
Biochemical Assessment of Coenzyme Q10 Deficiency


Abstract
:1. Introduction
2. CoQ10 Deficiency Syndrome
3. Primary CoQ10 Deficiency Therapy
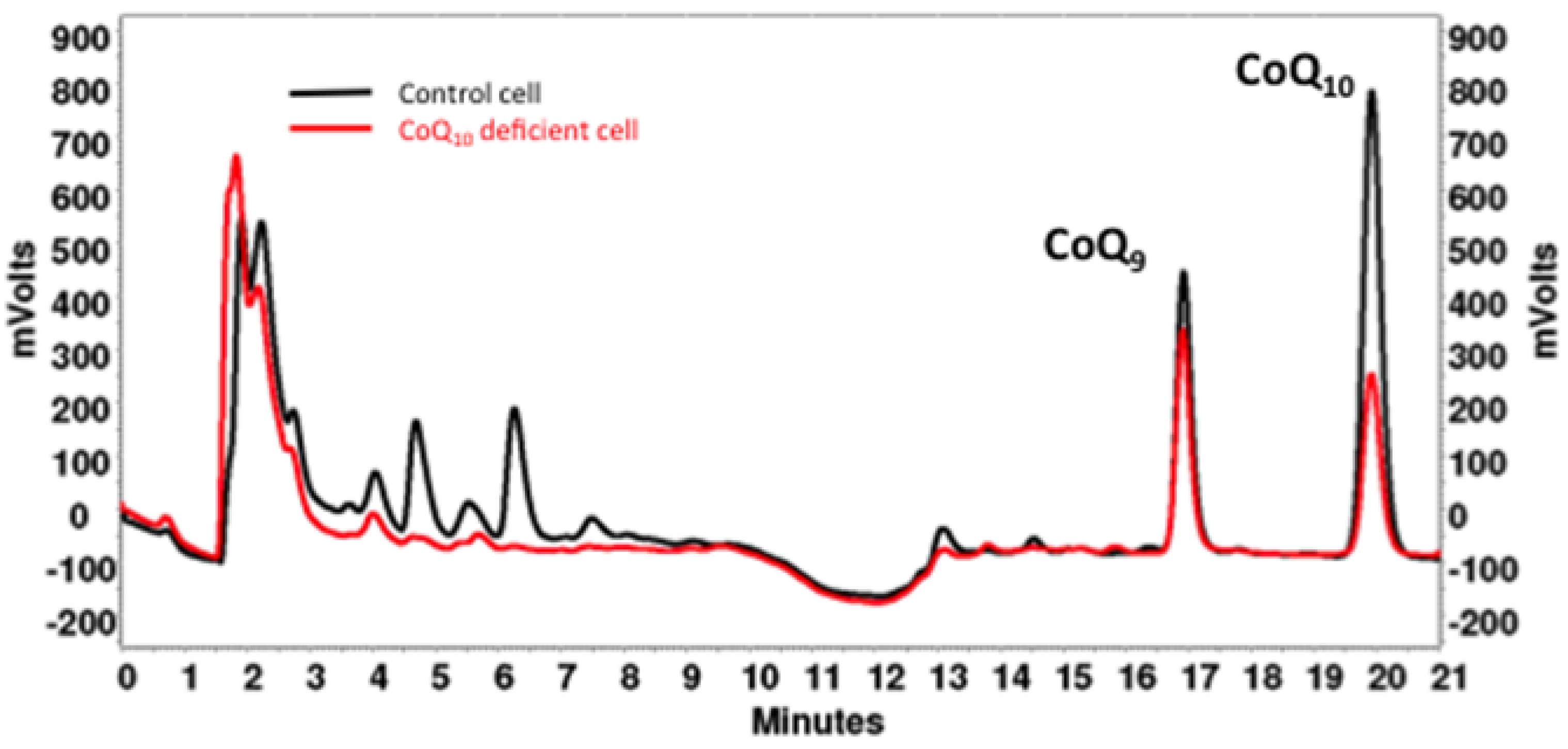
4. CoQ10 Determination in Cells and Tissues
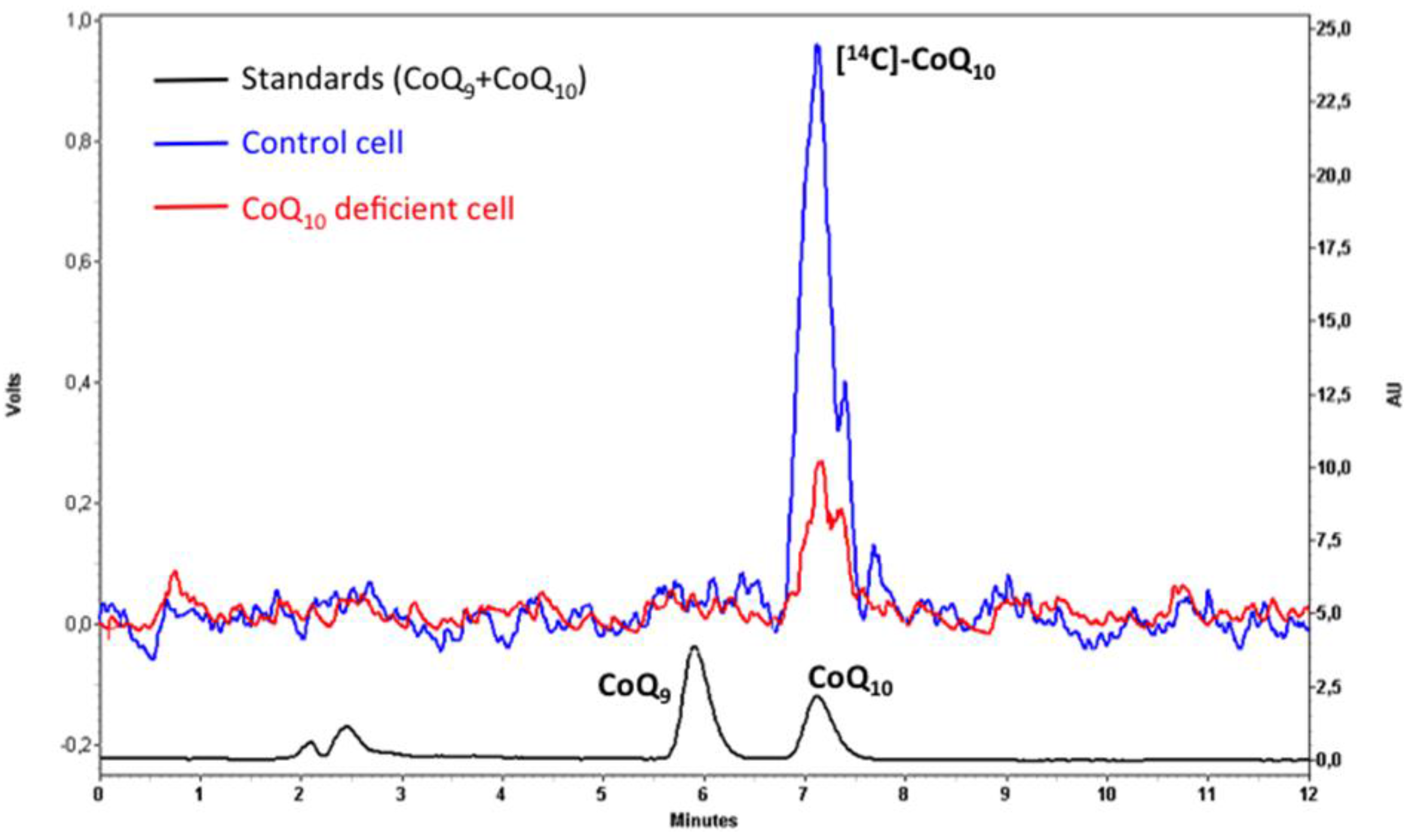
5. Analysis of CoQ10 Biosynthesis
6. Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial diseases, Nature reviews. Dis. Prim. 2016, 2, 16080. [Google Scholar] [CrossRef] [PubMed]
- Lapuente-Brun, E.; Moreno-Loshuertos, R.; Acin-Perez, R.; Latorre-Pellicer, A.; Colas, C.; Balsa, E.; Perales-Clemente, E.; Quiros, P.M.; Calvo, E.; Rodriguez-Hernandez, M.A.; et al. Supercomplex assembly determines electron flux in the mitochondrial electron transport chain. Science 2013, 340, 1567–1570. [Google Scholar] [CrossRef] [PubMed]
- Alcazar-Fabra, M.; Navas, P.; Brea-Calvo, G. Coenzyme Q biosynthesis and its role in the respiratory chain structure. Biochim. Biophys. Acta 2016, 1857, 1073–1078. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Lluch, G.; Rodriguez-Aguilera, J.C.; Santos-Ocana, C.; Navas, P. Is coenzyme Q a key factor in aging? Mech. Ageing Dev. 2010, 131, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Bentinger, M.; Tekle, M.; Dallner, G. Coenzyme Q—Biosynthesis and functions. Biochem. Biophys. Res. Commun. 2010, 396, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Mariscal, I.; Garcia-Teston, E.; Padilla, S.; Martin-Montalvo, A.; Viciana, T.P.; Vazquez-Fonseca, L.; Dominguez, P.G.; Santos-Ocana, C. The regulation of coenzyme q biosynthesis in eukaryotic cells: All that yeast can tell us. Mol. Syndromol. 2014, 5, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Turunen, M.; Peters, J.M.; Gonzalez, F.J.; Schedin, S.; Dallner, G. Influence of peroxisome proliferator-activated receptor alpha on ubiquinone biosynthesis. J. Mol. Biol. 2000, 297, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Bentinger, M.; Tekle, M.; Brismar, K.; Chojnacki, T.; Swiezewska, E.; Dallner, G. Stimulation of coenzyme Q synthesis. Biofactors 2008, 32, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Brea-Calvo, G.; Siendones, E.; Sanchez-Alcazar, J.A.; de Cabo, R.; Navas, P. Cell survival from chemotherapy depends on NF-kappaB transcriptional up-regulation of coenzyme Q biosynthesis. PLoS ONE 2009, 4, e5301. [Google Scholar] [CrossRef] [PubMed]
- Cascajo, M.V.; Abdelmohsen, K.; Noh, J.H.; Fernandez-Ayala, D.J.; Willers, I.M.; Brea, G.; Lopez-Lluch, G.; Valenzuela-Villatoro, M.; Cuezva, J.M.; Gorospe, M.; et al. RNA-binding proteins regulate cell respiration and coenzyme Q biosynthesis by post-transcriptional regulation of COQ7. RNA Biol. 2016, 13, 622–634. [Google Scholar] [CrossRef] [PubMed]
- Martin-Montalvo, A.; Gonzalez-Mariscal, I.; Padilla, S.; Ballesteros, M.; Brautigan, D.L.; Navas, P.; Santos-Ocana, C. Respiratory-induced coenzyme Q biosynthesis is regulated by a phosphorylation cycle of Cat5p/Coq7p. Biochem. J. 2011, 440, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Martín-Montalvo, A.; González-Mariscal, I.; Pomares-Viciana, T.; Padilla-López, S.; Ballesteros, M.; Vazquez-Fonseca, L.; Gandolfo, P.; Brautigan, D.L.; Navas, P.; Santos-Ocaña, C. The phosphatase Ptc7 induces coenzyme Q biosynthesis by activating the hydroxylase Coq7 in yeast. J. Biol. Chem. 2013, 288, 28126–28137. [Google Scholar] [CrossRef] [PubMed]
- Lanning, N.J.; Looyenga, B.D.; Kauffman, A.L.; Niemi, N.M.; Sudderth, J.; DeBerardinis, R.J.; MacKeigan, J.P. A mitochondrial RNAi screen defines cellular bioenergetic determinants and identifies an adenylate kinase as a key regulator of ATP levels. Cell Rep. 2014, 7, 907–917. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Mariscal, I.; Garcia-Teston, E.; Padilla, S.; Martin-Montalvo, A.; Pomares-Viciana, T.; Vazquez-Fonseca, L.; Gandolfo-Dominguez, P.; Santos-Ocana, C. Regulation of coenzyme Q biosynthesis in yeast: A new complex in the block. IUBMB Life 2014, 66, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Padilla, S.; Tran, U.C.; Jimenez-Hidalgo, M.; Lopez-Martin, J.M.; Martin-Montalvo, A.; Clarke, C.F.; Navas, P.; Santos-Ocana, C. Hydroxylation of demethoxy-Q6 constitutes a control point in yeast coenzyme Q6 biosynthesis. Cell. Mol. Life Sci. 2009, 66, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Lohman, D.C.; Forouhar, F.; Beebe, E.T.; Stefely, M.S.; Minogue, C.E.; Ulbrich, A.; Stefely, J.A.; Sukumar, S.; Luna-Sanchez, M.; Jochem, A.; et al. Mitochondrial COQ9 is a lipid-binding protein that associates with COQ7 to enable coenzyme Q biosynthesis. Proc. Natl. Acad. Sci. USA 2014, 111, E4697–E4705. [Google Scholar] [CrossRef] [PubMed]
- Stefely, J.A.; Licitra, F.; Laredj, L.; Reidenbach, A.G.; Kemmerer, Z.A.; Grangeray, A.; Jaeg-Ehret, T.; Minogue, C.E.; Ulbrich, A.; Hutchins, P.D.; et al. Cerebellar Ataxia and Coenzyme Q Deficiency through Loss of Unorthodox Kinase Activity. Mol. Cell 2016, 63, 608–620. [Google Scholar] [CrossRef] [PubMed]
- Stefely, J.A.; Reidenbach, A.G.; Ulbrich, A.; Oruganty, K.; Floyd, B.J.; Jochem, A.; Saunders, J.M.; Johnson, I.E.; Minogue, C.E.; Wrobel, R.L.; et al. Mitochondrial ADCK3 Employs an Atypical Protein Kinase-like Fold to Enable Coenzyme Q Biosynthesis. Mol. Cell 2015, 57, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Parrado-Fernandez, C.; Lopez-Lluch, G.; Rodriguez-Bies, E.; Santa-Cruz, S.; Navas, P.; Ramsey, J.J.; Villalba, J.M. Calorie restriction modifies ubiquinone and COQ transcript levels in mouse tissues. Free Radic. Biol. Med. 2011, 50, 1728–1736. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Lluch, G.; Santos-Ocana, C.; Sanchez-Alcazar, J.A.; Fernandez-Ayala, D.J.; Asencio-Salcedo, C.; Rodriguez-Aguilera, J.C.; Navas, P. Mitochondrial responsibility in ageing process: Innocent, suspect or guilty. Biogerontology 2015, 16, 599–620. [Google Scholar] [CrossRef] [PubMed]
- Schon, E.A.; DiMauro, S.; Hirano, M.; Gilkerson, R.W. Therapeutic prospects for mitochondrial disease. Trends Mol. Med. 2010, 16, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Quinzii, C.M.; Emmanuele, V.; Hirano, M. Clinical presentations of coenzyme q10 deficiency syndrome. Mol. Syndromol. 2014, 5, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Doimo, M.; Desbats, M.A.; Cerqua, C.; Cassina, M.; Trevisson, E.; Salviati, L. Genetics of coenzyme q10 deficiency. Mol. Syndromol. 2014, 5, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Desbats, M.A.; Lunardi, G.; Doimo, M.; Trevisson, E.; Salviati, L. Genetic bases and clinical manifestations of coenzyme Q10 (CoQ 10) deficiency. J. Inherit. Metab. Dis. 2015, 38, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Yubero, D.; Montero, R.; Martin, M.A.; Montoya, J.; Ribes, A.; Grazina, M.; Trevisson, E.; Rodriguez-Aguilera, J.C.; Hargreaves, I.P.; Salviati, L.; et al. Secondary coenzyme Q10 deficiencies in oxidative phosphorylation (OXPHOS) and non-OXPHOS disorders. Mitochondrion 2016, 30, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Montero, R.; Sanchez-Alcazar, J.A.; Briones, P.; Navarro-Sastre, A.; Gallardo, E.; Bornstein, B.; Herrero-Martin, D.; Rivera, H.; Martin, M.A.; Marti, R.; et al. Coenzyme Q10 deficiency associated with a mitochondrial DNA depletion syndrome: A case report. Clin. Biochem. 2009, 42, 742–745. [Google Scholar] [CrossRef] [PubMed]
- Quinzii, C.M.; Kattah, A.G.; Naini, A.; Akman, H.O.; Mootha, V.K.; DiMauro, S.; Hirano, M. Coenzyme Q deficiency and cerebellar ataxia associated with an aprataxin mutation. Neurology 2005, 64, 539–541. [Google Scholar] [CrossRef] [PubMed]
- Gempel, K.; Topaloglu, H.; Talim, B.; Schneiderat, P.; Schoser, B.G.; Hans, V.H.; Palmafy, B.; Kale, G.; Tokatli, A.; Quinzii, C.; et al. The myopathic form of coenzyme Q10 deficiency is caused by mutations in the electron-transferring-flavoprotein dehydrogenase (ETFDH) gene. Brain 2007, 130, 2037–2044. [Google Scholar] [CrossRef] [PubMed]
- Miles, M.V.; Putnam, P.E.; Miles, L.; Tang, P.H.; DeGrauw, A.J.; Wong, B.L.; Horn, P.S.; Foote, H.L.; Rothenberg, M.E. Acquired coenzyme Q10 deficiency in children with recurrent food intolerance and allergies. Mitochondrion 2011, 11, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Haas, D.; Niklowitz, P.; Horster, F.; Baumgartner, E.R.; Prasad, C.; Rodenburg, R.J.; Hoffmann, G.F.; Menke, T.; Okun, J.G. Coenzyme Q(10) is decreased in fibroblasts of patients with methylmalonic aciduria but not in mevalonic aciduria. J. Inherit. Metab. Dis. 2009, 32, 570–575. [Google Scholar] [CrossRef] [PubMed]
- Maes, M.; Mihaylova, I.; Kubera, M.; Uytterhoeven, M.; Vrydags, N.; Bosmans, E. Coenzyme Q10 deficiency in myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS) is related to fatigue, autonomic and neurocognitive symptoms and is another risk factor explaining the early mortality in ME/CFS due to cardiovascular disorder. Neuro Endocrinol. Lett. 2009, 30, 470–476. [Google Scholar] [PubMed]
- Fragaki, K.; Cano, A.; Benoist, J.F.; Rigal, O.; Chaussenot, A.; Rouzier, C.; Bannwarth, S.; Caruba, C.; Chabrol, B.; Paquis-Flucklinger, V. Fatal heart failure associated with CoQ10 and multiple OXPHOS deficiency in a child with propionic acidemia. Mitochondrion 2011, 11, 533–536. [Google Scholar] [CrossRef] [PubMed]
- Ogasahara, S.; Engel, A.G.; Frens, D.; Mack, D. Muscle coenzyme Q deficiency in familial mitochondrial encephalomyopathy. Proc. Natl. Acad. Sci. USA 1989, 86, 2379–2382. [Google Scholar] [CrossRef] [PubMed]
- Lopez, L.C.; Schuelke, M.; Quinzii, C.M.; Kanki, T.; Rodenburg, R.J.; Naini, A.; Dimauro, S.; Hirano, M. Leigh syndrome with nephropathy and CoQ10 deficiency due to decaprenyl diphosphate synthase subunit 2 (PDSS2) mutations. Am. J. Hum. Genet. 2006, 79, 1125–1129. [Google Scholar] [CrossRef] [PubMed]
- Quinzii, C.; Naini, A.; Salviati, L.; Trevisson, E.; Navas, P.; Dimauro, S.; Hirano, M. A mutation in para-hydroxybenzoate-polyprenyl transferase (COQ2) causes primary coenzyme Q10 deficiency. Am. J. Hum. Genet. 2006, 78, 345–349. [Google Scholar] [CrossRef] [PubMed]
- Heeringa, S.F.; Chernin, G.; Chaki, M.; Zhou, W.; Sloan, A.J.; Ji, Z.; Xie, L.X.; Salviati, L.; Hurd, T.W.; Vega-Warner, V.; et al. COQ6 mutations in human patients produce nephrotic syndrome with sensorineural deafness. J. Clin. Investig. 2011, 121, 2013–2024. [Google Scholar] [CrossRef] [PubMed]
- Ashraf, S.; Gee, H.Y.; Woerner, S.; Xie, L.X.; Vega-Warner, V.; Lovric, S.; Fang, H.; Song, X.; Cattran, D.C.; Avila-Casado, C.; et al. ADCK4 mutations promote steroid-resistant nephrotic syndrome through CoQ10 biosynthesis disruption. J. Clin. Investig. 2013, 123, 5179–5189. [Google Scholar] [CrossRef] [PubMed]
- Mollet, J.; Giurgea, I.; Schlemmer, D.; Dallner, G.; Chretien, D.; Delahodde, A.; Bacq, D.; de Lonlay, P.; Munnich, A.; Rotig, A. Prenyldiphosphate synthase, subunit 1 (PDSS1) and OH-benzoate polyprenyltransferase (COQ2) mutations in ubiquinone deficiency and oxidative phosphorylation disorders. J. Clin. Investig. 2007, 117, 765–772. [Google Scholar] [CrossRef] [PubMed]
- Duncan, A.J.; Bitner-Glindzicz, M.; Meunier, B.; Costello, H.; Hargreaves, I.P.; Lopez, L.C.; Hirano, M.; Quinzii, C.M.; Sadowski, M.I.; Hardy, J.; et al. A nonsense mutation in COQ9 causes autosomal-recessive neonatal-onset primary coenzyme Q10 deficiency: A potentially treatable form of mitochondrial disease. Am. J. Hum. Genet. 2009, 84, 558–566. [Google Scholar] [CrossRef] [PubMed]
- Freyer, C.; Stranneheim, H.; Naess, K.; Mourier, A.; Felser, A.; Maffezzini, C.; Lesko, N.; Bruhn, H.; Engvall, M.; Wibom, R.; et al. Rescue of primary ubiquinone deficiency due to a novel COQ7 defect using 2,4-dihydroxybensoic acid. J. Med. Genet. 2015, 52, 779–783. [Google Scholar] [CrossRef]
- Mollet, J.; Delahodde, A.; Serre, V.; Chretien, D.; Schlemmer, D.; Lombes, A.; Boddaert, N.; Desguerre, I.; de Lonlay, P.; de Baulny, H.O.; et al. CABC1 gene mutations cause ubiquinone deficiency with cerebellar ataxia and seizures. Am. J. Hum. Genet. 2008, 82, 623–630. [Google Scholar] [CrossRef] [PubMed]
- Lagier-Tourenne, C.; Tazir, M.; Lopez, L.C.; Quinzii, C.M.; Assoum, M.; Drouot, N.; Busso, C.; Makri, S.; Ali-Pacha, L.; Benhassine, T.; et al. ADCK3, an ancestral kinase, is mutated in a form of recessive ataxia associated with coenzyme Q10 deficiency. Am. J. Hum. Genet. 2008, 82, 661–672. [Google Scholar] [CrossRef] [PubMed]
- Horvath, R.; Czermin, B.; Gulati, S.; Demuth, S.; Houge, G.; Pyle, A.; Dineiger, C.; Blakely, E.L.; Hassani, A.; Foley, C.; et al. Adult-onset cerebellar ataxia due to mutations in CABC1/ADCK3. J. Neurol. Neurosurg. Psychiatry 2012, 83, 174–178. [Google Scholar] [CrossRef] [PubMed]
- Mignot, C.; Apartis, E.; Durr, A.; Lourenco, C.M.; Charles, P.; Devos, D.; Moreau, C.; de Lonlay, P.; Drouot, N.; Burglen, L.; et al. Phenotypic variability in ARCA2 and identification of a core ataxic phenotype with slow progression. Orphanet J. Rare Dis. 2013, 8, 173. [Google Scholar] [CrossRef]
- Liu, Y.T.; Hersheson, J.; Plagnol, V.; Fawcett, K.; Duberley, K.E.; Preza, E.; Hargreaves, I.P.; Chalasani, A.; Laura, M.; Wood, N.W.; et al. Autosomal-recessive cerebellar ataxia caused by a novel ADCK3 mutation that elongates the protein: Clinical, genetic and biochemical characterisation. J. Neurol. Neurosurg. Psychiatry 2014, 85, 493–498. [Google Scholar] [CrossRef]
- Blumkin, L.; Leshinsky-Silver, E.; Zerem, A.; Yosovich, K.; Lerman-Sagie, T.; Lev, D. Heterozygous Mutations in the ADCK3 Gene in Siblings with Cerebellar Atrophy and Extreme Phenotypic Variability. JIMD Rep. 2014, 12, 103–107. [Google Scholar] [PubMed]
- Hikmat, O.; Tzoulis, C.; Knappskog, P.M.; Johansson, S.; Boman, H.; Sztromwasser, P.; Lien, E.; Brodtkorb, E.; Ghezzi, D.; Bindoff, L.A. ADCK3 mutations with epilepsy, stroke-like episodes and ataxia: A POLG mimic? Eur. J. Neurol. 2016, 23, 1188–1194. [Google Scholar] [CrossRef] [PubMed]
- Salviati, L.; Trevisson, E.; Hernandez, M.A.R.; Casarin, A.; Pertegato, V.; Doimo, M.; Cassina, M.; Agosto, C.; Desbats, M.A.; Sartori, G.; et al. Haploinsufficiency of COQ4 causes coenzyme Q10 deficiency. J. Med. Genet. 2012, 49, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Brea-Calvo, G.; Haack, T.B.; Karall, D.; Ohtake, A.; Invernizzi, F.; Carrozzo, R.; Kremer, L.; Dusi, S.; Fauth, C.; Scholl-Burgi, S.; et al. COQ4 Mutations Cause a Broad Spectrum of Mitochondrial Disorders Associated with CoQ10 Deficiency. Am. J. Hum. Genet. 2015, 96, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Chung, W.K.; Martin, K.; Jalas, C.; Braddock, S.R.; Juusola, J.; Monaghan, K.G.; Warner, B.; Franks, S.; Yudkoff, M.; Lulis, L.; et al. Mutations in COQ4, an essential component of coenzyme Q biosynthesis, cause lethal neonatal mitochondrial encephalomyopathy. J. Med. Genet. 2015, 52, 627–635. [Google Scholar] [CrossRef] [PubMed]
- Public Health. Community Register of Orphan Medicinal Products. Available online: http://ec.europa.eu/health/documents/community-register/html/o1765.htm (accessed on 4 March 2017).
- Montini, G.; Malaventura, C.; Salviati, L. Early coenzyme Q10 supplementation in primary coenzyme Q10 deficiency. N. Engl. J. Med. 2008, 358, 2849–2850. [Google Scholar] [CrossRef] [PubMed]
- Pineda, M.; Montero, R.; Aracil, A.; O’Callaghan, M.M.; Mas, A.; Espinos, C.; Martinez-Rubio, D.; Palau, F.; Navas, P.; Briones, P.; et al. Coenzyme Q(10)-responsive ataxia: 2-year-treatment follow-up. Mov. Disord. 2010, 25, 1262–1268. [Google Scholar] [CrossRef] [PubMed]
- Saiki, R.; Lunceford, A.L.; Shi, Y.; Marbois, B.; King, R.; Pachuski, J.; Kawamukai, M.; Gasser, D.L.; Clarke, C.F. Coenzyme Q10 supplementation rescues renal disease in Pdss2kd/kd mice with mutations in prenyl diphosphate synthase subunit 2, American journal of physiology. Ren. Physiol. 2008, 295, F1535–F1544. [Google Scholar] [CrossRef] [PubMed]
- Lopez, L.C.; Quinzii, C.M.; Area, E.; Naini, A.; Rahman, S.; Schuelke, M.; Salviati, L.; Dimauro, S.; Hirano, M. Treatment of CoQ(10) deficient fibroblasts with ubiquinone, CoQ analogs, and vitamin C: Time- and compound-dependent effects. PLoS ONE 2010, 5, e11897. [Google Scholar] [CrossRef] [PubMed]
- Yubero, D.; Montero, R.; Armstrong, J.; Espinos, C.; Palau, F.; Santos-Ocana, C.; Salviati, L.; Navas, P.; Artuch, R. Molecular diagnosis of coenzyme Q10 deficiency. Expert Rev. Mol. Diagn. 2015, 15, 1049–1059. [Google Scholar] [CrossRef] [PubMed]
- Yubero, D.; Montero, R.; Artuch, R.; Land, J.M.; Heales, S.J.; Hargreaves, I.P. Biochemical diagnosis of coenzyme q10 deficiency. Mol. Syndromol. 2014, 5, 147–155. [Google Scholar]
- Trevisson, E.; DiMauro, S.; Navas, P.; Salviati, L. Coenzyme Q deficiency in muscle. Curr. Opin. Neurol. 2011, 24, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Yubero, D.; Montero, R.; Ramos, M.; Neergheen, V.; Navas, P.; Artuch, R.; Hargreaves, I. Determination of urinary coenzyme Q10 by HPLC with electrochemical detection: Reference values for a paediatric population. Biofactors 2015, 41, 424–430. [Google Scholar] [CrossRef] [PubMed]
- Bujan, N.; Arias, A.; Montero, R.; Garcia-Villoria, J.; Lissens, W.; Seneca, S.; Espinos, C.; Navas, P.; de Meirleir, L.; Artuch, R.; et al. Characterization of CoQ(1)(0) biosynthesis in fibroblasts of patients with primary and secondary CoQ(1)(0) deficiency. J. Inherit. Metab. Dis. 2014, 37, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Ayala, D.J.; Lopez-Lluch, G.; Garcia-Valdes, M.; Arroyo, A.; Navas, P. Specificity of coenzyme Q10 for a balanced function of respiratory chain and endogenous ubiquinone biosynthesis in human cells. Biochim. Biophys. Acta 2005, 1706, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Arias, A.; Garcia-Villoria, J.; Rojo, A.; Bujan, N.; Briones, P.; Ribes, A. Analysis of coenzyme Q(10) in lymphocytes by HPLC-MS/MS. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2012, 908, 23–26. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Yeast | Human | Function |
|---|---|---|
| COQ1 | PDSS1 */PDSS2 * | Synthesis of polyprenyl-diphosphate |
| COQ2 | COQ2 * | pHB-prenyl-transferase |
| COQ3 | COQ3 * | Methyltransferase |
| COQ4 | COQ4 * | Organization of the multi-enzyme complex |
| COQ5 | COQ5 | Methyltransferase |
| COQ6 | COQ6 * | Mono-oxygenase |
| COQ7 | COQ7 * | Hydroxylase |
| COQ8 | ADCK3 */ADCK4 * | Unorthodox kinase (regulatory) |
| COQ9 | COQ9 * | Lipid binding protein |
| COQ10 | COQ10A/COQ10B | CoQ chaperone |
| PTC7 | PPTC7 | Phosphatase (regulatory) |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodríguez-Aguilera, J.C.; Cortés, A.B.; Fernández-Ayala, D.J.M.; Navas, P. Biochemical Assessment of Coenzyme Q10 Deficiency. J. Clin. Med. 2017, 6, 27. https://doi.org/10.3390/jcm6030027
Rodríguez-Aguilera JC, Cortés AB, Fernández-Ayala DJM, Navas P. Biochemical Assessment of Coenzyme Q10 Deficiency. Journal of Clinical Medicine. 2017; 6(3):27. https://doi.org/10.3390/jcm6030027
Chicago/Turabian StyleRodríguez-Aguilera, Juan Carlos, Ana Belén Cortés, Daniel J. M. Fernández-Ayala, and Plácido Navas. 2017. "Biochemical Assessment of Coenzyme Q10 Deficiency" Journal of Clinical Medicine 6, no. 3: 27. https://doi.org/10.3390/jcm6030027
APA StyleRodríguez-Aguilera, J. C., Cortés, A. B., Fernández-Ayala, D. J. M., & Navas, P. (2017). Biochemical Assessment of Coenzyme Q10 Deficiency. Journal of Clinical Medicine, 6(3), 27. https://doi.org/10.3390/jcm6030027