, C.S.; Johansson, B.; Bouzas, B.; Mullen, M.J.; Poole-Wilson, P.A.; et al. Exercise intolerance in adult congenital heart disease: Comparative severity, correlates, and prognostic implication. Circulation 2005, 112, 828–835. [Google Scholar] [CrossRef] [PubMed]
- Diller, G.P.; Kempny, A.; Alonso-Gonzalez, R.; Swan, L.; Uebing, A.; Li, W.; Babu-Narayan, S.; Wort, S.J.; Dimopoulos, K.; Gatzoulis, M.A. Survival Prospects and Circumstances of Death in Contemporary Adult Congenital Heart Disease Patients Under Follow-Up at a Large Tertiary Centre. Circulation 2015, 132, 2118–2125. [Google Scholar] [CrossRef] [PubMed]
- Diller, G.P.; Alonso-Gonzalez, R.; Kempny, A.; Dimopoulos, K.; Inuzuka, R.; Giannakoulas, G.; Castle, L.; Lammers, A.E.; Hooper, J.; Uebing, A.; et al. B-type natriuretic peptide concentrations in contemporary Eisenmenger syndrome patients: Predictive value and response to disease targeting therapy. Heart 2012, 98, 736–742. [Google Scholar] [CrossRef] [PubMed]
- Giannakoulas, G.; Mouratoglou, S.A.; Gatzoulis, M.A.; Karvounis, H. Blood biomarkers and their potential role in pulmonary arterial hypertension associated with congenital heart disease. A systematic review. Int. J. Cardiol. 2014, 174, 618–623. [Google Scholar] [CrossRef] [PubMed]
- Blok, I.M.; van Riel, A.C.; Schuuring, M.J.; de Bruin-Bon, R.H.; van Dijk, A.P.; Hoendermis, E.S.; Zwinderman, A.H.; Mulder, B.J.; Bouma, B.J. The role of cystatin C as a biomarker for prognosis in pulmonary arterial hypertension due to congenital heart disease. Int. J. Cardiol. 2016, 209, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Mylotte, D.; Pilote, L.; Ionescu-Ittu, R.; Abrahamowicz, M.; Khairy, P.; Therrien, J.; Mackie, A.S.; Marelli, A. Specialized adult congenital heart disease care: The impact of policy on mortality. Circulation 2014, 129, 1804–1812. [Google Scholar] [CrossRef] [PubMed]
- Diller, G.P.; Korten, M.A.; Bauer, U.M.; Miera, O.; Tutarel, O.; Kaemmerer, H.; Berger, F.; Baumgartner, H. Current therapy and outcome of Eisenmenger syndrome: Data of the German National Register for congenital heart defects. Eur. Heart J. 2016, 37, 1449–1455. [Google Scholar] [CrossRef] [PubMed]
- Ladouceur, M.; Benoit, L.; Radojevic, J.; Basquin, A.; Dauphin, C.; Hascoet, S.; Moceri, P.; Bredy, C.; Iserin, L.; Gouton, M.; et al. Pregnancy outcomes in patients with pulmonary arterial hypertension associated with congenital heart disease. Heart 2017, 103, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Sliwa, K.; van Hagen, I.M.; Budts, W.; Swan, L.; Sinagra, G.; Caruana, M.; Blanco, M.V.; Wagenaar, L.J.; Johnson, M.R.; Webb, G.; et al. Pulmonary hypertension and pregnancy outcomes: Data from the Registry Of Pregnancy and Cardiac Disease (ROPAC) of the European Society of Cardiology. Eur. J. Heart Fail. 2016, 18, 1119–1128. [Google Scholar] [CrossRef] [PubMed]
- D’Alto, M.; Diller, G.P. Pulmonary hypertension in adults with congenital heart disease and Eisenmenger syndrome: Current advanced management strategies. Heart 2014, 100, 1322–1328. [Google Scholar] [CrossRef] [PubMed]
- Tay, E.L.; Peset, A.; Papaphylactou, M.; Inuzuka, R.; Alonso-Gonzalez, R.; Giannakoulas, G.; Tzifa, A.; Goletto, S.; Broberg, C.; Dimopoulos, K.; et al. Replacement therapy for iron deficiency improves exercise capacity and quality of life in patients with cyanotic congenital heart disease and/or the Eisenmenger syndrome. Int. J. Cardiol. 2011, 151, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Inuzuka, R.; Diller, G.P.; Borgia, F.; Benson, L.; Tay, E.L.; Alonso-Gonzalez, R.; Silva, M.; Charalambides, M.; Swan, L.; Dimopoulos, K.; et al. Comprehensive use of cardiopulmonary exercise testing identifies adults with congenital heart disease at increased mortality risk in the medium term. Circulation 2012, 125, 250–259. [Google Scholar] [CrossRef] [PubMed]
- Van De Bruaene, A.; De Meester, P.; Voigt, J.U.; Delcroix, M.; Pasquet, A.; De Backer, J.; De Pauw, M.; Naeije, R.; Vachiery, J.L.; Paelinck, B.P.; et al. Worsening in oxygen saturation and exercise capacity predict adverse outcome in patients with Eisenmenger syndrome. Int. J. Cardiol. 2013, 168, 1386–1392. [Google Scholar] [CrossRef] [PubMed]
- Kempny, A.; Dimopoulos, K.; Alonso-Gonzalez, R.; Alvarez-Barredo, M.; Tutarel, O.; Uebing, A.; Piatek, P.; Marino, P.; Swan, L.; Diller, G.P.; et al. Six-minute walk test distance and resting oxygen saturations but not functional class predict outcome in adult patients with Eisenmenger syndrome. Int. J. Cardiol. 2013, 168, 4784–4789. [Google Scholar] [CrossRef] [PubMed]
- Sandoval, J.; Aguirre, J.S.; Pulido, T.; Martinez-Guerra, M.L.; Santos, E.; Alvarado, P.; Rosas, M.; Bautista, E. Nocturnal oxygen therapy in patients with the Eisenmenger syndrome. Am. J. Respir. Crit. Care Med. 2001, 164, 1682–1687. [Google Scholar] [CrossRef] [PubMed]
- Broberg, C.S.; Ujita, M.; Prasad, S.; Li, W.; Rubens, M.; Bax, B.E.; Davidson, S.J.; Bouzas, B.; Gibbs, J.S.; Burman, J.; et al. Pulmonary arterial thrombosis in eisenmenger syndrome is associated with biventricular dysfunction and decreased pulmonary flow velocity. J. Am. Coll. Cardiol. 2007, 50, 634–642. [Google Scholar] [CrossRef] [PubMed]
- Klepetko, W.; Mayer, E.; Sandoval, J.; Trulock, E.P.; Vachiery, J.L.; Dartevelle, P.; Pepke-Zaba, J.; Jamieson, S.W.; Lang, I.; Corris, P. Interventional and surgical modalities of treatment for pulmonary arterial hypertension. J. Am. Coll. Cardiol. 2004, 43, 73s–80s. [Google Scholar] [CrossRef] [PubMed]
- Adriaenssens, T.; Delcroix, M.; Van Deyk, K.; Budts, W. Advanced therapy may delay the need for transplantation in patients with the Eisenmenger syndrome. Eur. Heart J. 2006, 27, 1472–1477. [Google Scholar] [CrossRef] [PubMed]
- Dimopoulos, K.; Inuzuka, R.; Goletto, S.; Giannakoulas, G.; Swan, L.; Wort, S.J.; Gatzoulis, M.A. Improved survival among patients with Eisenmenger syndrome receiving advanced therapy for pulmonary arterial hypertension. Circulation 2010, 121, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Simonneau, G.; Barst, R.J.; Galie, N.; Naeije, R.; Rich, S.; Bourge, R.C.; Keogh, A.; Oudiz, R.; Frost, A.; Blackburn, S.D.; et al. Continuous subcutaneous infusion of treprostinil, a prostacyclin analogue, in patients with pulmonary arterial hypertension: A double-blind, randomized, placebo-controlled trial. Am. J. Respir. Crit. Care Med. 2002, 165, 800–804. [Google Scholar] [CrossRef] [PubMed]
- Galie, N.; Rubin, L.; Hoeper, M.; Jansa, P.; Al-Hiti, H.; Meyer, G.; Chiossi, E.; Kusic-Pajic, A.; Simonneau, G. Treatment of patients with mildly symptomatic pulmonary arterial hypertension with bosentan (EARLY study): A double-blind, randomised controlled trial. Lancet 2008, 371, 2093–2100. [Google Scholar] [CrossRef]
- Galie, N.; Brundage, B.H.; Ghofrani, H.A.; Oudiz, R.J.; Simonneau, G.; Safdar, Z.; Shapiro, S.; White, R.J.; Chan, M.; Beardsworth, A.; et al. Tadalafil therapy for pulmonary arterial hypertension. Circulation 2009, 119, 2894–2903. [Google Scholar] [CrossRef] [PubMed]
- Ghofrani, H.A.; Galie, N.; Grimminger, F.; Grunig, E.; Humbert, M.; Jing, Z.C.; Keogh, A.M.; Langleben, D.; Kilama, M.O.; Fritsch, A.; et al. Riociguat for the treatment of pulmonary arterial hypertension. N. Engl. J. Med. 2013, 369, 330–340. [Google Scholar] [CrossRef] [PubMed]
- Simonneau, G.; Rubin, L.J.; Galiè, N.; Barst, R.J.; Fleming, T.R.; Frost, A.E.; Engel, P.J.; Kramer, M.R.; Burgess, G.; Collings, L.; et al. Addition of sildenafil to long-term intravenous epoprostenol therapy in patients with pulmonary arterial hypertension: A randomized trial. Ann. Intern. Med. 2008, 149, 521–530. [Google Scholar] [CrossRef] [PubMed]
- Galie, N.; Ghofrani, H.A.; Torbicki, A.; Barst, R.J.; Rubin, L.J.; Badesch, D.; Fleming, T.; Parpia, T.; Burgess, G.; Branzi, A.; et al. Sildenafil citrate therapy for pulmonary arterial hypertension. N. Engl. J. Med. 2005, 353, 2148–2157. [Google Scholar] [CrossRef] [PubMed]
- Tapson, V.F.; Torres, F.; Kermeen, F.; Keogh, A.M.; Allen, R.P.; Frantz, R.P.; Badesch, D.B.; Frost, A.E.; Shapiro, S.M.; Laliberte, K.; et al. Oral treprostinil for the treatment of pulmonary arterial hypertension in patients on background endothelin receptor antagonist and/or phosphodiesterase type 5 inhibitor therapy (the FREEDOM-C study): A randomized controlled trial. Chest 2012, 142, 1383–1390. [Google Scholar] [CrossRef] [PubMed]
- Tapson, V.F.; Jing, Z.C.; Xu, K.F.; Pan, L.; Feldman, J.; Kiely, D.G.; Kotlyar, E.; McSwain, C.S.; Laliberte, K.; Arneson, C.; et al. Oral treprostinil for the treatment of pulmonary arterial hypertension in patients receiving background endothelin receptor antagonist and phosphodiesterase type 5 inhibitor therapy (the FREEDOM-C2 study): A randomized controlled trial. Chest 2013, 144, 952–958. [Google Scholar] [CrossRef] [PubMed]
- Pulido, T.; Adzerikho, I.; Channick, R.N.; Delcroix, M.; Galie, N.; Ghofrani, H.A.; Jansa, P.; Jing, Z.C.; Le Brun, F.O.; Mehta, S.; et al. Macitentan and morbidity and mortality in pulmonary arterial hypertension. N. Engl. J. Med. 2013, 369, 809–818. [Google Scholar] [CrossRef] [PubMed]
- Jing, Z.C.; Parikh, K.; Pulido, T.; Jerjes-Sanchez, C.; White, R.J.; Allen, R.; Torbicki, A.; Xu, K.F.; Yehle, D.; Laliberte, K.; et al. Efficacy and safety of oral treprostinil monotherapy for the treatment of pulmonary arterial hypertension: A randomized, controlled trial. Circulation 2013, 127, 624–633. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, V.; Channick, R.N.; Ghofrani, H.A.; Lemarie, J.C.; Naeije, R.; Packer, M.; Souza, R.; Tapson, V.F.; Tolson, J.; Al Hiti, H.; et al. Bosentan added to sildenafil therapy in patients with pulmonary arterial hypertension. Eur. Respir. J. 2015, 46, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Galiè, N.; Barberà, J.A.; Frost, A.E.; Ghofrani, H.A.; Hoeper, M.M.; McLaughlin, V.V.; Peacock, A.J.; Simonneau, G.; Vachiery, J.-L.; Grünig, E.; et al. Initial Use of Ambrisentan plus Tadalafil in Pulmonary Arterial Hypertension. N. Engl. J. Med. 2015, 373, 834–844. [Google Scholar] [CrossRef] [PubMed]
- Sitbon, O.; Channick, R.; Chin, K.M.; Frey, A.; Gaine, S.; Galie, N.; Ghofrani, H.A.; Hoeper, M.M.; Lang, I.M.; Preiss, R.; et al. Selexipag for the Treatment of Pulmonary Arterial Hypertension. N. Engl. J. Med. 2015, 373, 2522–2533. [Google Scholar] [CrossRef] [PubMed]
- Galie, N.; Beghetti, M.; Gatzoulis, M.A.; Granton, J.; Berger, R.M.; Lauer, A.; Chiossi, E.; Landzberg, M. Bosentan therapy in patients with Eisenmenger syndrome: A multicenter, double-blind, randomized, placebo-controlled study. Circulation 2006, 114, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Singh, T.P.; Rohit, M.; Grover, A.; Malhotra, S.; Vijayvergiya, R. A randomized, placebo-controlled, double-blind, crossover study to evaluate the efficacy of oral sildenafil therapy in severe pulmonary artery hypertension. Am. Heart J. 2006, 151, 851.e1–851.e5. [Google Scholar] [CrossRef] [PubMed]
- Iversen, K.; Jensen, A.S.; Jensen, T.V.; Vejlstrup, N.G.; Sondergaard, L. Combination therapy with bosentan and sildenafil in Eisenmenger syndrome: A randomized, placebo-controlled, double-blinded trial. Eur. Heart J. 2010, 31, 1124–1131. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S.; Nathani, S.; Yusuf, J.; Shrimal, D.; Tyagi, S. Clinical efficacy of phosphodiesterase-5 inhibitor tadalafil in Eisenmenger syndrome—A randomized, placebo-controlled, double-blind crossover study. Congenit. Heart Dis. 2011, 6, 424–431. [Google Scholar] [CrossRef] [PubMed]
- Gatzoulis, M.A.; Beghetti, M.; Galie, N.; Granton, J.; Berger, R.M.; Lauer, A.; Chiossi, E.; Landzberg, M. Longer-term bosentan therapy improves functional capacity in Eisenmenger syndrome: Results of the BREATHE-5 open-label extension study. Int. J. Cardiol. 2008, 127, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Berger, R.M.; Beghetti, M.; Galie, N.; Gatzoulis, M.A.; Granton, J.; Lauer, A.; Chiossi, E.; Landzberg, M. Atrial septal defects versus ventricular septal defects in BREATHE-5, a placebo-controlled study of pulmonary arterial hypertension related to Eisenmenger’s syndrome: A subgroup analysis. Int. J. Cardiol. 2010, 144, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Diller, G.P.; Dimopoulos, K.; Kaya, M.G.; Harries, C.; Uebing, A.; Li, W.; Koltsida, E.; Gibbs, J.S.; Gatzoulis, M.A. Long-term safety, tolerability and efficacy of bosentan in adults with pulmonary arterial hypertension associated with congenital heart disease. Heart 2007, 93, 974–976. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Caraballo, E.; Gonzalez-Garcia, A.E.; Renones, M.; Sanchez-Recalde, A.; Garcia-Rio, F.; Oliver-Ruiz, J.M. Long-term bosentan treatment of complex congenital heart disease and Eisenmenger’s syndrome. Rev. Esp. Cardiol. 2009, 62, 1046–1049. [Google Scholar] [CrossRef]
- Vis, J.C.; Duffels, M.G.; Mulder, P.; de Bruin-Bon, R.H.; Bouma, B.J.; Berger, R.M.; Hoendermis, E.S.; van Dijk, A.P.; Mulder, B.J. Prolonged beneficial effect of bosentan treatment and 4-year survival rates in adult patients with pulmonary arterial hypertension associated with congenital heart disease. Int. J. Cardiol. 2013, 164, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Zuckerman, W.A.; Leaderer, D.; Rowan, C.A.; Mituniewicz, J.D.; Rosenzweig, E.B. Ambrisentan for pulmonary arterial hypertension due to congenital heart disease. Am. J. Cardiol. 2011, 107, 1381–1385. [Google Scholar] [CrossRef] [PubMed]
- Condliffe, R.; Elliot, C.A.; Hurdman, J.; Sabroe, I.; Billings, C.; Kiely, D.G.; Hamilton, N. Ambrisentan therapy in pulmonary hypertension: Clinical use and tolerability in a referral centre. Ther. Adv. Respir. Dis. 2014, 8, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Blok, I.M.; van Riel, A.C.; van Dijk, A.P.; Mulder, B.J.; Bouma, B.J. From bosentan to macitentan for pulmonary arterial hypertension and adult congenital heart disease: Further improvement? Int. J. Cardiol. 2017, 227, 51–52. [Google Scholar] [CrossRef] [PubMed]
- Tay, E.L.; Papaphylactou, M.; Diller, G.P.; Alonso-Gonzalez, R.; Inuzuka, R.; Giannakoulas, G.; Harries, C.; Wort, S.J.; Swan, L.; Dimopoulos, K.; et al. Quality of life and functional capacity can be improved in patients with Eisenmenger syndrome with oral sildenafil therapy. Int. J. Cardiol. 2011, 149, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.N.; Jiang, X.; Zhang, R.; Li, X.L.; Wu, B.X.; Zhao, Q.H.; Wang, Y.; Dai, L.Z.; Pan, L.; Gomberg-Maitland, M.; et al. Oral sildenafil treatment for Eisenmenger syndrome: A prospective, open-label, multicentre study. Heart 2011, 97, 1876–1881. [Google Scholar] [CrossRef] [PubMed]
- Chau, E.M.; Fan, K.Y.; Chow, W.H. Effects of chronic sildenafil in patients with Eisenmenger syndrome versus idiopathic pulmonary arterial hypertension. Int. J. Cardiol. 2007, 120, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S.; Sharma, M.; Ramakrishnan, S.; Yusuf, J.; Gupta, M.D.; Bhamri, N.; Trehan, V.; Tyagi, S. Phosphodiesterase-5 inhibitor in Eisenmenger syndrome: A preliminary observational study. Circulation 2006, 114, 1807–1810. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, S.M.; Newburger, J.W.; Lang, P.; Pearson, D.D.; Feinstein, J.A.; Gauvreau, K.; Landzberg, M.J. Usefulness of epoprostenol therapy in the severely ill adolescent/adult with Eisenmenger physiology. Am. J. Cardiol. 2003, 91, 632–635. [Google Scholar] [CrossRef]
- Rosenzweig, E.B.; Kerstein, D.; Barst, R.J. Long-term prostacyclin for pulmonary hypertension with associated congenital heart defects. Circulation 1999, 99, 1858–1865. [Google Scholar] [CrossRef] [PubMed]
- Thomas, I.C.; Glassner-Kolmin, C.; Gomberg-Maitland, M. Long-term effects of continuous prostacyclin therapy in adults with pulmonary hypertension associated with congenital heart disease. Int. J. Cardiol. 2013, 168, 4117–4121. [Google Scholar] [CrossRef] [PubMed]
- Cha, K.S.; Cho, K.I.; Seo, J.S.; Choi, J.H.; Park, Y.H.; Yang, D.H.; Hong, G.R.; Kim, D.S. Effects of inhaled iloprost on exercise capacity, quality of life, and cardiac function in patients with pulmonary arterial hypertension secondary to congenital heart disease (the Eisenmenger syndrome) (from the EIGER Study). Am. J. Cardiol. 2013, 112, 1834–1839. [Google Scholar] [CrossRef] [PubMed]
- Fox, B.D.; Shtraichman, O.; Langleben, D.; Shimony, A.; Kramer, M.R. Combination Therapy for Pulmonary Arterial Hypertension: A Systematic Review and Meta-analysis. Can. J. Cardiol. 2016, 32, 1520–1530. [Google Scholar] [CrossRef] [PubMed]
- Farber, H.W.; Miller, D.P.; Meltzer, L.A.; McGoon, M.D. Treatment of patients with pulmonary arterial hypertension at the time of death or deterioration to functional class IV: Insights from the REVEAL Registry. J. Heart Lung Transplant. 2013, 32, 1114–1122. [Google Scholar] [CrossRef] [PubMed]
- Hoeper, M.M.; Leuchte, H.; Halank, M.; Wilkens, H.; Meyer, F.J.; Seyfarth, H.J.; Wensel, R.; Ripken, F.; Bremer, H.; Kluge, S.; et al. Combining inhaled iloprost with bosentan in patients with idiopathic pulmonary arterial hypertension. Eur. Respir. J. 2006, 28, 691–694. [Google Scholar] [CrossRef] [PubMed]
- Humbert, M.; Barst, R.J.; Robbins, I.M.; Channick, R.N.; Galiè, N.; Boonstra, A.; Rubin, L.J.; Horn, E.M.; Manes, A.; Simonneau, G. Combination of bosentan with epoprostenol in pulmonary arterial hypertension: BREATHE-2. Eur. Respir. J. 2004, 24, 353–359. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, V.V.; Benza, R.L.; Rubin, L.J.; Channick, R.N.; Voswinckel, R.; Tapson, V.F.; Robbins, I.M.; Olschewski, H.; Rubenfire, M.; Seeger, W. Addition of Inhaled Treprostinil to Oral Therapy for Pulmonary Arterial Hypertension: A Randomized Controlled Clinical Trial. J. Am. Coll. Cardiol. 2010, 55, 1915–1922. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, V.V.; Oudiz, R.J.; Frost, A.; Tapson, V.F.; Murali, S.; Channick, R.N.; Badesch, D.B.; Barst, R.J.; Hsu, H.H.; Rubin, L.J. Randomized study of adding inhaled iloprost to existing bosentan in pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2006, 174, 1257–1263. [Google Scholar] [CrossRef] [PubMed]
- D’Alto, M.; Romeo, E.; Argiento, P.; Sarubbi, B.; Santoro, G.; Grimaldi, N.; Correra, A.; Scognamiglio, G.; Russo, M.G.; Calabro, R. Bosentan-sildenafil association in patients with congenital heart disease-related pulmonary arterial hypertension and Eisenmenger physiology. Int. J. Cardiol. 2012, 155, 378–382. [Google Scholar] [CrossRef] [PubMed]
- Sitbon, O.; Jais, X.; Savale, L.; Cottin, V.; Bergot, E.; Macari, E.A.; Bouvaist, H.; Dauphin, C.; Picard, F.; Bulifon, S.; et al. Upfront triple combination therapy in pulmonary arterial hypertension: A pilot study. Eur. Respir. J. 2014, 43, 1691–1697. [Google Scholar] [CrossRef] [PubMed]
- Warnes, C.A.; Williams, R.G.; Bashore, T.M.; Child, J.S.; Connolly, H.M.; Dearani, J.A.; del Nido, P.; Fasules, J.W.; Graham, J.T.P.; Hijazi, Z.M.; et al. ACC/AHA 2008 Guidelines for the Management of Adults with Congenital Heart DiseaseA Report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Develop Guidelines on the Management of Adults with Congenital Heart Disease) Developed in Collaboration with the American Society of Echocardiography, Heart Rhythm Society, International Society for Adult Congenital Heart Disease, Society for Cardiovascular Angiography and Interventions, and Society of Thoracic Surgeons. J. Am. Coll. Cardiol. 2008, 52, e143–e263. [Google Scholar] [PubMed]
- Frost, A.E.; Quinones, M.A.; Zoghbi, W.A.; Noon, G.P. Reversal of pulmonary hypertension and subsequent repair of atrial septal defect after treatment with continuous intravenous epoprostenol. J. Heart Lung Transplant. 2005, 24, 501–503. [Google Scholar] [CrossRef] [PubMed]
- Ussia, G.P.; Mule, M.; Caruso, E.; Aiello, R.; Tamburino, C. Combined endothelin receptor antagonist and transcatheter interventional therapy of patent ductus arteriosus with severe pulmonary artery hypertension. Int. J. Cardiol. 2007, 116, 427–429. [Google Scholar] [CrossRef] [PubMed]
- Mitropoulos, F.A.; Apostolopoulou, S.C.; Kanakis, M.A.; Rammos, S.; Anagnostopoulos, C.E. Bosentan treatment in an adult with pulmonary hypertension due to patent ductus arteriosus permits surgical repair. J. Heart Lung Transplant. 2007, 26, 1345–1346. [Google Scholar] [CrossRef] [PubMed]
- Talwar, S.; Choudhary, S.K.; Saxena, A.; Kothari, S.S.; Juneja, R.; Airan, B. Unidirectional valved patches for closure of septal defects in patients with severe pulmonary hypertension. Ann. Pediatr. Cardiol. 2008, 1, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Althoff, T.F.; Knebel, F.; Panda, A.; McArdle, J.; Gliech, V.; Franke, I.; Witt, C.; Baumann, G.; Borges, A.C. Long-term follow-up of a fenestrated Amplatzer atrial septal occluder in pulmonary arterial hypertension. Chest 2008, 133, 283–285. [Google Scholar] [CrossRef] [PubMed]
- Van Riel, A.C.; Blok, I.M.; Zwinderman, A.H.; Wajon, E.M.; Sadee, A.S.; Bakker-de Boo, M.; van Dijk, A.P.; Hoendermis, E.S.; Riezebos, R.K.; Mulder, B.J.; et al. Lifetime Risk of Pulmonary Hypertension for All Patients After Shunt Closure. J. Am. Coll. Cardiol. 2015, 66, 1084–1086. [Google Scholar] [CrossRef] [PubMed]
- D’Alto, M.; Romeo, E.; Argiento, P.; Correra, A.; Santoro, G.; Gaio, G.; Sarubbi, B.; Calabro, R.; Russo, M.G. Hemodynamics of patients developing pulmonary arterial hypertension after shunt closure. Int. J. Cardiol. 2013, 168, 3797–3801. [Google Scholar] [CrossRef] [PubMed]
- Kerstein, D.; Levy, P.S.; Hsu, D.T.; Hordof, A.J.; Gersony, W.M.; Barst, R.J. Blade balloon atrial septostomy in patients with severe primary pulmonary hypertension. Circulation 1995, 91, 2028–2035. [Google Scholar] [CrossRef] [PubMed]
- Blanc, J.; Vouhe, P.; Bonnet, D. Potts shunt in patients with pulmonary hypertension. N. Engl. J. Med. 2004, 350, 623. [Google Scholar] [CrossRef] [PubMed]
- Baruteau, A.E.; Belli, E.; Boudjemline, Y.; Laux, D.; Levy, M.; Simonneau, G.; Carotti, A.; Humbert, M.; Bonnet, D. Palliative Potts shunt for the treatment of children with drug-refractory pulmonary arterial hypertension: Updated data from the first 24 patients. Eur. J. Cardio Thorac. Surg. 2015, 47, e105–e110. [Google Scholar] [CrossRef] [PubMed]
- Smadja, D.M.; Gaussem, P.; Mauge, L.; Israel-Biet, D.; Dignat-George, F.; Peyrard, S.; Agnoletti, G.; Vouhe, P.R.; Bonnet, D.; Levy, M. Circulating endothelial cells: A new candidate biomarker of irreversible pulmonary hypertension secondary to congenital heart disease. Circulation 2009, 119, 374–381. [Google Scholar] [CrossRef] [PubMed]
- Schermuly, R.T.; Pullamsetti, S.S.; Kwapiszewska, G.; Dumitrascu, R.; Tian, X.; Weissmann, N.; Ghofrani, H.A.; Kaulen, C.; Dunkern, T.; Schudt, C.; et al. Phosphodiesterase 1 upregulation in pulmonary arterial hypertension: Target for reverse-remodeling therapy. Circulation 2007, 115, 2331–2339. [Google Scholar] [CrossRef] [PubMed]
- Dumitrascu, R.; Weissmann, N.; Ghofrani, H.A.; Dony, E.; Beuerlein, K.; Schmidt, H.; Stasch, J.P.; Gnoth, M.J.; Seeger, W.; Grimminger, F.; et al. Activation of soluble guanylate cyclase reverses experimental pulmonary hypertension and vascular remodeling. Circulation 2006, 113, 286–295. [Google Scholar] [CrossRef] [PubMed]
- McMurtry, M.S.; Bonnet, S.; Wu, X.; Dyck, J.R.; Haromy, A.; Hashimoto, K.; Michelakis, E.D. Dichloroacetate prevents and reverses pulmonary hypertension by inducing pulmonary artery smooth muscle cell apoptosis. Circ. Res. 2004, 95, 830–840. [Google Scholar] [CrossRef] [PubMed]
- Vis, J.C.; van der Velde, E.T.; Schuuring, M.J.; Engelfriet-Rijk, L.C.; Harms, I.M.; Mantels, S.; Bouma, B.J.; Mulder, B.J. Wanted! 8000 heart patients: Identification of adult patients with a congenital heart defect lost to follow-up. Int. J. Cardiol. 2011, 149, 246–247. [Google Scholar] [CrossRef] [PubMed]
- Mackie, A.S.; Ionescu-Ittu, R.; Therrien, J.; Pilote, L.; Abrahamowicz, M.; Marelli, A.J. Children and Adults with Congenital Heart Disease Lost to Follow-Up. Circulation 2009, 120, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Gatzoulis, M.A.; Beghetti, M.; Landzberg, M.J.; Galie, N. Pulmonary arterial hypertension associated with congenital heart disease: Recent advances and future directions. Int. J. Cardiol. 2014, 177, 340–347. [Google Scholar] [CrossRef] [PubMed]
- Diller, G.P.; Kempny, A.; Inuzuka, R.; Radke, R.; Wort, S.J.; Baumgartner, H.; Gatzoulis, M.A.; Dimopoulos, K. Survival prospects of treatment naive patients with Eisenmenger: A systematic review of the literature and report of own experience. Heart 2014, 100, 1366–1372. [Google Scholar] [CrossRef] [PubMed]
- Cantor, W.J.; Harrison, D.A.; Moussadji, J.S.; Connelly, M.S.; Webb, G.D.; Liu, P.; McLaughlin, P.R.; Siu, S.C. Determinants of survival and length of survival in adults with Eisenmenger syndrome. Am. J. Cardiol. 1999, 84, 677–681. [Google Scholar] [CrossRef]
- Schuuring, M.J.; van Riel, A.C.; Vis, J.C.; Duffels, M.G.; van Dijk, A.P.; de Bruin-Bon, R.H.; Zwinderman, A.H.; Mulder, B.J.; Bouma, B.J. New predictors of mortality in adults with congenital heart disease and pulmonary hypertension: Midterm outcome of a prospective study. Int. J. Cardiol. 2015, 181, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Greenwood, R.D.; Nadas, A.S. The clinical course of cardiac disease in Down’s syndrome. Pediatrics 1976, 58, 893–897. [Google Scholar] [PubMed]
- Weijerman, M.E.; van Furth, A.M.; van der Mooren, M.D.; van Weissenbruch, M.M.; Rammeloo, L.; Broers, C.J.; Gemke, R.J. Prevalence of congenital heart defects and persistent pulmonary hypertension of the neonate with Down syndrome. Eur. J. Pediatr. 2010, 169, 1195–1199. [Google Scholar] [CrossRef] [PubMed]
- Duffels, M.G.; Vis, J.C.; van Loon, R.L.; Berger, R.M.; Hoendermis, E.S.; van Dijk, A.P.; Bouma, B.J.; Mulder, B.J. Down patients with Eisenmenger syndrome: Is bosentan treatment an option? Int. J. Cardiol. 2009, 134, 378–383. [Google Scholar] [CrossRef] [PubMed]
- D’Alto, M.; Romeo, E.; Argiento, P.; D’Andrea, A.; Sarubbi, B.; Correra, A.; Scognamiglio, G.; Papa, S.; Bossone, E.; Calabro, R.; et al. Therapy for pulmonary arterial hypertension due to congenital heart disease and Down’s syndrome. Int. J. Cardiol. 2013, 164, 323–326. [Google Scholar] [CrossRef] [PubMed]
- Schuuring, M.J.; Vis, J.C.; van Dijk, A.P.; van Melle, J.P.; Vliegen, H.W.; Pieper, P.G.; Sieswerda, G.T.; de Bruin-Bon, R.H.; Mulder, B.J.; Bouma, B.J. Impact of bosentan on exercise capacity in adults after the Fontan procedure: A randomized controlled trial. Eur. J. Heart Fail. 2013, 15, 690–698. [Google Scholar] [CrossRef] [PubMed]
- Hebert, A.; Mikkelsen, U.R.; Thilen, U.; Idorn, L.; Jensen, A.S.; Nagy, E.; Hanseus, K.; Sorensen, K.E.; Sondergaard, L. Bosentan improves exercise capacity in adolescents and adults after Fontan operation: The TEMPO (Treatment with Endothelin Receptor Antagonist in Fontan Patients, a Randomized, Placebo-Controlled, Double-Blind Study Measuring Peak Oxygen Consumption) study. Circulation 2014, 130, 2021–2030. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, D.J.; French, B.; McBride, M.G.; Marino, B.S.; Mirarchi, N.; Hanna, B.D.; Wernovsky, G.; Paridon, S.M.; Rychik, J. Impact of Oral Sildenafil on Exercise Performance in Children and Young Adults After the Fontan Operation: A Randomized, Double-Blind, Placebo-Controlled, Crossover Trial. Circulation 2011, 123, 1185–1193. [Google Scholar] [CrossRef] [PubMed]
- Sharif Kashani, B.; Tahmaseb Pour, P.; Malekmohammad, M.; Behzadnia, N.; Sheybani-Afshar, F.; Fakhri, M.; Chaibakhsh, S.; Naghashzadeh, F.; Aidenlou, S. Oral l-citrulline malate in patients with idiopathic pulmonary arterial hypertension and Eisenmenger Syndrome: A clinical trial. J. Cardiol. 2014, 64, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Ghofrani, H.A.; Seeger, W.; Grimminger, F. Imatinib for the treatment of pulmonary arterial hypertension. N. Engl. J. Med. 2005, 353, 1412–1413. [Google Scholar] [CrossRef] [PubMed]
- Ghofrani, H.A.; Morrell, N.W.; Hoeper, M.M.; Olschewski, H.; Peacock, A.J.; Barst, R.J.; Shapiro, S.; Golpon, H.; Toshner, M.; Grimminger, F.; et al. Imatinib in pulmonary arterial hypertension patients with inadequate response to established therapy. Am. J. Respir. Crit. Care Med. 2010, 182, 1171–1177. [Google Scholar] [CrossRef] [PubMed]
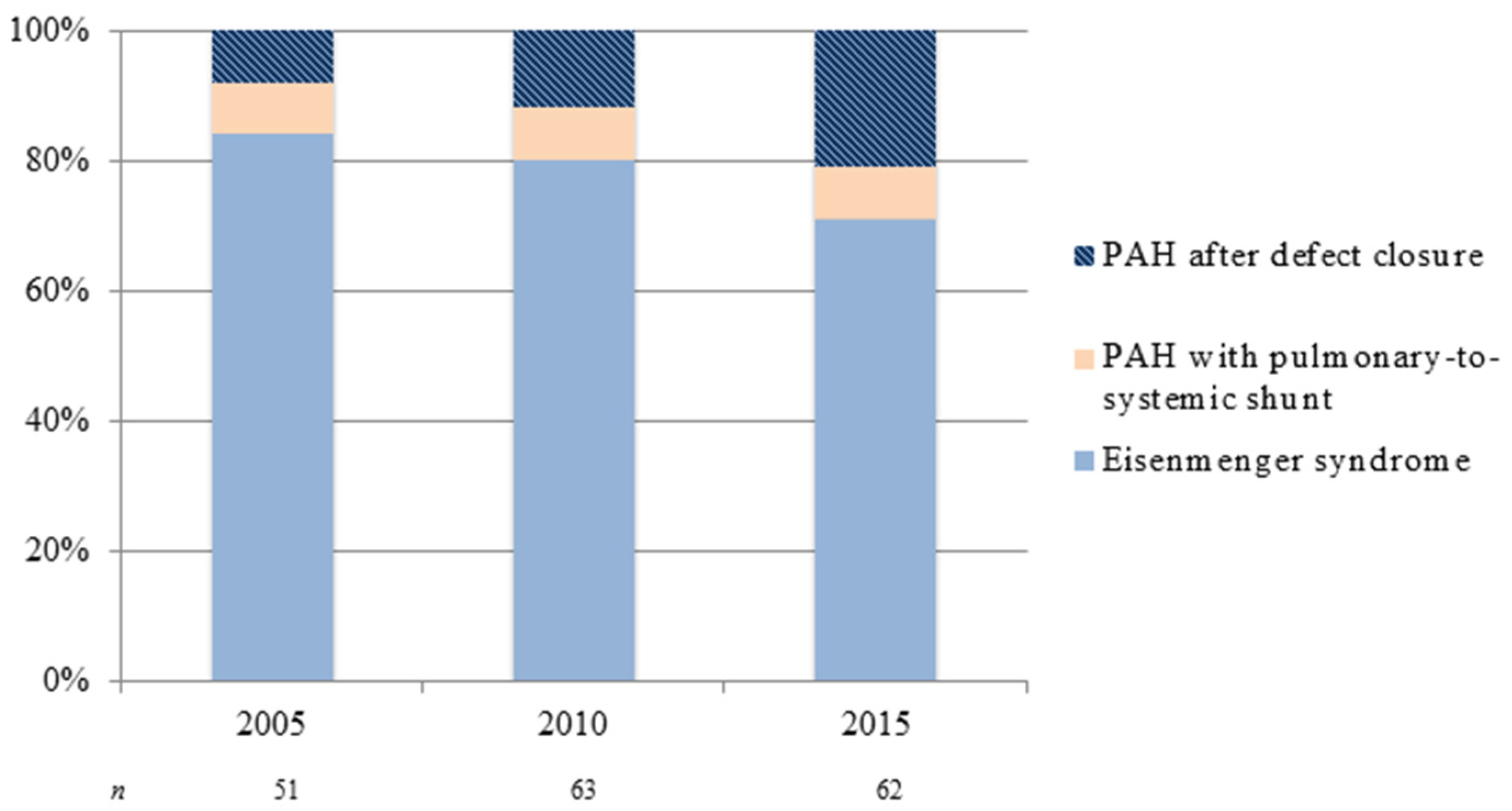
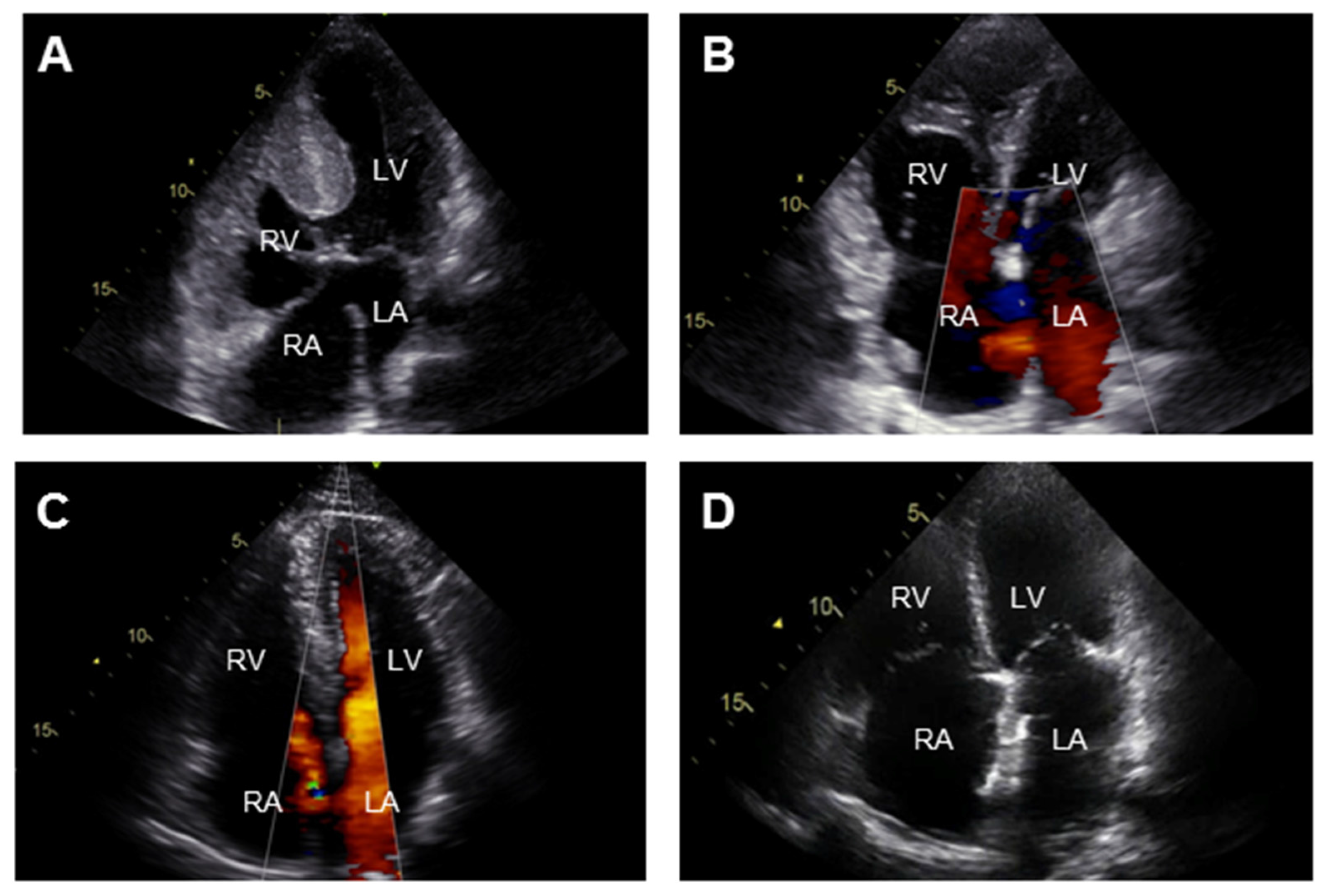
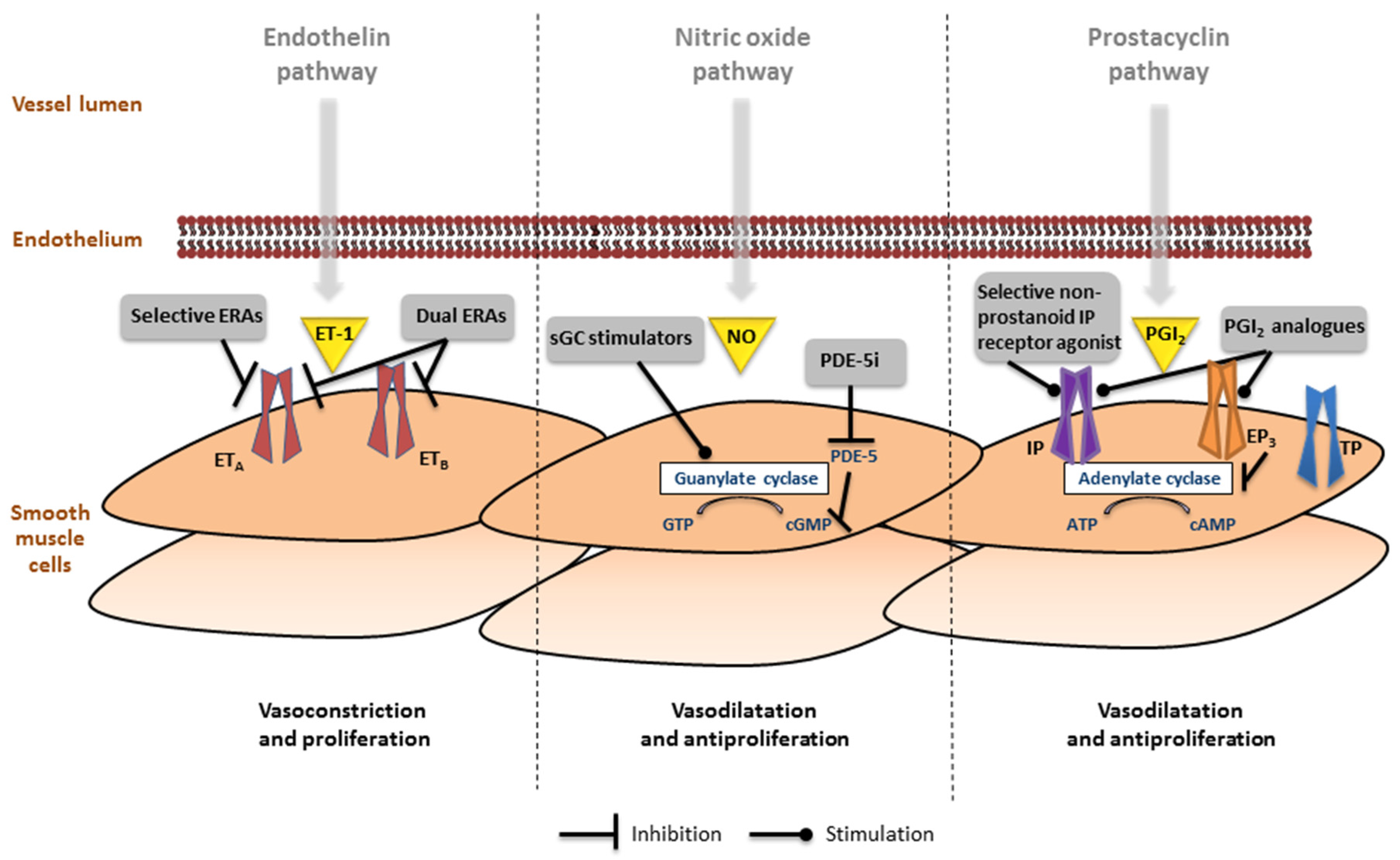

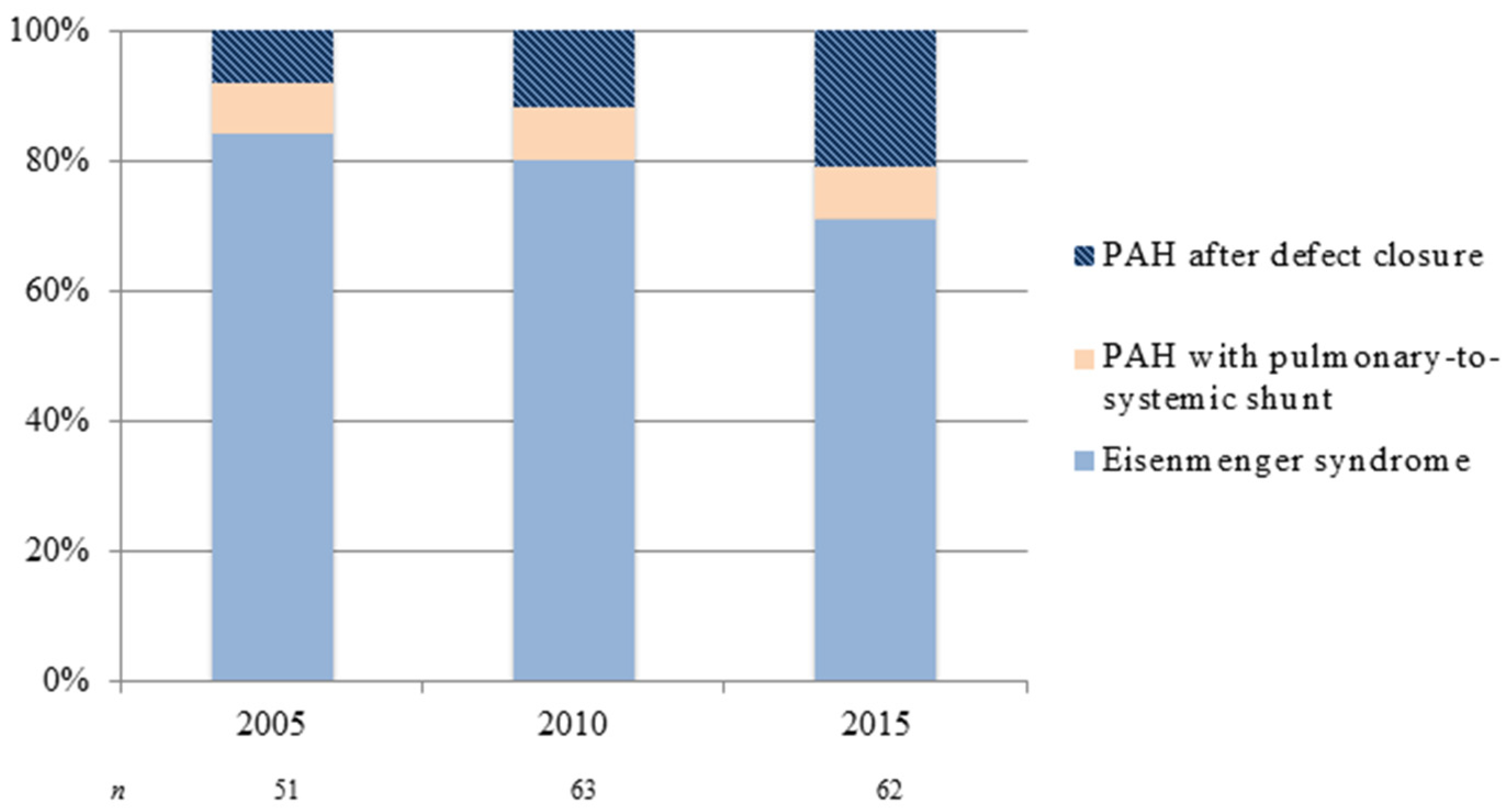{kind=link}
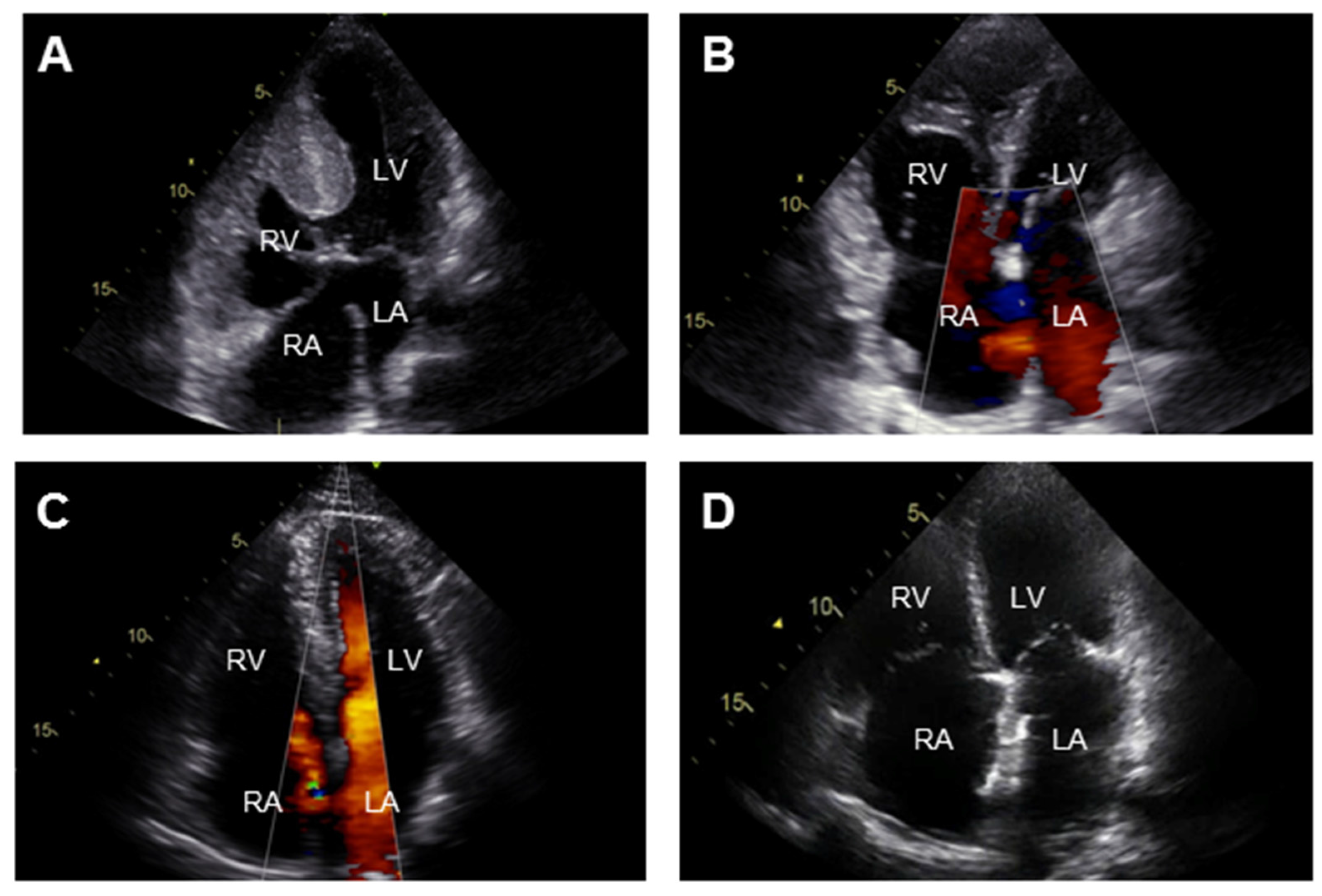{kind=link}
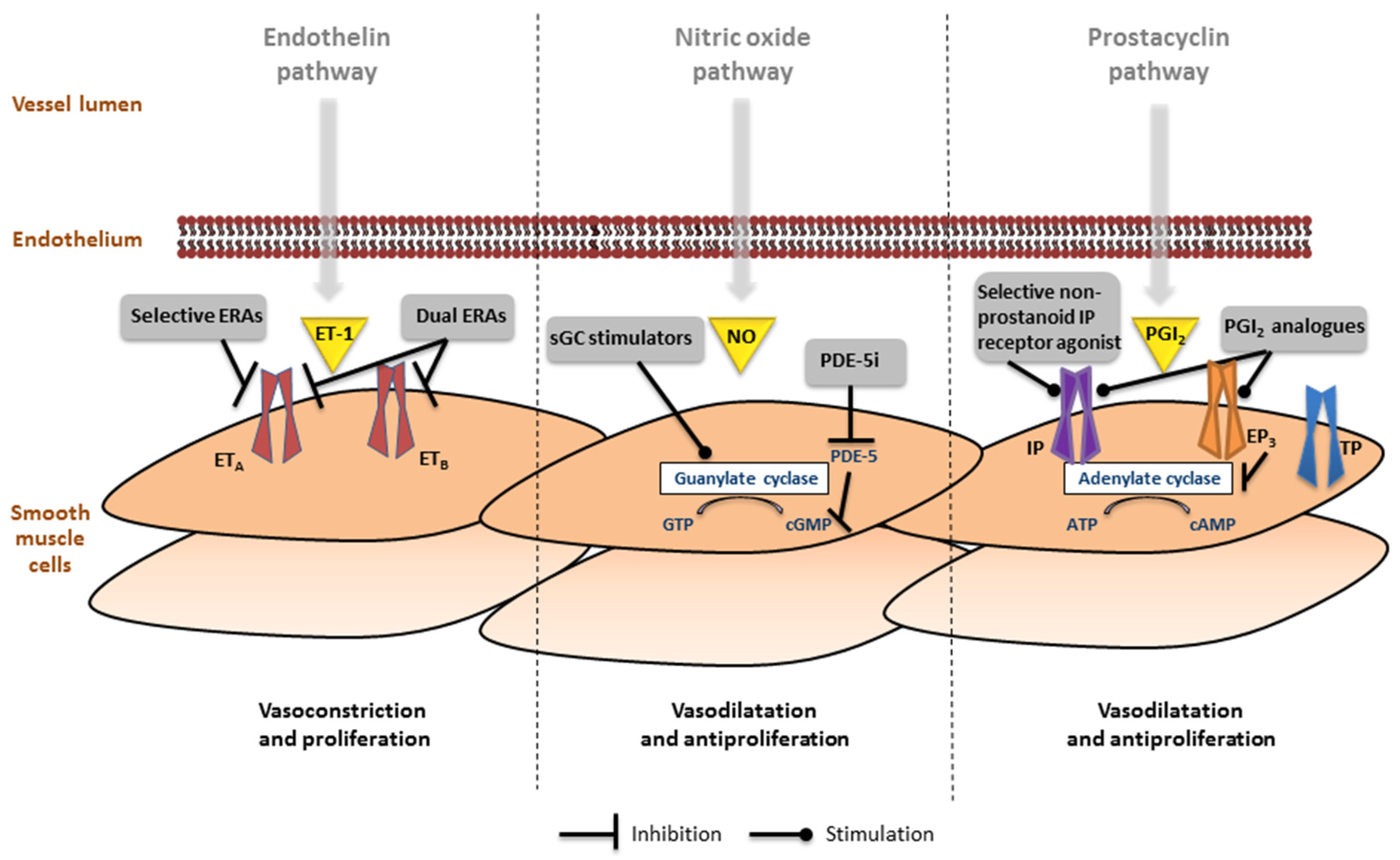{kind=link}
| Subgroup | Clinical Classification |
|---|---|
| (i) | Eisenmenger syndrome Includes intra- and extracardiac defects with initial systemic-to-pulmonary shunts leading to elevated PVR with ultimate reversal or bidirectional shunting and development of central cyanosis at rest. |
| (ii) | PAH associated with prevalent systemic-to-pulmonary shunt lesions PVR is mild to moderately elevated in presence of moderate to large systemic-to-pulmonary shunt, either correctable or not. No cyanosis at rest. |
| (iii) | PAH with small or coincidental cardiac defects The small cardiac defect (usually ASD < 2 cm and VSD < 1 cm of effective diameter assessed by echocardiography) is generally considered coincidental and unrelated to the marked elevation in PVR. The clinical picture is very similar to idiopathic PAH. |
| (iv) | PAH after defect closure PAH is either persisting or recurring immediately or months to years after closure of the cardiac defect in the absence of relevant postoperative hemodynamic lesions. |
| First Author (Study Acronym) | n with CHD (% of Study Population) | Background Therapy | Intervention | Follow-Up (Weeks) | Primary Outcome | Study Conclusion |
|---|---|---|---|---|---|---|
| Study Population Including Mixed Group of PAH-CHD | ||||||
| Simonneau et al. [42] | 109 (23) | None | Treprostinil | 12 | Δ 6MWD | ↑ PVR, symptoms |
| Galiè et al. [43] (EARLY) | 32 (17) | None | Bosentan | 26 | Δ 6MWD and PVR | ↑ |
| Galiè et al. [44] (PHIRST) | 47 (12) | None or bosentan | Tadalafil | 16 | Δ 6MWD | ↑ TTCW |
| Ghofrani et al. [45] (PATENT-1) | 35 (8) | None, ERA or prostanoids | Riociguat | 12 | Δ 6MWD | ↑ PVR, WHO, TTCW, NT-proBNP |
| Study Population Including PAH with Closed Defects | ||||||
| Simonneau et al. [46] (PACES) | 10 (4) | Epoprostenol | Sildenafil | 16 | Δ 6MWD | ↑ PVR, TTCW, QoL |
| Galiè et al. [47] (SUPER-1) | 18 (6) | None | Sildenafil | 12 | Δ 6MWD | ↑ WHO class, and hemodynamics |
| Tapson et al. [48] (FREEDOM-C) | 22 (6) | ERA, PDE-5i or both | Treprostinil | 16 | Δ 6MWD | = |
| Tapson et al. [49] (FREEDOM-C2) | 4 (1) | ERA, PDE-5i or both | Treprostinil | 16 | Δ 6MWD | = |
| Pulido et al. [50] (SERAPHIN) | 62 (8) | PDE-5i or prostanoids | Macitentan | 85–104 | TTCW | ↑ |
| Jing et al. [51] (FREEDOM-M) | 18 (5) | None | Treprostinil | 12 | Δ 6MWD | ↑ |
| McLaughlin et al. [52] (COMPASS-2) | 20 (6) | Sildenafil | Bosentan | +/− 170 | TTCW | = |
| Galiè et al. [53] (AMBITION) | 13 (3) | None | Ambrisentan + tadalafil vs. ambrisentan vs. tadalafil | 74 | TTCW | ↑ with initial combination therapy > ambrisentan or tadalafil monotherapy |
| Sitbon et al. [54] (GRIPHON) | 110 (10) | None, ERA, PDE-5i or both | Selexipag | 67 | TTCW | ↑ in all baseline treatment groups |
| Study Population Including Eisenmenger Syndrome | ||||||
| Galiè et al. [55] (BREATHE-5) | 54 (100) | None | Bosentan | 16 | Δ Spo2 and PVR | ↑ without compromising SpO2 |
| Singh et al. [56] | 10 (50) | None | Sildenafil | 6 | Δ 6MWD | ↑ NYHA class, PVR |
| Iversen et al. [57] | 21 (100) | None | Bosentan, add sildenafil cross-over | 39 | Δ 6MWD | Bosentan alone ↑ with addition of sildenafil =, but ↑ SO2 |
| Mukhopadhyay et al. [58] | 28 (100) | None | Tadalafil | 6 | Δ 6MWD | ↑ WHO, SO2, and PVR |
| Study (Clinicaltrial.gov) | Phase | Patients | Intervention | Primary Outcome | Anticipated Completion |
|---|---|---|---|---|---|
| Combination Therapy with Currently Available PAH-Specific Therapy | |||||
| BEAT (NCT01908699) | III | PAH on inhaled treprostinil | Beraprost vs. placebo | TTCW | 2017 |
| TRITON (NCT02558231) | III | PAH diagnosis <6 months | Macitentan + tadalafil + selexipag vs. macitentan + tadalafil + placebo | PVR | 2018 |
| FREEDOM-Ev (NCT01560624) | III | PAH on monotherapy | Oral treprostinil vs. placebo | TTCW | 2018 |
| INOvation-1 (NCT02725372) | III | PAH on therapy | Inhaled NO vs. placebo | Δ 6MWD | 2018 |
| REPLACE (NCT02891850) | IV | PAH on PDE-5i | Switch to riociguat vs. standard-of-care | Δ 6MWD, WHO class, NT-proBNP | 2018 |
| Triple vs. dual therapy (NCT02999906) | III | PAH on ambrisentan + tadalafil | Treprostinil vs. placebo | Δ 6MWD | 2022 |
| Heart Failure Therapy | |||||
| CAPS-PAH (NCT02253394) | IV | PAH on ambrisentan | Spironolactone vs. placebo | Δ 6MWD, pVO2 | 2017 |
| Spironolactone (NCT01712620) | II | PAH | Spironolactone vs. placebo | Δ 6MWD, TTCW | 2018 |
| Beta-blockers (NCT02507011) | II | PAH on therapy | Carvedilol vs. placebo | Δ RVEF | 2018 |
| New Candidate Drugs | |||||
| LIBERTY (NCT02664558) | II | PAH, on therapy | Ubenimex vs. placebo | PVR | 2017 |
| APD811 in PAH (NCT02279160) | II | PAH, on therapy | Ralinepag vs. placebo | PVR, Δ 6MWD | 2017 |
| LARIAT (NCT02036970) | II | PAH | Bardoxolone methyl vs. placebo | Δ 6MWD | 2018 |
| QCC374 in PAH (NCT02927366) | II | PAH on therapy | QCC374 vs. placebo | PVR | 2019 |
| SAPPHIRE (NCT03001414) | II | PAH on therapy | Autologous progenitor cell-based gene therapy vs. placebo | Δ 6MWD | 2020 |
| Type | Correctable? | AHA/ACC CHD Guidelines, 2008 [83] | ESC GUCH Guidelines, 2010 [15] | ESC/ERS PH Guidelines, 2015 [13] * |
|---|---|---|---|---|
| ASD | Yes |
|
|
|
| No |
|
|
| |
| Individual patient evaluation |
|
|
| |
| VSD | Yes |
|
|
|
| No |
|
|
| |
| Individual patient evaluation |
|
|
|
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Van Dissel, A.C.; Mulder, B.J.M.; Bouma, B.J. The Changing Landscape of Pulmonary Arterial Hypertension in the Adult with Congenital Heart Disease. J. Clin. Med. 2017, 6, 40. https://doi.org/10.3390/jcm6040040
Van Dissel AC, Mulder BJM, Bouma BJ. The Changing Landscape of Pulmonary Arterial Hypertension in the Adult with Congenital Heart Disease. Journal of Clinical Medicine. 2017; 6(4):40. https://doi.org/10.3390/jcm6040040
Chicago/Turabian StyleVan Dissel, Alexandra C., Barbara J. M. Mulder, and Berto J. Bouma. 2017. "The Changing Landscape of Pulmonary Arterial Hypertension in the Adult with Congenital Heart Disease" Journal of Clinical Medicine 6, no. 4: 40. https://doi.org/10.3390/jcm6040040
APA StyleVan Dissel, A. C., Mulder, B. J. M., & Bouma, B. J. (2017). The Changing Landscape of Pulmonary Arterial Hypertension in the Adult with Congenital Heart Disease. Journal of Clinical Medicine, 6(4), 40. https://doi.org/10.3390/jcm6040040