Abstract
Immune thrombocytopenia (ITP) is a complex autoimmune disease characterized by low platelet counts. The pathogenesis of ITP remains unclear although both antibody-mediated and/or T cell-mediated platelet destruction are key processes. In addition, impairment of T cells, cytokine imbalances, and the contribution of the bone marrow niche have now been recognized to be important. Treatment strategies are aimed at the restoration of platelet counts compatible with adequate hemostasis rather than achieving physiological platelet counts. The first line treatments focus on the inhibition of autoantibody production and platelet degradation, whereas second-line treatments include immunosuppressive drugs, such as Rituximab, and splenectomy. Finally, third-line treatments aim to stimulate platelet production by megakaryocytes. This review discusses the pathophysiology of ITP and how the different treatment modalities affect the pathogenic mechanisms.
1. Introduction
Primary immune thrombocytopenia (ITP) is an acquired immune disorder characterized by an isolated thrombocytopenia (peripheral blood platelet count <100 × 109/L) [1] due to pathogenic anti-platelet autoantibodies [2,3], T cell-mediated platelet destruction [4], and impaired megakaryocyte (MK) function [5,6,7]. It can be observed in both adults and children, with both sexes being affected [8]; however, the underlying mechanisms of pediatric ITP compared to adult ITP may be different [9,10,11]. On the other hand, secondary ITP is triggered by inherited or acquired predisposing diseases such as chronic infections, including Helicobacter pylori and human immunodeficiency virus (HIV), or autoimmune diseases such as systemic lupus erythematosus or rheumatoid arthritis. In the infectious cases, it may be that a viral antigen is recognized as being similar to a platelet antigen, a process termed molecular mimicry, which then gives rise to cross-reactive anti-platelet autoantibodies [12,13,14,15]. Treatment of or the natural elimination of the infection eventually contributes to an increase the platelet count accompanied by a decrease in autoantibody titers [13]. Thus, these cases are usually associated with a better outcome; such is the case with most children with newly diagnosed ITP. In adults, primary ITP constitutes approximately 80% of the diagnosed patients, whereas the remaining 20% are affected by secondary ITP [16]. Primary ITP has a prevalence of up to 9.5 per 100,000 adults and an incidence of about 3.3/100,000 adults per year [17], and this increases with age [18,19]. If symptoms occur they can manifest as petechiae; purpura; mucosal bleeding in the urinary tract or in the gastrointestinal and/or oral cavities, including epistaxis [20]; and a reduced quality of life [21,22,23,24,25,26]. In the worst cases, fatal intracranial haemorrhages can occur, but this is only in about 0.2% of cases [27]. The bleeding diatheses are, however, very heterogenous, and it is still unclear why patients with similar platelet counts can present with different clinical bleeding manifestations [9].
ITP is mainly due to IgG autoantibodies, which bind to platelets and MKs [28,29,30], targeting very abundant surface antigens such as glycoprotein (GP) αIIbβ3 (GPIIbIIIA) and GPIb-IX-V [31,32]. Platelets with bound autoantibodies are subsequently recognized by phagocytes bearing Fcγ-receptors (FcγRs), which results in enhanced antibody-mediated platelet phagocytosis and destruction primarily in the spleen [2,3,33]. Moreover, autoantibody binding to MKs can inhibit their maturation or can lead to their destruction [34,35,36], and thrombopoietin (TPO), a liver derived glycoprotein hormone that drives thrombopoiesis, cannot normalize the platelet count [37]. In fact, approximately two-thirds of patients with ITP present with normal or decreased TPO plasma levels, adding a novel functional deficit of TPO to the pathophysiology of the disease [38,39,40]. In addition, autoreactive T cells are also involved in platelet [4,41] and MK destruction [42,43], and, despite an increased MK number in the bone marrow of some patients, many present signs of morphological abnormalities including apoptotic ultrastructure as well as activation of Caspase-3 [44,45]. Superimposed on these cellular impairments, the cytokine profile of patients with ITP is also imbalanced with, for example, higher serum levels of interleukin (IL)-2, interferon (IFN)-γ, and IL-17 [46,47,48].
ITP can be clinically classified into 3 phases [1] with the first phase, called newly diagnosed, occurring within the first 3 months post-diagnosis. The second phase is termed persistent ITP and refers to symptoms lasting between 3 and 12 months, and the third phase is termed chronic ITP, in which symptoms remain present beyond 12 months [1]. Acute ITP, a term originally used primarily for children, is now considered newly diagnosed. ITP is termed severe when it is characterised by the necessity of active intervention to treat bleeding symptoms. The majority of the adult patients will progress to the chronic stage [49], and several treatment modalities are now utilized, which target various aspects of ITP pathophysiology such as the inhibition of autoantibody production, the decrease of platelet destruction, the modulation of T cell activity, or the stimulation of platelet production [50]. In this review, we will give an overview of the pathological mechanisms involved in ITP and the effects of the different therapeutic regimens.
2. Molecular and Cellular Mechanisms of the Pathogenesis of ITP
2.1. B Cells and Autoantibodies
Patients with ITP produce anti-platelet IgG antibodies (and more rarely IgM or IgA antibodies) [28,29,30,51,52] which bind to platelets and mark them for phagocytic breakdown in the spleen and liver [39]. These antibodies often bind to very abundant glycoproteins on the platelet surface, particularly GPαIIbβ3 (GPIIbIIIA) and GPIb-IX-V molecules [31,32]. However, in as many as 30% to 40% of the patients, no detectible antibodies can be found [53,54]. Whether the lack of antibodies in patients is due to the robustness of the antibody tests used or perhaps due to a purely T cell-mediated mechanism is still unknown. Of interest, in those patients positive for anti-platelet antibodies, other antibody specificities beside the classic surface glycoproteins have been found, including cytosolic proteins [55], which may suggest that platelets undergo protein degradation by antigen presenting cells (APC) followed by antigen presentation to T cells [56]. Moreover, other mechanisms have been proposed to be involved in antibody production in ITP including antigenic cross-reactivity (mimicry), somatic mutation [16,53], and defects in the elimination of autoreactive B-cell clones [16]. In addition, oxidative stress, which favours the production of autoantibodies, may also be involved [57]. The type of epitope targeted by autoantibodies may also be a marker of disease severity and, to some extent, of response to treatment, in mice at least [58,59]. Indeed, it has been hypothesised that certain antibody specificities are more prone to induce platelet clearance [58] and apoptosis [60,61,62] or to inhibit megakaryopoiesis [35]. For example, anti-GPIb antibodies appear to induce a stronger platelet destruction by increasing the release of CD62P and phosphatidylserine and the clustering of GPIb receptors [63], and, in mice, these antibodies tend to be more resistant to the effects of intravenous immunoglobulin (IVIg) treatment [58]. Antibody-mediated platelet destruction has also been shown to be enhanced by the acute phase protein C-reactive protein (CRP) both in vitro and in vivo [64]. Interestingly, increased levels of CRP at the diagnosis of childhood ITP predicted a slower platelet count recovery, but after IVIg treatment, the levels of CRP dropped, accompanied by a recovery in the platelet count and decreased bleeding severity [64]. Furthermore, platelet opsonisation by autoreactive antibodies can affect platelet reactivity by modulating agonist stimulation and platelet secretory granule release [65]. This observation may partially explain the variability of ITP bleeding severity as well as the differences in response to treatment observed in some patients with similar platelet counts. Furthermore, the presence of anti-platelet autoantibodies increases the risk of thrombotic events [66,67,68], perhaps due to procoagulant microparticles released by activated platelets [69,70] or associated predispositions [37,63,71].
Autoreactive antibodies are secreted by plasma cells, which have been reported to be present at higher levels in patients with ITP [72], as well as the B cell regulator, and B cell-activating factor (BAFF, also called B cell stimulator (BlyS)), which is an important factor in B cell selection, survival, and proliferation. Indeed, BAFF promoter region polymorphisms as well as its up-regulation in the plasma have been strongly associated with ITP in humans and in a murine ITP model [73,74,75,76]. B cells were also shown to be increased in the red pulp of the spleens from patients with ITP [77] and they appear to have higher proliferative rates in these splenic areas [78]. Moreover CD19+CD41hiCD38hi B-regulatory cells (Bregs), which promote peripheral tolerance, are also impaired in ITP [79,80]. They fail to reduce CD4+ T cell activation and trigger the recruitment of CD4+CD25+FoxP3+ T regulatory cells (Tregs), a subtype of CD4+ T cells crucial for immune suppression and tolerance [81] via IL-10 secretion [82]. The CD19+CD24+ FOXP3+ Breg subpopulation has also been recently shown to be significantly increased in the spleens of patients with ITP compared with control trauma patients [83]. These studies suggest that, like Tregs, the peripheral deficiency of Bregs may be due to sequestration of these cells within lymphoid compartments. Nonetheless, there appears to be a central role for Bregs and IL-10 secretion in ITP and their modulating effects on Tregs in the pathogenesis of the disorder.
Taken together, these studies demonstrate that ITP patients present with impaired plasma cells, Bregs, and B cells, leading to the production of pathogenic antibodies. These antibodies, via platelet and MK opsonisation, trigger platelet destruction in the spleen and liver as well as defective megakaryopoiesis.
2.2. T-Cell Imbalance in ITP
Abnormal T cells have been described in patients with ITP, including a higher T helper cell reactivity against platelets, a lower frequency of circulating CD4+CD25+FoxP3+ Tregs and CD4+ Th0, and Th1 activation patterns [33,41,46,81,84,85,86,87]. Only about 60% of patients with ITP have detectible plasma and/or platelet-bound autoantibodies [54,56], suggesting a non-antibody-mediated mechanism of ITP. Related to this, cytotoxic CD8+ T cells were found in the circulation of patients [4,88,89] and a similar finding was observed in an active murine model of ITP [34,87]. These CD8+ T cells are able to directly lyse platelets in vitro [4] and can accumulate in the bone marrow, where they are able to inhibit thrombopoiesis [43]. Furthermore, compared with healthy individuals, CD3+ T cells from patients with ITP have a lower rate of apoptosis and a higher clonal expansion rate, leading to abnormal cytokine secretion, including IL-2, INF-γ, and IL-10 [46], which may be responsible for the lower CD4+CD25+FoxP3+ Treg levels and function observed in patients with active disease [43,90,91,92,93,94,95,96,97,98,99,100,101,102,103,104,105,106,107].
Tregs, via their critical functions in maintaining self-tolerance by interacting with APC and decreasing CD19+ B cell and CD8+ T cell responses [108,109], appear to be the key cell types that may be responsible for the initiation of ITP [110,111,112]. This T cell subpopulation exists in 5% to 10% of all the circulating CD4+ T cells and their lower levels, and function in patients with chronic ITP [43,90,91,92,93,95,96,97,98,99,100,101,102,103,104,105,106,113,114] may reflect a dysregulated cellular immunity, leading to the autoimmune destruction of platelets. Furthermore, Tregs were shown to be significantly decreased in the spleen of thrombocytopenic ITP mice but concomitantly accumulated in the thymus [34,115]. Like Bregs, it is possible that the peripheral deficiency of Tregs is due to the sequestration of this cell type. In addition, Catani and colleagues showed that impaired communication between dendritic cells (DC) and Tregs also resulted in less tolerogenic DCs [116]. These results were supported by Daridon et al. [78], who observed a lower percentage of Tregs in the splenic proliferative lymphoid nodules in patients with ITP, confirming an impaired Treg development and suggesting that autoreactive antibody production may take place in these nodules [116]. On the other hand, Tregs are also modulated by CD16+ monocytes that were shown to be increased in patients with active ITP [117]. It appears that CD16+ monocytes release IL-12, which promotes T cell differentiation into Th1 cells and inhibits the secretion of IL-17 thereby down-modulating Tregs [118]. In addition, CD4+ T cell proliferation is promoted in vitro by CD16+ monocytes via INF-γ [117].
Tregs are not the only affected T cell population in ITP. For example, CD4+ Th cells have also been shown to be defective in patients with chronic ITP, resulting in increased IL-2 secretion [46] via a mechanism involving platelet GPIIbIIIa presentation and Th cell stimulation in vitro [119,120,121]. Th22 levels were also found to be elevated in patients with ITP as well as their associated cytokines, TNF-α and IL-22 [122,123,124,125,126]. In addition, other cytokines such as INF-γ have been found to be increased in adult patients with ITP [127]. Taken together, these results suggest that a Th cell defect and highly imbalanced cytokine secretion patterns exist, which may promote B cell activation [46]. Indeed, this observation is supported by several studies showing a Th1/Th2 deregulation, which may be mediated by the reduced secretion of platelet-derived chemokines and cytokines, including epidermal growth factor (EFG), chemokine C-C motif ligand 5 (CCL5), and chemokine C-X-C motif ligand 5 (CXCL5) [20,47]. Feng and co-workers showed that these chemokine/cytokine levels were associated with the severity of ITP in a passive model of ITP further supporting a crucial immunomodulatory role for platelets in ITP [47].
On the other hand, Zang et al. [99,100] and others [106,124,125,128,129,130,131] reported that Th17 cells and their associated cytokines IL-6 and TGF-β were significantly upregulated in patients with ITP, which may, in association with Treg impairment, promote a Th1-mediated immune response, triggering the disease in both humans and mice. Th17, like Tregs, differentiate from CD4+ T cells in the presence of IL-6 and TGF-β [132], and the secretion of IL-17 stimulates the production of pro-inflammatory cytokines such as IL-1, IL-6, and IFN-γ leading to anti-platelet antibody production in mice [131] and in patients with ITP [99,123,129,133,134,135]. This hypothesis is, however, not without controversy as others have not demonstrated a modulation of these cytokines in the plasma of patients with ITP [134].
In summary, T cells also play a crucial role in ITP. Indeed, abnormal T cell subsets, including lower Tregs and unbalanced Th17, Th0, and Th1 profiles, as well as the presence of cytotoxic CD8+ T cells constitute the cellular mechanisms of ITP pathogenesis.
2.3. Dendritic Cells in ITP
APC including DCs, macrophages and, in certain conditions, B cells, are continuously scanning their environment to process and present foreign antigens to immune cells [136,137,138,139,140,141]. In certain circumstances such as during inflammation, their function can be altered, and abnormal processing as well as enhanced self-antigen presentation can be observed, contributing to the development of autoimmune diseases. DCs are the most efficient APC [142,143], and several studies have shown their impairment in ITP [116,144,145]. For example, Catani et al. showed that DCs from patients with ITP were able to stimulate T cell proliferation upon platelet antigen presentation in vitro, probably via an increased DC CD86 expression [144]. Moreover, plasmacytoid DCs (pDCs), which are a particular subset of DCs specialized in type I interferon production (INF-α and INF-β) [146,147], are also affected in ITP. The pDCs levels were found to be lower in patients affected by either primary ITP or H. pylori-mediated secondary ITP [145]. It appeared that platelet counts in these patients were highly correlated with the number of circulating pDCs supporting their role in ITP pathology [145]. On the other hand, the DC enzyme indoleamine 2,3-dioxygenase 1 (IDO1) was found to be lower in patients with ITP, which would hamper the transition of CD4+ T cells to Tregs and therefore contribute to a Treg deficiency which may stimulate the onset of disease [116].
In addition to antigen presentation, Toll-like receptor (TLR)-mediated recognition of infectious agents has also been associated with ITP. For example, TLR4, which recognizes and binds lipopolysaccharide (LPS), a gram-negative bacterial endotoxin, and O-linked mannosyl residues of fungi, has been shown to be involved in LPS-induced ITP [74,148,149,150]. In addition, DC-associated TLR7 was also shown to induce B cell proliferation as well as anti-platelet autoantibody production in vitro via BAFF (BlyS) production [76]. Taken together, APC function, particularly in DC subsets, is abnormal and may significantly contribute to lymphocyte activation and the autoimmune pathology in ITP.
Thus, APCs, primarily DCs, are also impaired in ITP, which may suggest that abnormal self-antigen presentation takes place, which contributes to stimulating pathogenic antibody production thereby contributing to the progression of the disease.
2.4. Megakaryocytes in ITP
Megakaryopoiesis predominantly takes place in the bone marrow (BM) niche, a very restricted micro-environment, where specific cytokines, adhesion molecules, and growth factors regulate MK-maturation and pro-platelet release [38,151]. MKs are strongly affected in ITP, as demonstrated by impaired development (decreased ploidy and granularity) and platelet release [6]. In addition, approximately two-thirds of patients with ITP have plasma autoantibodies that are able to significantly inhibit MK maturation from TPO-treated CD34+ hematopoietic progenitor cells [36] and induce apoptosis [44] in vitro. It may be that different autoantibodies have different affinities for MKs and thus trigger different morphological and kinetic changes in these cells [152]. Indeed, several studies have shown that MKs were directly cleared by neutrophils and macrophages [44] despite normal or slightly elevated plasma levels of TPO [153]. Plasma TPO levels are, however, generally normal in patients with ITP compared to those with other thrombocytopenic diseases while the levels of other MK regulatory cytokines, such as IL-6 and IL-11, are increased [154,155]. The causes for these decreased TPO levels are still not clear, but TPO may be degraded along with the increased destruction of antibody-bearing platelets [153].
In ITP, MKs are clearly targeted by anti-platelet autoantibodies binding GPIb and GPIIbIIIa, and this induces both morphological and physiological changes [6,35,156]. These changes include a reduction of granules with a vacuolisation of the cytoplasm and a smoothing of the plasma membrane [6,7]. In addition, immature MKs as well as mesenchymal stem cells (MSC), which sustain MK maturation and pro-platelet formation [157], are also affected and appear to be apoptotic [44,45]. On the other hand, MKs regulate other cells in the BM niche including plasma cells, which produce antibodies [158,159,160,161], and may thus indirectly contribute to the pathophysiology of the disease. For example, the whole BM niche may be affected in ITP via MSC and MKs and their important immunomodulatory roles including the inhibition of T-cell activation and the production of IL-10 [5,162]. In patients with ITP, in addition to defective megakaryopoiesis [6,35,39,152], MSC do not appear to multiply and lose their ability to prevent CD8 T cell proliferation [157]. It seems that patients with chronic ITP present a defective vascular niche in the bone marrow, thereby reducing the interaction of MKs with the niche microenvironment, including endothelial cells [163] and plasma cells [160,161]. These studies indicate that MKs are directly affected in ITP, and the whole BM is impaired, enhancing the progression of the disease. The exact mechanisms, particularly the role of TPO, however, still remain to be unravelled.
MKs and the whole BM niche are thus also damaged in ITP. It is targeted by autoantibodies and T cells, which leads to impaired MK maturation and platelet production despite relatively normal TPO levels.
Figure 1 summarizes the multiple defects associated with ITP pathogenesis including B- and plasma cell-defects as well as T cell-impairment. These abnormalities lead to both platelet and MK damage, resulting in platelet degradation and inefficient thrombopoiesis.
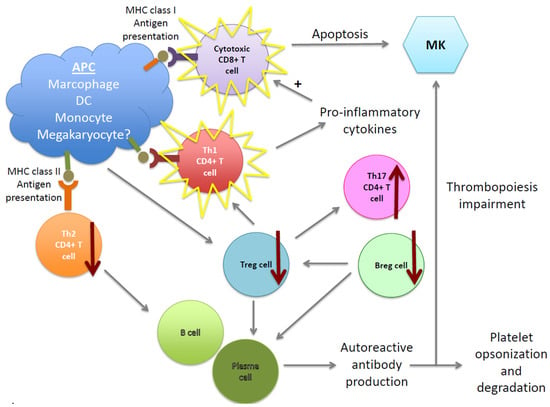
Figure 1.
Cellular pathogenic mechanisms in immune thrombocytopenia (ITP). Multiple cells are involved in the pathogenesis of ITP. B cells and plasma cells are abnormally regulated and produce autoantibodies, which bind platelets and megakaryocytes (MKs), inducing their impairment and/or degradation in the spleen and liver. The cellular immune response is also affected, leading to a decrease of Tregs and Bregs, which contributes to autoreactive plasma cell survival (supporting autoantibody production) and unbalanced Th CD4+ T cell subsets. Moreover, cytotoxic CD8+ T cells are also activated, inducing platelet and MK apoptosis as well as the dysregulation of BM niche homeostasis. Therefore, ITP pathogenesis does not only results in platelet destruction, but also in a megakayopoiesis and thrombopoiesis defect.
3. Therapies of ITP
Primary ITP in adults usually progresses towards chronic disease and therefore necessitates therapy aiming to restore a durable platelet count allowing sufficient hemostasis. Historically, the best way to achieve this goal was splenectomy. Indeed, 60% of the splenectomized patients presented with a normalized platelet count 5 years post-splenectomy [164,165]. Considering the potential surgical, vascular, and infectious complications, medical therapies are often considered less invasive, despite the fact that they don’t always succeed in providing long-term remission. The treatment strategies consist in stimulating platelet production to increase the platelet counts, increasing platelet half-life, and decreasing the autoreactive nature of the immune response by targeting the autoreactive antibody production and the platelet destruction.
First-line treatments include corticosteroids with or without intravenous IVIg and anti-D [166,167]. Second-line therapies consist of splenectomy and/or immune-suppressive agents such as the B cell-depleting anti-CD20 agent Rituximab, and TPO-receptor agonists such as Romiplostim and Eltrombopag are considered third-line treatments [168] (Figure 2). Recently, several studies (reviewed in [49]) have suggested that an early aggressive treatment of chronic ITP may improve the long-term outcome of patients. Indeed, the effectiveness of the treatment depends on the initial trigger of the thrombocytopenia, which is often multifactorial and targets components of adaptive immunity (T and B cells) as well as inflammatory factors (cytokines and the presence of autoantigens), as described above. Therefore, therapies targeting these two aspects of the disease may be more efficient than administrating a treatment that affects one or the other [169,170]. In addition, it is also relevant to take into account the duration of the disease in terms of response to the treatment. The chronic aspect of the disease may be the result of a B and T cell memory response and may therefore be more difficult to resolve than the primary immune response responsible for the induction of ITP. B cell clonal expansion and its positive selection may lead to a higher titer of autoreactive anti-platelet antibodies with a higher affinity for platelets and may be more difficult to combat with conventional therapies [171,172]. Further investigations are, however, still needed to better understand the transition towards chronicity and its implication for the treatment response, outcome, and cure for ITP.
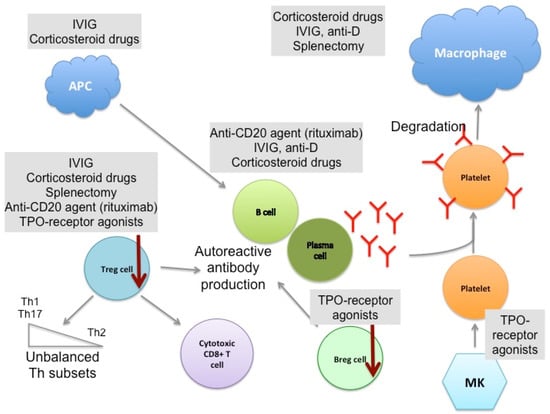
Figure 2.
Therapeutic mechanisms of current ITP treatments. Several drugs are used to treat chronic ITP. The first line of treatment consists of corticosteroids alone or in combination with intravenous immunoglobulin (IVIg) or anti-D, which aim to decrease platelet destruction and platelet antigen presentation by antigen presenting cells (APC) to restore a normal immune response. They also act on B cells and plasma cells, thus decreasing autoantibody production, and rescue impaired Treg function. Second-line therapies include immunosupressive drugs such as Rituximab, which directly targets B cells, and splenectomy. Both treatments also modulate the T cell compartment, notably increasing Tregs. Thrombopoietin (TPO)-receptor agonists (Romiplostim and Eltrombopag), which stimulate platelet production by MKs, are third-line treatments and are used for patients who do not respond to other therapies. Here again, TPO-agonists present indirect immunomodulatory effects on Bregs and Tregs. Combining multiple therapeutic approaches is often required to ensure the restoration of a physiological platelet count.
3.1. First-Line Treatments
The primary effect of first-line treatments is to decrease autoantibody-mediated platelet clearance, presumably through Fcγ receptors [53]. Corticosteroids are pharmacological derivatives of the glucocorticoid steroid hormones, and they bind cytosolic receptors and modulate a large variety of genes, triggering many physiological changes [173]. Immunosuppressive agents (e.g., high-dose dexamethasone and low-dose prednisone together with rapamycin or rituximab) were shown in patients with chronic ITP to modulate T cells by increasing the number of peripheral Tregs, restoring the Th1/Th2 ratio, and normalizing the Th17 subpopulation consistent with an increase of IL-10 and TGF-β [90,93,174,175,176]. Immunosuppressive drugs such as prednisolone or dexamethasone also modulate B cell activation via a decrease of BAFF (BlyS) [73] and modulate DCs [96].
IVIg is used as a treatment for ITP as well as for other autoimmune diseases [177]. The working-mechanisms of IVIg are incompletely understood although several modes of action have been suggested. These include, for example, blocking antibody-mediated platelet clearance by saturating Fc receptors on macrophages; promoting the expression of inhibitory FcγRIIb via sialylated IgG Fc fragments; saturation of the neonatal FcR, which increases the clearance of autoreactive antibodies; modulation of DC maturation; and/or the modulation of T cell subsets towards a higher proportion of Tregs and a lower proportion of Th17 [177,178,179]. In addition, IVIg may also affect several other pathways such as inhibition of autoantibody production and regulation of the B cell repertoire, modulation of inflammatory cytokines such as IFN-γ [180], neutralization of autoreactive antibodies by anti-idiotype antibodies, and inhibition of the complement cascade pathway [115,178,179,181,182,183,184,185,186,187,188]. This multitude of theories on the mechanism of action of how IVIg works in ITP are somewhat enigmatic; however, the treatment is a very effective means to temporarily raise platelet counts in patients with ITP.
Patients with ITP who are RhD antigen positive and have an intact spleen can also be treated with polyclonal anti-D [189]. This treatment is prepared from the plasma of RhD negative subjects immunized against the D antigen [181]. However, like IVIg, there are more questions than answers about how this medication exactly works, and several attempts to produce monoclonal versions of anti-D have remained unsuccessful [166]. In a murine ITP model, it appeared that anti-D-coated erythrocytes competed with antibody-opsonized platelets for FcγIIIA–mediated degradation by splenic macrophages [190], and they were suggested to have a similar mode of action in patients with ITP [189,191]. It has, however, also been associated with a decrease in autoreactive antibody production in patients with chronic ITP [192], suggesting an additional effect of anti-D on B cells. Although some patients with ITP have had serious hemolytic events, this therapy, like IVIg, is very effective in temporarily raising platelet counts.
3.2. Second-Line Treatments
If patients with ITP fail first-line treatments or relapse, second-line treatments are necessary to manage the disease. For example, because the spleen is the primary site for platelet-reactive T and B cell activation and platelet destruction in ITP [193], it is not surprising that a splenectomy is still the gold standard for restoring physiological platelet counts in patients with ITP, and it remains the method of choice in refractory patients with ITP [49,194]. A complete remission is indeed achieved in about 60% of the patients, and another fifth of them show a partial response [164,195]. As with any surgical procedure, however, splenectomy is not without risk, and surgery-related complications have been reported in up to approximately 25% of the cases [195], including about a 1% mortality rate [165]. For example, it is well known that splenectomy is associated with an increased risk of sepsis and increased incidence of vascular complications [165]. Despite these risks, this surgical procedure is still considered the best treatment modality for the long-term increase in platelet counts in patients with ITP.
Rituximab is a chimeric antibody directed against the CD20 antigen on B cells and, upon administration, results in their virtual elimination in vivo [90,93,196,197,198,199]. It is thought to induce either B cell apoptosis or destruction in the spleen via either complement-dependant cytotoxicity or antibody-dependent cellular cytotoxicity (ADCC) [196,198,200,201]. The resulting depletion of B cells results in decreases of anti-platelet antibody titers and, interestingly, in the normalization of the T cell impairments observed in patients with chronic ITP [90,92,199] and in murine ITP [197]. These studies suggest that the mechanism of action of rituximab may in fact be an indirect regulation of the T cell compartment. However, the real benefit of this treatment remains controversial. Indeed, about 50% of resistant chronic ITP patients present a short-term response to Rituximab, and this platelet rise lasts for 6 months or more in about 30% of the cases [202,203,204,205]. Clinical variables, such as gender, duration of the disease, or co-treatment were also shown to affect the short-term efficacy of the medication [204]. In addition, recent studies have shown that only a fifth of the patients maintained a physiological platelet count 5 years post-treatment [206], and no clear long-term improvement was observed in a cohort of more than 100 patients in comparison to corticosteroid drugs [207].
3.3. Third-Line Treatment
Patients who fail splenectomy or Rituximab can be treated with TPO-receptor agonists. Both Eltrombopag and Romiplostim activate TPO receptors on MKs and induce platelet production via the JAK2 and STAT5 kinase pathways [208,209], and both therapies have proven efficacious in most refractory patients with ITP. In addition, it appears that approximately one third of Romiplostim-treated patients remain in remission even after 24 weeks off TPO treatment [210]. In addition to its obvious role in enhancing MK proliferation, it appears that Romiplostim can also rescue the Treg deficiency observed during active disease [91]. It was shown that Treg function was increased, and the platelet counts correlated with circulating TGF-β levels [91], which may be due the increase in the platelet mass. Similarly, Bregs were also shown to be increased in non-splenectomized patients with ITP under this therapy, concomitant with a reduction in pro-inflammatory monocytes and the enhancement of B cell immune-modulatory activity by CD16+ monocytes [118]. These studies suggest that TPO-receptor agonists may not only directly induce thrombopoiesis but also modulate the immune system, perhaps by modulating both Tregs and Bregs [9].
4. Conclusions and Perspectives
ITP is a highly complex autoimmune disease and its etiology and pathogenesis remain to be fully understood. Indeed, despite extensive genetic research pointing out the genes involved in T cell activation or the single nucleotide polymorphisms (SNP) associated with cytokines (for instance reviewed in [211]), the pre-existing factors which may promote the loss of tolerance toward platelet antigens and affect patient outcome are yet to be identified. They are multifactorial in nature and may consist of a combination of genetic predispositions, environmental conditions (including various forms of infection), and responses to treatment. ITP is characterized by autoreactive antibodies associated with impaired T and B cells, leading to the destruction of platelets and defects in thrombopoiesis and megakaryopoiesis. This pathogenic pattern is enhanced by a pro-inflammatory cytokine profile that consists of increased IFN-y, IL-2 and IL-17 as well as decreased immunosuppressive IL-10, TGF-β and IL-4, promoting autoantibody development. Underlying these defects is a central deficiency of immune tolerance due to defects in both Tregs and Bregs [212,213]. Of interest is that, although the diverse array of therapies are available for ITP that can increase platelet counts, they all seem to affect Tregs and Bregs in a similar fashion by rescuing their peripheral deficiency. Since platelets are not only hemostatic but also have significant immunomodulatory functions [20,214,215,216], perhaps increasing their counts by whatever means also sets up an anti-inflammatory milieu that alters the pathogenic immune responses seen in ITP. Thus, it is possible that although platelets are the target of autoimmune attack in ITP, they may also be able to regulate the autoimmunity against themselves. Further research will be required in order to test this interesting concept.
Acknowledgments
This work was supported by a grant from Health Canada and the Canadian Blood Services (J.W.S.). A.Z. is the recipient of a postdoctoral Fellowship from the Swiss National Science Foundation (P300PB-164760). R.K. is the recipient of a Postdoctoral Fellowship from the Canadian Blood Services (CBS). A.Z.’s current address is Centre de Recherche du CHU de Québec, CHUL-UL, Quebec City, QC, Canada.
Author Contributions
A.Z. wrote the first draft and edited the paper. R.K. edited the paper. J.W.S. provided the financial resources and edited the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Rodeghiero, F.; Stasi, R.; Gernsheimer, T.; Michel, M.; Provan, D.; Arnold, D.M.; Bussel, J.B.; Cines, D.B.; Chong, B.H.; Cooper, N.; et al. Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children: Report from an international working group. Blood 2009, 113, 2386–2393. [Google Scholar] [CrossRef] [PubMed]
- Harrington, W.J.; Minnich, V.; Hollingsworth, J.W.; Moore, C.V. Demonstration of a thrombocytopenic factor in the blood of patients with thrombocytopenic purpura. J. Lab. Clin. Med. 1951, 38, 1–10. [Google Scholar] [PubMed]
- Shulman, N.R.; Marder, V.J.; Weinrach, R.S. Similarities between known antiplatelet antibodies and the factor responsible for thrombocytopenia in idiopathic purpura. Physiologic, serologic and isotopic studies. Ann. N. Y. Acad. Sci. 1965, 124, 499–542. [Google Scholar] [CrossRef] [PubMed]
- Olsson, B.; Andersson, P.O.; Jernas, M.; Jacobsson, S.; Carlsson, B.; Carlsson, L.M.; Wadenvik, H. T-cell-mediated cytotoxicity toward platelets in chronic idiopathic thrombocytopenic purpura. Nat. Med. 2003, 9, 1123–1124. [Google Scholar] [CrossRef] [PubMed]
- Khodadi, E.; Asnafi, A.A.; Shahrabi, S.; Shahjahani, M.; Saki, N. Bone marrow niche in immune thrombocytopenia: A focus on megakaryopoiesis. Ann. Hematol. 2016, 95, 1765–1776. [Google Scholar] [CrossRef] [PubMed]
- Dameshek, W.; Miller, E.B. The megakaryocytes in idiopathic thrombocytopenic purpura, a form of hypersplenism. Blood 1946, 1, 27–50. [Google Scholar] [PubMed]
- Pisciotta, A.V.; Stefanini, M.; Dameshek, W. Studies on platelets. X. Morphologic characteristics of megakaryocytes by phase contrast microscopy in normals and in patients with idiopathic thrombocytopenic purpura. Blood 1953, 8, 703–723. [Google Scholar] [PubMed]
- Neylon, A.J.; Saunders, P.W.; Howard, M.R.; Proctor, S.J.; Taylor, P.R.; Northern Region Haematology Group. Clinically significant newly presenting autoimmune thrombocytopenic purpura in adults: A prospective study of a population-based cohort of 245 patients. Br. J. Haematol. 2003, 122, 966–974. [Google Scholar] [CrossRef] [PubMed]
- Perera, M.; Garrido, T. Advances in the pathophysiology of primary immune thrombocytopenia. Hematology 2016, 22, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Schulze, H.; Gaedicke, G. Immune thrombocytopenia in children and adults: What’s the same, what’s different? Haematologica 2011, 96, 1739–1741. [Google Scholar] [CrossRef] [PubMed]
- Provan, D.; Newland, A.C. Current Management of Primary Immune Thrombocytopenia. Adv. Ther. 2015, 32, 875–887. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Nardi, M.A.; Borkowsky, W.; Li, Z.; Karpatkin, S. Role of molecular mimicry of hepatitis C virus protein with platelet GPIIIa in hepatitis C-related immunologic thrombocytopenia. Blood 2009, 113, 4086–4093. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.F.; Blanchette, V.S.; Wang, H.; Arya, N.; Petric, M.; Semple, J.W.; Chia, W.K.; Freedman, J. Characterization of platelet-reactive antibodies in children with varicella-associated acute immune thrombocytopenic purpura (ITP). Br. J. Haematol. 1996, 95, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Yujiri, T.; Shinohara, K.; Inoue, Y.; Sato, Y.; Fujii, Y.; Okubo, M.; Zaitsu, Y.; Ariyoshi, K.; Nakamura, Y.; et al. Molecular mimicry by Helicobacter pylori CagA protein may be involved in the pathogenesis of H. pylori-associated chronic idiopathic thrombocytopenic purpura. Br. J. Haematol. 2004, 124, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Nardi, M.A.; Karpatkin, S. Role of molecular mimicry to HIV-1 peptides in HIV-1-related immunologic thrombocytopenia. Blood 2005, 106, 572–576. [Google Scholar] [CrossRef] [PubMed]
- Cines, D.B.; Bussel, J.B.; Liebman, H.A.; Luning Prak, E.T. The ITP syndrome: Pathogenic and clinical diversity. Blood 2009, 113, 6511–6521. [Google Scholar] [CrossRef] [PubMed]
- Terrell, D.R.; Beebe, L.A.; Vesely, S.K.; Neas, B.R.; Segal, J.B.; George, J.N. The incidence of immune thrombocytopenic purpura in children and adults: A critical review of published reports. Am. J. Hematol. 2010, 85, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Frederiksen, H.; Schmidt, K. The incidence of idiopathic thrombocytopenic purpura in adults increases with age. Blood 1999, 94, 909–913. [Google Scholar] [PubMed]
- Fogarty, P.F.; Segal, J.B. The epidemiology of immune thrombocytopenic purpura. Curr. Opin. Hematol. 2007, 14, 515–519. [Google Scholar] [CrossRef] [PubMed]
- Semple, J.W.; Italiano, J.E., Jr.; Freedman, J. Platelets and the immune continuum. Nat. Rev. Immunol. 2011, 11, 264–274. [Google Scholar] [CrossRef] [PubMed]
- Lazarus, A.H.; Semple, J.W.; Cines, D.B. Innate and adaptive immunity in immune thrombocytopenia. Semin. Hematol. 2013, 50, S68–S70. [Google Scholar] [CrossRef] [PubMed]
- Heitink-Polle, K.M.; Haverman, L.; Annink, K.V.; Schep, S.J.; de Haas, M.; Bruin, M.C. Health-related quality of life in children with newly diagnosed immune thrombocytopenia. Haematologica 2014, 99, 1525–1531. [Google Scholar] [CrossRef] [PubMed]
- George, J.N.; Mathias, S.D.; Go, R.S.; Guo, M.; Henry, D.H.; Lyons, R.; Redner, R.L.; Rice, L.; Schipperus, M.R. Improved quality of life for romiplostim-treated patients with chronic immune thrombocytopenic purpura: Results from two randomized, placebo-controlled trials. Br. J. Haematol. 2009, 144, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Efficace, F.; Mandelli, F.; Fazi, P.; Santoro, C.; Gaidano, G.; Cottone, F.; Borchiellini, A.; Carpenedo, M.; Simula, M.P.; Di Giacomo, V.; et al. Health-related quality of life and burden of fatigue in patients with primary immune thrombocytopenia by phase of disease. Am. J. Hematol. 2016, 91, 995–1001. [Google Scholar] [CrossRef] [PubMed]
- Hill, Q.A.; Newland, A.C. Fatigue in immune thrombocytopenia. Br. J. Haematol. 2015, 170, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Newton, J.L.; Reese, J.A.; Watson, S.I.; Vesely, S.K.; Bolton-Maggs, P.H.; George, J.N.; Terrell, D.R. Fatigue in adult patients with primary immune thrombocytopenia. Eur. J. Haematol. 2011, 86, 420–429. [Google Scholar] [CrossRef] [PubMed]
- Neunert, C.; Lim, W.; Crowther, M.; Cohen, A.; Solberg, L., Jr.; Crowther, M.A.; American Society of Hematology. The American Society of Hematology 2011 evidence-based practice guideline for immune thrombocytopenia. Blood 2011, 117, 4190–4207. [Google Scholar] [CrossRef] [PubMed]
- Karpatkin, S.; Siskind, G.W. In vitro detection of platelet antibody in patients with idiopathic thrombocytopenic purpura and systemic lupus erythematosus. Blood 1969, 33, 795–812. [Google Scholar] [PubMed]
- Karpatkin, S.; Strick, N.; Karpatkin, M.B.; Siskind, G.W. Cumulative experience in the detection of antiplatelet antibody in 234 patients with idiopathic thrombocytopenic purpura, systemic lupus erythematosus and other clinical disorders. Am. J. Med. 1972, 52, 776–785. [Google Scholar] [CrossRef]
- Lightsey, A.L., Jr.; McMillan, R.; Koenig, H.M.; Schanberger, J.E.; Lang, J.E. In vitro production of platelet-binding IgG in childhood idiopathic thrombocytopenic purpura. J. Pediatr. 1976, 88, 415–418. [Google Scholar] [CrossRef]
- He, R.; Reid, D.M.; Jones, C.E.; Shulman, N.R. Spectrum of Ig classes, specificities, and titers of serum antiglycoproteins in chronic idiopathic thrombocytopenic purpura. Blood 1994, 83, 1024–1032. [Google Scholar] [PubMed]
- Boylan, B.; Chen, H.; Rathore, V.; Paddock, C.; Salacz, M.; Friedman, K.D.; Curtis, B.R.; Stapleton, M.; Newman, D.K.; Kahn, M.L.; et al. Anti-GPVI-associated ITP: An acquired platelet disorder caused by autoantibody-mediated clearance of the GPVI/FcRgamma-chain complex from the human platelet surface. Blood 2004, 104, 1350–1355. [Google Scholar] [CrossRef] [PubMed]
- Saleh, M.N.; Moore, D.L.; Lee, J.Y.; LoBuglio, A.F. Monocyte-platelet interaction in immune and nonimmune thrombocytopenia. Blood 1989, 74, 1328–1331. [Google Scholar] [PubMed]
- Chow, L.; Aslam, R.; Speck, E.R.; Kim, M.; Cridland, N.; Webster, M.L.; Chen, P.; Sahib, K.; Ni, H.; Lazarus, A.H.; et al. A murine model of severe immune thrombocytopenia is induced by antibody- and CD8+ T cell-mediated responses that are differentially sensitive to therapy. Blood 2010, 115, 1247–1253. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.; Nakagawa, P.A.; Williams, S.A.; Schwartz, M.R.; Imfeld, K.L.; Buzby, J.S.; Nugent, D.J. Immune thrombocytopenic purpura (ITP) plasma and purified ITP monoclonal autoantibodies inhibit megakaryocytopoiesis in vitro. Blood 2003, 102, 887–895. [Google Scholar] [CrossRef] [PubMed]
- McMillan, R.; Wang, L.; Tomer, A.; Nichol, J.; Pistillo, J. Suppression of in vitro megakaryocyte production by antiplatelet autoantibodies from adult patients with chronic ITP. Blood 2004, 103, 1364–1369. [Google Scholar] [CrossRef] [PubMed]
- Aledort, L.M.; Hayward, C.P.; Chen, M.G.; Nichol, J.L.; Bussel, J.; Group, I.T.P.S. Prospective screening of 205 patients with ITP, including diagnosis, serological markers, and the relationship between platelet counts, endogenous thrombopoietin, and circulating antithrombopoietin antibodies. Am. J. Hematol. 2004, 76, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Kaushansky, K. The molecular mechanisms that control thrombopoiesis. J. Clin. Investig. 2005, 115, 3339–3347. [Google Scholar] [CrossRef] [PubMed]
- Ballem, P.J.; Segal, G.M.; Stratton, J.R.; Gernsheimer, T.; Adamson, J.W.; Slichter, S.J. Mechanisms of thrombocytopenia in chronic autoimmune thrombocytopenic purpura. Evidence of both impaired platelet production and increased platelet clearance. J. Clin. Investig. 1987, 80, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Emmons, R.V.; Reid, D.M.; Cohen, R.L.; Meng, G.; Young, N.S.; Dunbar, C.E.; Shulman, N.R. Human thrombopoietin levels are high when thrombocytopenia is due to megakaryocyte deficiency and low when due to increased platelet destruction. Blood 1996, 87, 4068–4071. [Google Scholar] [PubMed]
- Semple, J.W.; Freedman, J. Increased antiplatelet T helper lymphocyte reactivity in patients with autoimmune thrombocytopenia. Blood 1991, 78, 2619–2625. [Google Scholar] [PubMed]
- Stasi, R. Immune thrombocytopenia: Pathophysiologic and clinical update. Semin. Thromb. Hemost. 2012, 38, 454–462. [Google Scholar] [CrossRef] [PubMed]
- Olsson, B.; Ridell, B.; Carlsson, L.; Jacobsson, S.; Wadenvik, H. Recruitment of T cells into bone marrow of ITP patients possibly due to elevated expression of VLA-4 and CX3CR1. Blood 2008, 112, 1078–1084. [Google Scholar] [CrossRef] [PubMed]
- Houwerzijl, E.J.; Blom, N.R.; van der Want, J.J.; Esselink, M.T.; Koornstra, J.J.; Smit, J.W.; Louwes, H.; Vellenga, E.; de Wolf, J.T. Ultrastructural study shows morphologic features of apoptosis and para-apoptosis in megakaryocytes from patients with idiopathic thrombocytopenic purpura. Blood 2004, 103, 500–506. [Google Scholar] [CrossRef] [PubMed]
- Houwerzijl, E.J.; Blom, N.R.; van der Want, J.J.; Vellenga, E.; de Wolf, J.T. Megakaryocytic dysfunction in myelodysplastic syndromes and idiopathic thrombocytopenic purpura is in part due to different forms of cell death. Leukemia 2006, 20, 1937–1942. [Google Scholar] [CrossRef] [PubMed]
- Semple, J.W.; Milev, Y.; Cosgrave, D.; Mody, M.; Hornstein, A.; Blanchette, V.; Freedman, J. Differences in serum cytokine levels in acute and chronic autoimmune thrombocytopenic purpura: Relationship to platelet phenotype and antiplatelet T-cell reactivity. Blood 1996, 87, 4245–4254. [Google Scholar] [PubMed]
- Feng, X.; Scheinberg, P.; Samsel, L.; Rios, O.; Chen, J.; McCoy, J.P., Jr.; Ghanima, W.; Bussel, J.B.; Young, N.S. Decreased plasma cytokines are associated with low platelet counts in aplastic anemia and immune thrombocytopenic purpura. J. Thromb. Haemost. 2012, 10, 1616–1623. [Google Scholar] [CrossRef] [PubMed]
- Talaat, R.M.; Elmaghraby, A.M.; Barakat, S.S.; El-Shahat, M. Alterations in immune cell subsets and their cytokine secretion profile in childhood idiopathic thrombocytopenic purpura (ITP). Clin. Exp. Immunol. 2014, 176, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Cuker, A.; Prak, E.T.; Cines, D.B. Can immune thrombocytopenia be cured with medical therapy? Semin. Thromb. Hemost. 2015, 41, 395–404. [Google Scholar] [PubMed]
- Nomura, S. Advances in Diagnosis and Treatments for Immune Thrombocytopenia. Clin. Med. Insights Blood Disord. 2016, 9, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, O.H.; Tuckuviene, R.; Nielsen, K.R.; Rosthoj, S. Flow cytometric measurement of platelet-associated immunoglobulin in children with newly diagnosed Immune Thrombocytopenia. Eur. J. Haematol. 2016, 96, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Arnason, J.E.; Campigotto, F.; Neuberg, D.; Bussel, J.B. Abnormalities in IgA and IgM are associated with treatment-resistant ITP. Blood 2012, 119, 5016–5020. [Google Scholar] [CrossRef] [PubMed]
- Cines, D.B.; Cuker, A.; Semple, J.W. Pathogenesis of immune thrombocytopenia. Presse Med. 2014, 43, e49–e59. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.Y.; Hou, M.; Zhang, X.H.; Guan, X.H.; Sun, G.Z. The diagnostic value of platelet glycoprotein-specific autoantibody detection in idiopathic thrombocytopenic purpura. Zhongguo Shi Yan Xue Ye Xue Za Zhi 2004, 12, 204–206. [Google Scholar] [PubMed]
- Fujisawa, K.; O’Toole, T.E.; Tani, P.; Loftus, J.C.; Plow, E.F.; Ginsberg, M.H.; McMillan, R. Autoantibodies to the presumptive cytoplasmic domain of platelet glycoprotein IIIa in patients with chronic immune thrombocytopenic purpura. Blood 1991, 77, 2207–2213. [Google Scholar] [PubMed]
- Kuwana, M.; Kaburaki, J.; Ikeda, Y. Autoreactive T cells to platelet GPIIb-IIIa in immune thrombocytopenic purpura. Role in production of anti-platelet autoantibody. J. Clin. Investig. 1998, 102, 1393–1402. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.Q.; Dong, H.X.; Cheng, P.P.; Zhou, J.W.; Zheng, B.Y.; Liu, F. Antioxidant status and oxidative stress in patients with chronic ITP. Scand. J. Immunol. 2013, 77, 482–487. [Google Scholar] [CrossRef] [PubMed]
- Nieswandt, B.; Bergmeier, W.; Rackebrandt, K.; Gessner, J.E.; Zirngibl, H. Identification of critical antigen-specific mechanisms in the development of immune thrombocytopenic purpura in mice. Blood 2000, 96, 2520–2527. [Google Scholar] [PubMed]
- Webster, M.L.; Sayeh, E.; Crow, M.; Chen, P.; Nieswandt, B.; Freedman, J.; Ni, H. Relative efficacy of intravenous immunoglobulin G in ameliorating thrombocytopenia induced by antiplatelet GPIIbIIIa versus GPIbalpha antibodies. Blood 2006, 108, 943–946. [Google Scholar] [CrossRef] [PubMed]
- Mason, K.D.; Carpinelli, M.R.; Fletcher, J.I.; Collinge, J.E.; Hilton, A.A.; Ellis, S.; Kelly, P.N.; Ekert, P.G.; Metcalf, D.; Roberts, A.W.; et al. Programmed anuclear cell death delimits platelet life span. Cell 2007, 128, 1173–1186. [Google Scholar] [CrossRef] [PubMed]
- Leytin, V.; Mykhaylov, S.; Starkey, A.F.; Allen, D.J.; Lau, H.; Ni, H.; Semple, J.W.; Lazarus, A.H.; Freedman, J. Intravenous immunoglobulin inhibits anti-glycoprotein IIb-induced platelet apoptosis in a murine model of immune thrombocytopenia. Br. J. Haematol. 2006, 133, 78–82. [Google Scholar] [CrossRef] [PubMed]
- Deng, G.; Yu, S.; Li, Q.; He, Y.; Liang, W.; Yu, L.; Xu, D. Investigation of platelet apoptosis in adult patients with chronic immune thrombocytopenia. Hematology 2016, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Urbanus, R.T.; van der Wal, D.E.; Koekman, C.A.; Huisman, A.; van den Heuvel, D.J.; Gerritsen, H.C.; Deckmyn, H.; Akkerman, J.N.; Schutgens, R.E.G.; Gitz, E. Patient autoantibodies induce platelet destruction signals via raft-associated glycoprotein Ibalpha and Fc RIIa in immune thrombocytopenia. Haematologica 2013, 98, e70–e72. [Google Scholar] [CrossRef] [PubMed]
- Kapur, R.; Heitink-Polle, K.M.; Porcelijn, L.; Bentlage, A.E.; Bruin, M.C.; Visser, R.; Roos, D.; Schasfoort, R.B.; de Haas, M.; van der Schoot, C.E.; et al. C-reactive protein enhances IgG-mediated phagocyte responses and thrombocytopenia. Blood 2015, 125, 1793–1802. [Google Scholar] [CrossRef] [PubMed]
- Weiss, H.J.; Rosove, M.H.; Lages, B.A.; Kaplan, K.L. Acquired storage pool deficiency with increased platelet-associated IgG. Report of five cases. Am. J. Med. 1980, 69, 711–717. [Google Scholar] [CrossRef]
- Sarpatwari, A.; Bennett, D.; Logie, J.W.; Shukla, A.; Beach, K.J.; Newland, A.C.; Sanderson, S.; Proven, D. Thromboembolic events among adult patients with primary immune thrombocytopenia in the United Kingdom General Practice Research Database. Haematologica 2010, 95, 1167–1175. [Google Scholar] [CrossRef] [PubMed]
- Severinsen, M.T.; Engebjerg, M.C.; Farkas, D.K.; Jensen, A.O.; Norgaard, M.; Zhao, S.; Sorensen, H.T. Risk of venous thromboembolism in patients with primary chronic immune thrombocytopenia: A Danish population-based cohort study. Br. J. Haematol. 2011, 152, 360–362. [Google Scholar] [CrossRef] [PubMed]
- Norgaard, M.; Severinsen, M.T.; Lund Maegbaek, M.; Jensen, A.O.; Cha, S.; Sorensen, H.T. Risk of arterial thrombosis in patients with primary chronic immune thrombocytopenia: A Danish population-based cohort study. Br. J. Haematol. 2012, 159, 109–111. [Google Scholar] [CrossRef] [PubMed]
- Jy, W.; Horstman, L.L.; Arce, M.; Ahn, Y.S. Clinical significance of platelet microparticles in autoimmune thrombocytopenias. J. Lab. Clin. Med. 1992, 119, 334–345. [Google Scholar] [PubMed]
- Alvarez-Roman, M.T.; Fernandez-Bello, I.; Jimenez-Yuste, V.; Martin-Salces, M.; Arias-Salgado, E.G.; Rivas Pollmar, M.I.; Justo Sanz, R.; Butta, N.V. Procoagulant profile in patients with immune thrombocytopenia. Br. J. Haematol. 2016, 175, 925–934. [Google Scholar] [CrossRef] [PubMed]
- Yanabu, M.; Nomura, S.; Fukuroi, T.; Suzuki, M.; Kawakatsu, T.; Kido, H.; Yamaguchi, K. Platelet activation induced by an antiplatelet autoantibody against CD9 antigen and its inhibition by another autoantibody in immune thrombocytopenic purpura. Br. J. Haematol. 1993, 84, 694–701. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.F.; Yang, L.H.; Chang, L.X.; Feng, J.J.; Liu, J.Q. The clinical significance of circulating B cells secreting anti-glycoprotein IIb/IIIa antibody and platelet glycoprotein IIb/IIIa in patients with primary immune thrombocytopenia. Hematology 2012, 17, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Emmerich, F.; Bal, G.; Barakat, A.; Milz, J.; Muhle, C.; Martinez-Gamboa, L.; Dorner, T.; Salama, A. High-level serum B-cell activating factor and promoter polymorphisms in patients with idiopathic thrombocytopenic purpura. Br. J. Haematol. 2007, 136, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.H.; Zhuang, L.; Li, X.Y.; Li, J.; Luo, S.K. The role of B cell-activating factor secreted by peripheral blood monocyte-derived dendritic cell in chronic idiopathic thrombocytopenic purpura. Zhonghua Xue Ye Xue Za Zhi 2010, 31, 599–602. [Google Scholar] [PubMed]
- Yang, Q.; Xu, S.; Li, X.; Wang, B.; Wang, X.; Ma, D.; Yang, L.; Peng, J.; Hou, M. Pathway of Toll-like receptor 7/B cell activating factor/B cell activating factor receptor plays a role in immune thrombocytopenia in vivo. PLoS ONE 2011, 6, e22708. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Liu, Y.; Han, J.; Yang, Z.; Sheng, W.; Dai, H.; Wang, Y.; Xia, T.; Hou, M. TLR7 regulates dendritic cell-dependent B-cell responses through BlyS in immune thrombocytopenic purpura. Eur. J. Haematol. 2011, 86, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Olsson, B.; Ridell, B.; Jernas, M.; Wadenvik, H. Increased number of B-cells in the red pulp of the spleen in ITP. Ann. Hematol. 2012, 91, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Daridon, C.; Loddenkemper, C.; Spieckermann, S.; Kuhl, A.A.; Salama, A.; Burmester, G.R.; Lipsky, P.E.; Dörner, T. Splenic proliferative lymphoid nodules distinct from germinal centers are sites of autoantigen stimulation in immune thrombocytopenia. Blood 2012, 120, 5021–5031. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhong, H.; Bao, W.; Boulad, N.; Evangelista, J.; Haider, M.A.; Bussel, J.; Yazdanbakhsh, K. Defective regulatory B-cell compartment in patients with immune thrombocytopenia. Blood 2012, 120, 3318–3325. [Google Scholar] [CrossRef] [PubMed]
- Semple, J.W. Bregging rights in ITP. Blood 2012, 120, 3169. [Google Scholar] [CrossRef] [PubMed]
- Nishimoto, T.; Kuwana, M. CD4+CD25+Foxp3+ regulatory T cells in the pathophysiology of immune thrombocytopenia. Semin. Hematol. 2013, 50, S43–S49. [Google Scholar] [CrossRef] [PubMed]
- Mauri, C.; Bosma, A. Immune regulatory function of B cells. Annu. Rev. Immunol. 2012, 30, 221–241. [Google Scholar] [CrossRef] [PubMed]
- Aslam, R.; Segel, G.B.; Burack, R.; Spence, S.A.; Speck, E.R.; Guo, L.; Semple, J.W. Splenic lymphocyte subtypes in immune thrombocytopenia: Increased presence of a subtype of B-regulatory cells. Br. J. Haematol. 2016, 173, 159–160. [Google Scholar] [CrossRef] [PubMed]
- Jansen, P.H.; Renier, W.O.; de Vaan, G.; Reekers, P.; Vingerhoets, D.M.; Gabreels, F.J. Effect of thymectomy on myasthenia gravis and autoimmune thrombocytopenic purpura in a 13-year-old girl. Eur. J. Pediatr. 1987, 146, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Stuart, M.J.; Tomar, R.H.; Miller, M.L.; Davey, F.R. Chronic idiopathic thrombocytopenic purpura. A familial immunodeficiency syndrome? JAMA 1978, 239, 939–942. [Google Scholar] [CrossRef] [PubMed]
- Waters, A.H. Autoimmune thrombocytopenia: Clinical aspects. Semin. Hematol. 1992, 29, 18–25. [Google Scholar] [PubMed]
- Qiu, J.; Liu, X.; Li, X.; Zhang, X.; Han, P.; Zhou, H.; Shao, L.; Hou, Y.; Min, Y.; Kong, Z.; et al. CD8(+) T cells induce platelet clearance in the liver via platelet desialylation in immune thrombocytopenia. Sci. Rep. 2016, 6, 27445. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Chu, X.; Wang, L.; Zhu, Y.; Li, L.; Ma, D.; Peng, J.; Hou, M. Cell-mediated lysis of autologous platelets in chronic idiopathic thrombocytopenic purpura. Eur. J. Haematol. 2006, 76, 427–431. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Li, X.; Zhang, F.; Wang, L.; Peng, J.; Hou, M. Increased cytotoxic T-lymphocyte-mediated cytotoxicity predominant in patients with idiopathic thrombocytopenic purpura without platelet autoantibodies. Haematologica 2008, 93, 1428–1430. [Google Scholar] [CrossRef] [PubMed]
- Stasi, R.; Cooper, N.; Del Poeta, G.; Stipa, E.; Laura Evangelista, M.; Abruzzese, E.; Amadori, S. Analysis of regulatory T-cell changes in patients with idiopathic thrombocytopenic purpura receiving B cell-depleting therapy with rituximab. Blood 2008, 112, 1147–1150. [Google Scholar] [CrossRef] [PubMed]
- Bao, W.; Bussel, J.B.; Heck, S.; He, W.; Karpoff, M.; Boulad, N.; Yazdanbakhsh, K. Improved regulatory T-cell activity in patients with chronic immune thrombocytopenia treated with thrombopoietic agents. Blood 2010, 116, 4639–4645. [Google Scholar] [CrossRef] [PubMed]
- Audia, S.; Samson, M.; Guy, J.; Janikashvili, N.; Fraszczak, J.; Trad, M.; Ciudad, M.; Leguy, V.; Berthier, S.; Petrella, T.; et al. Immunologic effects of rituximab on the human spleen in immune thrombocytopenia. Blood 2011, 118, 4394–4400. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Mou, W.; Lu, G.; Cao, J.; He, X.; Pan, X.; Xu, K. Low-dose rituximab combined with short-term glucocorticoids up-regulates Treg cell levels in patients with immune thrombocytopenia. Int. J. Hematol. 2011, 93, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Ge, J.; Zhao, H.; Du, W.; Xu, J.; Sui, T.; Ma, L.; Zhou, Z.; Qi, A.; Yang, R. Association of cytotoxic T-lymphocyte antigen 4 gene polymorphisms with idiopathic thrombocytopenic purpura in a Chinese population. Platelets 2011, 22, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Fahim, N.M.; Monir, E. Functional role of CD4+CD25+ regulatory T cells and transforming growth factor-beta1 in childhood immune thrombocytopenic purpura. Egypt. J. Immunol. 2006, 13, 173–187. [Google Scholar] [PubMed]
- Ling, Y.; Cao, X.; Yu, Z.; Ruan, C. Circulating dendritic cells subsets and CD4+Foxp3+ regulatory T cells in adult patients with chronic ITP before and after treatment with high-dose dexamethasome. Eur. J. Haematol. 2007, 79, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Ling, Y.; Cao, X.S.; Yu, Z.Q.; Luo, G.H.; Bai, X.; Su, J.; Dai, L.; Ruan, C.G. Alterations of CD4+ CD25+ regulatory T cells in patients with idiopathic thrombocytopenic purpura. Zhonghua Xue Ye Xue Za Zhi 2007, 28, 184–188. [Google Scholar] [PubMed]
- Sakakura, M.; Wada, H.; Tawara, I.; Nobori, T.; Sugiyama, T.; Sagawa, N.; Shiku, H. Reduced Cd4+Cd25+ T cells in patients with idiopathic thrombocytopenic purpura. Thromb. Res. 2007, 120, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ma, D.; Zhu, X.; Qu, X.; Ji, C.; Hou, M. Elevated profile of Th17, Th1 and Tc1 cells in patients with immune thrombocytopenic purpura. Haematologica 2009, 94, 1326–1329. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.L.; Peng, J.; Sun, J.Z.; Liu, J.J.; Guo, C.S.; Wang, Z.G.; Yu, Y.; Shi, Y.; Qin, P.; Li, S.G.; et al. De novo induction of platelet-specific CD4(+)CD25(+) regulatory T cells from CD4(+)CD25(−) cells in patients with idiopathic thrombocytopenic purpura. Blood 2009, 113, 2568–2577. [Google Scholar] [CrossRef] [PubMed]
- Abudureheman, A.; Yasen, H.; Zhao, F.; Zhang, X.; Ding, J.; Ma, X.; Guo, X. Expression of CD4+ CD25+ regulatory T cells and TGF-ss1 in patient with idiopathic thrombocytopenic purpura. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi 2010, 26, 895–897. [Google Scholar] [PubMed]
- Chang, D.Y.; Ouyang, J.; Zhou, R.F.; Xu, J.Y.; Chen, B.; Yang, Y.G.; Zhang, Q.G.; Shao, X.Y.; Guan, C.Y.; Xu, Y. Profiles of different subsets of CD(4)(+) T cells in chronic idiopathic thrombocytopenic purpura. Zhonghua Nei Ke Za Zhi 2010, 49, 213–216. [Google Scholar] [PubMed]
- Aboul-Fotoh Lel, M.; Abdel Raheem, M.M.; El-Deen, M.A.; Osman, A.M. Role of CD4+CD25+ T cells in children with idiopathic thrombocytopenic purpura. J. Pediatr. Hematol. Oncol. 2011, 33, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.P.; Qiu, Y.S.; Hao, G.P.; Zhu, L. Levels of regulatory T cells in peripheral blood of children with idiopathic thrombocytopenic purpura. Zhongguo Dang Dai Er Ke Za Zhi 2011, 13, 282–284. [Google Scholar] [PubMed]
- Park, S.H.; Kim, J.Y.; Kim, S.K.; Choe, J.Y.; Kim, S.G.; Ryoo, H.M. Regulatory T-cells in systemic lupus erythematosus-associated thrombocytopenia: A comparison with idiopathic thrombocytopenic purpura. Lupus 2010, 19, 888–889. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.; Zhan, Y.; Hua, F.; Li, F.; Zou, S.; Wang, W.; Song, D. The ratio of Treg/Th17 cells correlates with the disease activity of primary immune thrombocytopenia. PLoS ONE 2012, 7, e50909. [Google Scholar] [CrossRef] [PubMed]
- Olsson, B.; Andersson, P.O.; Jacobsson, S.; Carlsson, L.; Wadenvik, H. Disturbed apoptosis of T-cells in patients with active idiopathic thrombocytopenic purpura. Thromb. Haemost. 2005, 93, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Gratz, I.K.; Rosenblum, M.D.; Abbas, A.K. The life of regulatory T cells. Ann. N. Y. Acad. Sci. 2013, 1283, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, S.; Miyara, M.; Costantino, C.M.; Hafler, D.A. FOXP3+ regulatory T cells in the human immune system. Nat. Rev. Immunol. 2010, 10, 490–500. [Google Scholar] [CrossRef] [PubMed]
- Semple, J.W.; Provan, D.; Garvey, M.B.; Freedman, J. Recent progress in understanding the pathogenesis of immune thrombocytopenia. Curr. Opin. Hematol. 2010, 17, 590–595. [Google Scholar] [CrossRef] [PubMed]
- Kuwana, M.; Ikeda, Y. The role of autoreactive T-cells in the pathogenesis of idiopathic thrombocytopenic purpura. Int. J. Hematol. 2005, 81, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Andre, S.; Tough, D.F.; Lacroix-Desmazes, S.; Kaveri, S.V.; Bayry, J. Surveillance of antigen-presenting cells by CD4+ CD25+ regulatory T cells in autoimmunity: Immunopathogenesis and therapeutic implications. Am. J. Pathol. 2009, 174, 1575–1587. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Zhao, H.; Poon, M.C.; Han, Z.; Gu, D.; Xu, M.; Jia, H.; Yang, R.; Han, Z.C. Abnormality of CD4(+)CD25(+) regulatory T cells in idiopathic thrombocytopenic purpura. Eur. J. Haematol. 2007, 78, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Heck, S.; Patel, V.; Levan, J.; Yu, Y.; Bussel, J.B.; Yazdanbakhsh, K. Defective circulating CD25 regulatory T cells in patients with chronic immune thrombocytopenic purpura. Blood 2008, 112, 1325–1328. [Google Scholar] [CrossRef] [PubMed]
- Aslam, R.; Hu, Y.; Gebremeskel, S.; Segel, G.B.; Speck, E.R.; Guo, L.; Kim, M. Thymic retention of CD4+CD25+FoxP3+ T regulatory cells is associated with their peripheral deficiency and thrombocytopenia in a murine model of immune thrombocytopenia. Blood 2012, 120, 2127–2132. [Google Scholar] [CrossRef] [PubMed]
- Catani, L.; Sollazzo, D.; Trabanelli, S.; Curti, A.; Evangelisti, C.; Polverelli, N.; Palandri, F. Decreased expression of indoleamine 2,3-dioxygenase 1 in dendritic cells contributes to impaired regulatory T cell development in immune thrombocytopenia. Ann. Hematol. 2013, 92, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhang, X.; Zhang, D.; Li, H.; Ma, L.; Xuan, M.; Wang, H.; Yang, R. Abnormal Distribution and Function of Monocyte Subsets in Patients With Primary Immune Thrombocytopenia. Clin. Appl. Thromb. Hemost. 2016. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.; Bao, W.; Li, X.; Miller, A.; Seery, C.; Haq, N.; Bussel, J.; Yazdanbakhsh, K. CD16+ monocytes control T-cell subset development in immune thrombocytopenia. Blood 2012, 120, 3326–3335. [Google Scholar] [CrossRef] [PubMed]
- Ware, R.E.; Howard, T.A. Phenotypic and clonal analysis of T lymphocytes in childhood immune thrombocytopenic purpura. Blood 1993, 82, 2137–2142. [Google Scholar] [PubMed]
- Filion, M.C.; Bradley, A.J.; Devine, D.V.; Decary, F.; Chartrand, P. Autoreactive T cells in healthy individuals show tolerance in vitro with characteristics similar to but distinct from clonal anergy. Eur. J. Immunol. 1995, 25, 3123–3127. [Google Scholar] [CrossRef] [PubMed]
- Coopamah, M.D.; Garvey, M.B.; Freedman, J.; Semple, J.W. Cellular immune mechanisms in autoimmune thrombocytopenic purpura: An update. Transfus. Med. Rev. 2003, 17, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Eyerich, S.; Eyerich, K.; Pennino, D.; Carbone, T.; Nasorri, F.; Pallotta, S.; Cianfarani, F.; Odorisio, T.; Traidl-Hoffmann, C.; Behrendt, H. Th22 cells represent a distinct human T cell subset involved in epidermal immunity and remodeling. J. Clin. Investig. 2009, 119, 3573–3585. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Chen, C.; Li, L.; Zeng, L.; Li, Z.; Yan, Z.; Chen, W.; Cheng, H.; Sang, W.; Xu, K. Effects of high-dose dexamethasone on regulating interleukin-22 production and correcting Th1 and Th22 polarization in immune thrombocytopenia. J. Clin. Immunol. 2012, 32, 523–529. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Chen, C.; Zeng, L.; Li, L.; Li, X.; Li, Z.; Xu, K. Elevated plasma IL-22 levels correlated with Th1 and Th22 cells in patients with immune thrombocytopenia. Clin. Immunol. 2011, 141, 121–123. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Li, H.; Zhang, L.; Shan, B.; Xu, X.; Li, H.; Liu, X.; Xu, S.; Yu, S.; Ma, D.; et al. Elevated profiles of Th22 cells and correlations with Th17 cells in patients with immune thrombocytopenia. Hum. Immunol. 2012, 73, 629–635. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.M.; Zhang, X.X.; Zhao, G.S.; Si, Y.J.; Lin, G.Q.; Zhang, Y.M.; He, G.S.; Wu, D.P. Change of Th22 cells in peripheral blood of patients with primary immune thrombocytopenia and clinical implication. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi 2012, 28, 1314–1316. [Google Scholar] [PubMed]
- Guo, N.H.; Shi, Q.Z.; Hua, J.Y.; Li, Z.J.; Li, J.; He, W.F.; Wu, Q. Expression of regulatory T cells and Th17 cells in idiopathic thrombocytopenic purpura and its significance. Zhonghua Xue Ye Xue Za Zhi 2010, 31, 610–612. [Google Scholar] [PubMed]
- Hu, Y.; Ma, D.X.; Shan, N.N.; Zhu, Y.Y.; Liu, X.G.; Zhang, L.; Yu, S. Increased number of Tc17 and correlation with Th17 cells in patients with immune thrombocytopenia. PLoS ONE 2011, 6, e26522. [Google Scholar] [CrossRef] [PubMed]
- Rocha, A.M.; Souza, C.; Rocha, G.A.; de Melo, F.F.; Clementino, N.C.; Marino, M.C.; Bozzi, A.; Silva, M.L.; Martins Filho, O.A.; Queiroz, D.M. The levels of IL-17A and of the cytokines involved in Th17 cell commitment are increased in patients with chronic immune thrombocytopenia. Haematologica 2011, 96, 1560–1564. [Google Scholar] [CrossRef] [PubMed]
- Duan, X.J.; Yang, L.H.; Zhang, L.; Ren, F.G.; Zhang, R.J.; Chen, J.F.; Qin, X.Y.; Liang, H.Z. Expressions of Th17 cells and interleukin 17 in patients with primary immune thrombocytopenia and their clinical significance. Zhongguo Shi Yan Xue Ye Xue Za Zhi 2012, 20, 1154–1157. [Google Scholar] [PubMed]
- Yoh, K.; Morito, N.; Ojima, M.; Shibuya, K.; Yamashita, Y.; Morishima, Y.; Ishii, Y.; Kusakabe, M.; Nishikii, H.; Fujita, A.; et al. Overexpression of RORgammat under control of the CD2 promoter induces polyclonal plasmacytosis and autoantibody production in transgenic mice. Eur. J. Immunol. 2012, 42, 1999–2009. [Google Scholar] [CrossRef] [PubMed]
- Baeten, D.L.; Kuchroo, V.K. How Cytokine networks fuel inflammation: Interleukin-17 and a tale of two autoimmune diseases. Nat. Med. 2013, 19, 824–825. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.X.; Chen, Z.P.; Zheng, C.L.; Jia, H.R.; Ge, J.; Gu, D.S.; Du, W.T.; Wang, X.Y.; Zhao, H.F.; Yang, R.C. The role of Th17 cells in adult patients with chronic idiopathic thrombocytopenic purpura. Eur. J. Haematol. 2009, 82, 488–489. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Zhu, X.; Zhao, P.; Zhao, C.; Li, X.; Zhu, Y.; Li, L. Profile of Th17 cytokines (IL-17, TGF-beta, IL-6) and Th1 cytokine (IFN-gamma) in patients with immune thrombocytopenic purpura. Ann. Hematol. 2008, 87, 899–904. [Google Scholar] [CrossRef] [PubMed]
- Sollazzo, D.; Trabanelli, S.; Curti, A.; Vianelli, N.; Lemoli, R.M.; Catani, L. Circulating CD4+CD161+CD196+ Th17 cells are not increased in immune thrombocytopenia. Haematologica 2011, 96, 632–634. [Google Scholar] [CrossRef] [PubMed]
- Rock, K.L.; Benacerraf, B.; Abbas, A.K. Antigen presentation by hapten-specific B lymphocytes. I. Role of surface immunoglobulin receptors. J. Exp. Med. 1984, 160, 1102–1113. [Google Scholar] [CrossRef] [PubMed]
- Unanue, E.R. Antigen-presenting function of the macrophage. Annu. Rev. Immunol. 1984, 2, 395–428. [Google Scholar] [CrossRef] [PubMed]
- Amodio, G.; Gregori, S. Dendritic cells a double-edge sword in autoimmune responses. Front. Immunol. 2012, 3, 233. [Google Scholar] [CrossRef] [PubMed]
- Watts, C. Capture and processing of exogenous antigens for presentation on MHC molecules. Annu. Rev. Immunol. 1997, 15, 821–850. [Google Scholar] [CrossRef] [PubMed]
- Banchereau, J.; Briere, F.; Caux, C.; Davoust, J.; Lebecque, S.; Liu, Y.J.; Pulendran, B. Immunobiology of dendritic cells. Annu. Rev. Immunol. 2000, 18, 767–811. [Google Scholar] [CrossRef] [PubMed]
- Chaplin, D.D. Overview of the immune response. J. Allergy Clin. Immunol. 2010, 125, S3–23. [Google Scholar] [CrossRef] [PubMed]
- Hughes, C.E.; Benson, R.A.; Bedaj, M.; Maffia, P. Antigen-Presenting Cells and Antigen Presentation in Tertiary Lymphoid Organs. Front. Immunol. 2016, 7, 481. [Google Scholar] [CrossRef] [PubMed]
- Coutant, F.; Miossec, P. Altered dendritic cell functions in autoimmune diseases: Distinct and overlapping profiles. Nat. Rev. Rheumatol. 2016, 12, 703–715. [Google Scholar] [CrossRef] [PubMed]
- Catani, L.; Fagioli, M.E.; Tazzari, P.L.; Ricci, F.; Curti, A.; Rovito, M.; Preda, P. Dendritic cells of immune thrombocytopenic purpura (ITP) show increased capacity to present apoptotic platelets to T lymphocytes. Exp. Hematol. 2006, 34, 879–887. [Google Scholar] [CrossRef] [PubMed]
- Saito, A.; Yokohama, A.; Osaki, Y.; Ogawa, Y.; Nakahashi, H.; Toyama, K.; Mitsui, T.; Hashimoto, Y.; Koiso, H.; Uchiumi, H.; et al. Circulating plasmacytoid dendritic cells in patients with primary and Helicobacter pylori-associated immune thrombocytopenia. Eur. J. Haematol. 2012, 88, 340–349. [Google Scholar] [CrossRef] [PubMed]
- Swiecki, M.; Colonna, M. The multifaceted biology of plasmacytoid dendritic cells. Nat. Rev. Immunol. 2015, 15, 471–485. [Google Scholar] [CrossRef] [PubMed]
- Panda, S.K.; Kolbeck, R.; Sanjuan, M.A. Plasmacytoid dendritic cells in autoimmunity. Curr. Opin. Immunol. 2016, 44, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Andonegui, G.; Kerfoot, S.M.; McNagny, K.; Ebbert, K.V.; Patel, K.D.; Kubes, P. Platelets express functional Toll-like receptor-4. Blood 2005, 106, 2417–2423. [Google Scholar] [CrossRef] [PubMed]
- Aslam, R.; Speck, E.R.; Kim, M.; Crow, A.R.; Bang, K.W.; Nestel, F.P.; Ni, H.; Lazarus, A.H.; Freedman, J.; Semple, J.W. Platelet Toll-like receptor expression modulates lipopolysaccharide-induced thrombocytopenia and tumor necrosis factor-alpha production in vivo. Blood 2006, 107, 637–641. [Google Scholar] [CrossRef] [PubMed]
- Semple, J.W.; Aslam, R.; Kim, M.; Speck, E.R.; Freedman, J. Platelet-bound lipopolysaccharide enhances Fc receptor-mediated phagocytosis of IgG-opsonized platelets. Blood 2007, 109, 4803–4805. [Google Scholar] [CrossRef] [PubMed]
- Machlus, K.R.; Thon, J.N.; Italiano, J.E., Jr. Interpreting the developmental dance of the megakaryocyte: A review of the cellular and molecular processes mediating platelet formation. Br. J. Haematol. 2014, 165, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Stahl, C.P.; Zucker-Franklin, D.; McDonald, T.P. Incomplete antigenic cross-reactivity between platelets and megakaryocytes: Relevance to ITP. Blood 1986, 67, 421–428. [Google Scholar] [PubMed]
- Nugent, D.; McMillan, R.; Nichol, J.L.; Slichter, S.J. Pathogenesis of chronic immune thrombocytopenia: Increased platelet destruction and/or decreased platelet production. Br. J. Haematol. 2009, 146, 585–596. [Google Scholar] [CrossRef] [PubMed]
- Norol, F.; Vitrat, N.; Cramer, E.; Guichard, J.; Burstein, S.A.; Vainchenker, W.; Debili, N. Effects of cytokines on platelet production from blood and marrow CD34+ cells. Blood 1998, 91, 830–843. [Google Scholar] [PubMed]
- Hou, M.; Andersson, P.O.; Stockelberg, D.; Mellqvist, U.H.; Ridell, B.; Wadenvik, H. Plasma thrombopoietin levels in thrombocytopenic states: Implication for a regulatory role of bone marrow megakaryocytes. Br. J. Haematol. 1998, 101, 420–424. [Google Scholar] [CrossRef] [PubMed]
- Iraqi, M.; Perdomo, J.; Yan, F.; Choi, P.Y.; Chong, B.H. Immune thrombocytopenia: Antiplatelet autoantibodies inhibit proplatelet formation by megakaryocytes and impair platelet production in vitro. Haematologica 2015, 100, 623–632. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Li, H.; Ma, L.; Zhang, X.; Xue, F.; Zhou, Z.; Chi, Y. The defective bone marrow-derived mesenchymal stem cells in patients with chronic immune thrombocytopenia. Autoimmunity 2014, 47, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Bruns, I.; Lucas, D.; Pinho, S.; Ahmed, J.; Lambert, M.P.; Kunisaki, Y.; Scheiermann, C.; Schiff, L.; Poncz, M.; Bergman, A.; et al. Megakaryocytes regulate hematopoietic stem cell quiescence through CXCL4 secretion. Nat. Med. 2014, 20, 1315–1320. [Google Scholar] [CrossRef] [PubMed]
- Nakamura-Ishizu, A.; Takubo, K.; Kobayashi, H.; Suzuki-Inoue, K.; Suda, T. CLEC-2 in megakaryocytes is critical for maintenance of hematopoietic stem cells in the bone marrow. J. Exp. Med. 2015, 212, 2133–2146. [Google Scholar] [CrossRef] [PubMed]
- Malara, A.; Abbonante, V.; Di Buduo, C.A.; Tozzi, L.; Currao, M.; Balduini, A. The secret life of a megakaryocyte: Emerging roles in bone marrow homeostasis control. Cell. Mol. Life Sci. 2015, 72, 1517–1536. [Google Scholar] [CrossRef] [PubMed]
- Winter, O.; Moser, K.; Mohr, E.; Zotos, D.; Kaminski, H.; Szyska, M.; Roth, K.; Wong, D.M.; Dame, C.; Tarlinton, D.M.; et al. Megakaryocytes constitute a functional component of a plasma cell niche in the bone marrow. Blood 2010, 116, 1867–1875. [Google Scholar] [CrossRef] [PubMed]
- Uccelli, A.; Moretta, L.; Pistoia, V. Immunoregulatory function of mesenchymal stem cells. Eur. J. Immunol. 2006, 36, 2566–2573. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Hu, Y.; Zhang, X.H.; Wang, Y.Z.; Mo, X.D.; Zhang, Y.Y.; Wang, Y.; Han, W.; Xu, L.P.; Chang, Y.J.; et al. Association between an impaired bone marrow vascular microenvironment and prolonged isolated thrombocytopenia after allogeneic hematopoietic stem cell transplantation. Biol. Blood Marrow Transplant. 2014, 20, 1190–1197. [Google Scholar] [CrossRef] [PubMed]
- Kojouri, K.; Vesely, S.K.; Terrell, D.R.; George, J.N. Splenectomy for adult patients with idiopathic thrombocytopenic purpura: A systematic review to assess long-term platelet count responses, prediction of response, and surgical complications. Blood 2004, 104, 2623–2634. [Google Scholar] [CrossRef] [PubMed]
- Rodeghiero, F.; Ruggeri, M. Short- and long-term risks of splenectomy for benign haematological disorders: Should we revisit the indications? Br. J. Haematol. 2012, 158, 16–29. [Google Scholar] [CrossRef] [PubMed]
- Lazarus, A.H. Monoclonal versus polyclonal anti-D in the treatment of ITP. Expert Opin. Biol. Ther. 2013, 13, 1353–1356. [Google Scholar] [CrossRef] [PubMed]
- Crow, A.R.; Lazarus, A.H. Mechanistic properties of intravenous immunoglobulin in murine immune thrombocytopenia: Support for FcgammaRIIB falls by the wayside. Semin. Hematol. 2016, 53, S20–S22. [Google Scholar] [CrossRef] [PubMed]
- Provan, D.; Stasi, R.; Newland, A.C.; Blanchette, V.S.; Bolton-Maggs, P.; Bussel, J.B.; Chong, B.H. International consensus report on the investigation and management of primary immune thrombocytopenia. Blood 2010, 115, 168–186. [Google Scholar] [CrossRef] [PubMed]
- Bussel, J.B.; Lee, C.S.; Seery, C.; Imahiyerobo, A.A.; Thompson, M.V.; Catellier, D.; Turenne, I.G. Rituximab and three dexamethasone cycles provide responses similar to splenectomy in women and those with immune thrombocytopenia of less than two years duration. Haematologica 2014, 99, 1264–1271. [Google Scholar] [CrossRef] [PubMed]
- Zaja, F.; Baccarani, M.; Mazza, P.; Bocchia, M.; Gugliotta, L.; Zaccaria, A.; Vianelli, N.; Defina, M.; Tieghi, A.; Amadori, S.; et al. Dexamethasone plus rituximab yields higher sustained response rates than dexamethasone monotherapy in adults with primary immune thrombocytopenia. Blood 2010, 115, 2755–2762. [Google Scholar] [CrossRef] [PubMed]
- McHeyzer-Williams, L.J.; McHeyzer-Williams, M.G. Antigen-specific memory B cell development. Annu. Rev. Immunol. 2005, 23, 487–513. [Google Scholar] [CrossRef] [PubMed]
- Grimaldi, D.; Canoui-Poitrine, F.; Croisille, L.; Lee, K.; Roudot-Thoraval, F.; Languille, L.; Khellaf, M. Antiplatelet antibodies detected by the MAIPA assay in newly diagnosed immune thrombocytopenia are associated with chronic outcome and higher risk of bleeding. Ann. Hematol. 2014, 93, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Cole, T.J. Glucocorticoid action and the development of selective glucocorticoid receptor ligands. Biotechnol. Annu. Rev. 2006, 12, 269–300. [Google Scholar] [PubMed]
- Li, J.; Wang, Z.; Dai, L.; Cao, L.; Su, J.; Zhu, M.; Yu, Z.; Bai, X.; Ruan, C. Effects of rapamycin combined with low dose prednisone in patients with chronic immune thrombocytopenia. Clin. Dev. Immunol. 2013, 2013, 548085. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, Z.; Hu, S.; Zhao, X.; Cao, L. Correction of abnormal T cell subsets by high-dose dexamethasone in patients with chronic idiopathic thrombocytopenic purpura. Immunol. Lett. 2013, 154, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.H.; Zhao, F.; Shi, W.; Ma, X.M.; Xu, Q.; Patiguli, A.B.; Halida, Y.S. Detection and clinical significance of Th1/Th2 cytokines in patients with idiopathic thrombocytopenic purpura. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi 2012, 28, 1185–1187. [Google Scholar] [PubMed]
- Clynes, R. Immune complexes as therapy for autoimmunity. J. Clin. Investig. 2005, 115, 25–27. [Google Scholar] [CrossRef] [PubMed]
- Nagelkerke, S.Q.; Kuijpers, T.W. Immunomodulation by IVIg and the Role of Fc-Gamma Receptors: Classic Mechanisms of Action after all? Front. Immunol. 2014, 5, 674. [Google Scholar] [CrossRef] [PubMed]
- Gilardin, L.; Bayry, J.; Kaveri, S.V. Intravenous immunoglobulin as clinical immune-modulating therapy. CMAJ 2015, 187, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Park-Min, K.H.; Serbina, N.V.; Yang, W.; Ma, X.; Krystal, G.; Neel, B.G.; Nutt, S.L.; Hu, X.; Ivashkiv, L.B. FcgammaRIII-dependent inhibition of interferon-gamma responses mediates suppressive effects of intravenous immune globulin. Immunity 2007, 26, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Pang, S.J.; Lazarus, A.H. Mechanisms of platelet recovery in ITP associated with therapy. Ann. Hematol. 2010, 89, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Crow, A.R.; Lazarus, A.H. The mechanisms of action of intravenous immunoglobulin and polyclonal anti-d immunoglobulin in the amelioration of immune thrombocytopenic purpura: What do we really know? Transfus. Med. Rev. 2008, 22, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Fehr, J.; Hofmann, V.; Kappeler, U. Transient reversal of thrombocytopenia in idiopathic thrombocytopenic purpura by high-dose intravenous gamma globulin. N. Engl. J. Med. 1982, 306, 1254–1258. [Google Scholar] [CrossRef] [PubMed]
- Berchtold, P.; Dale, G.L.; Tani, P.; McMillan, R. Inhibition of autoantibody binding to platelet glycoprotein IIb/IIIa by anti-idiotypic antibodies in intravenous gammaglobulin. Blood 1989, 74, 2414–2417. [Google Scholar] [PubMed]
- Hansen, R.J.; Balthasar, J.P. Intravenous immunoglobulin mediates an increase in anti-platelet antibody clearance via the FcRn receptor. Thromb. Haemost. 2002, 88, 898–899. [Google Scholar] [PubMed]
- Bayry, J.; Lacroix-Desmazes, S.; Carbonneil, C.; Misra, N.; Donkova, V.; Pashov, A.; Chevailler, A.; Mouthon, L.; Weill, B.; Bruneval, P.; et al. Inhibition of maturation and function of dendritic cells by intravenous immunoglobulin. Blood 2003, 101, 758–765. [Google Scholar] [CrossRef] [PubMed]
- Siragam, V.; Crow, A.R.; Brinc, D.; Song, S.; Freedman, J.; Lazarus, A.H. Intravenous immunoglobulin ameliorates ITP via activating Fc gamma receptors on dendritic cells. Nat. Med. 2006, 12, 688–692. [Google Scholar] [CrossRef] [PubMed]
- Anthony, R.M.; Wermeling, F.; Karlsson, M.C.; Ravetch, J.V. Identification of a receptor required for the anti-inflammatory activity of IVIG. Proc. Natl. Acad. Sci. USA 2008, 105, 19571–19578. [Google Scholar] [CrossRef] [PubMed]
- Salama, A.; Kiefel, V.; Amberg, R.; Mueller-Eckhardt, C. Treatment of autoimmune thrombocytopenic purpura with rhesus antibodies (anti-Rho(D)). Blut 1984, 49, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Crow, A.R.; Siragam, V.; Freedman, J.; Lazarus, A.H. Monoclonal antibodies that mimic the action of anti-D in the amelioration of murine ITP act by a mechanism distinct from that of IVIg. Blood 2005, 105, 1546–1548. [Google Scholar] [CrossRef] [PubMed]
- Ambriz-Fernandez, R.; Martinez-Murillo, C.; Quintana-Gonzalez, S.; Collazo-Jaloma, J.; Bautista-Juarez, J. Fc receptor blockade in patients with refractory chronic immune thrombocytopenic purpura with anti-D IgG. Arch. Med. Res. 2002, 33, 536–540. [Google Scholar] [CrossRef]
- Boughton, B.J.; Cooke, R.M.; Smith, N.A.; Simpson, A.W. Autoimmune thrombocytopenia: Anti-glycoprotein IIb/IIIa auto antibodies are reduced after human anti-D immunoglobulin treatment. Autoimmunity 1994, 18, 141–144. [Google Scholar] [CrossRef] [PubMed]
- Kuwana, M.; Okazaki, Y.; Kaburaki, J.; Kawakami, Y.; Ikeda, Y. Spleen is a primary site for activation of platelet-reactive T and B cells in patients with immune thrombocytopenic purpura. J. Immunol. 2002, 168, 3675–3682. [Google Scholar] [CrossRef] [PubMed]
- Knobl, P. Inherited and acquired thrombotic thrombocytopenic purpura (TTP) in adults. Semin. Thromb. Hemost. 2014, 40, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; Wang, S.; Xue, F.; Liu, X.; Zhang, L.; Li, H.; Yang, R. Long-term results of splenectomy in adult chronic immune thrombocytopenia. Eur. J. Haematol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Golay, J.; Manganini, M.; Facchinetti, V.; Gramigna, R.; Broady, R.; Borleri, G.; Rambaldi, A.; Introna, M. Rituximab-mediated antibody-dependent cellular cytotoxicity against neoplastic B cells is stimulated strongly by interleukin-2. Haematologica 2003, 88, 1002–1012. [Google Scholar] [PubMed]
- Guo, L.; Kapur, R.; Aslam, R.; Speck, E.R.; Zufferey, A.; Zhao, Y.; Kim, M.; Lazarus, A.H.; Ni, H.; Semple, J.W. CD20+ B-cell depletion therapy suppresses murine CD8+ T-cell-mediated immune thrombocytopenia. Blood 2016, 127, 735–738. [Google Scholar] [CrossRef] [PubMed]
- Rudnicka, D.; Oszmiana, A.; Finch, D.K.; Strickland, I.; Schofield, D.J.; Lowe, D.C.; Sleeman, M.A. Rituximab causes a polarization of B cells that augments its therapeutic function in NK-cell-mediated antibody-dependent cellular cytotoxicity. Blood 2013, 121, 4694–4702. [Google Scholar] [CrossRef] [PubMed]
- Stasi, R.; Del Poeta, G.; Stipa, E.; Evangelista, M.L.; Trawinska, M.M.; Cooper, N.; Amadori, S. Response to B-cell depleting therapy with rituximab reverts the abnormalities of T-cell subsets in patients with idiopathic thrombocytopenic purpura. Blood 2007, 110, 2924–2930. [Google Scholar] [CrossRef] [PubMed]
- Eisenbeis, C.F.; Grainger, A.; Fischer, B.; Baiocchi, R.A.; Carrodeguas, L.; Roychowdhury, S.; Chen, L. Combination immunotherapy of B-cell non-Hodgkin’s lymphoma with rituximab and interleukin-2: A preclinical and phase I study. Clin. Cancer Res. 2004, 10, 6101–6110. [Google Scholar] [CrossRef] [PubMed]
- Gluck, W.L.; Hurst, D.; Yuen, A.; Levine, A.M.; Dayton, M.A.; Gockerman, J.P.; Lucas, J. Phase I studies of interleukin (IL)-2 and rituximab in B-cell non-hodgkin’s lymphoma: IL-2 mediated natural killer cell expansion correlations with clinical response. Clin. Cancer Res. 2004, 10, 2253–2264. [Google Scholar] [CrossRef] [PubMed]
- Stasi, R.; Pagano, A.; Stipa, E.; Amadori, S. Rituximab chimeric anti-CD20 monoclonal antibody treatment for adults with chronic idiopathic thrombocytopenic purpura. Blood 2001, 98, 952–957. [Google Scholar] [CrossRef] [PubMed]
- Chapin, J.; Lee, C.S.; Zhang, H.; Zehnder, J.L.; Bussel, J.B. Gender and duration of disease differentiate responses to rituximab-dexamethasone therapy in adults with immune thrombocytopenia. Am. J. Hematol. 2016, 91, 907–911. [Google Scholar] [CrossRef] [PubMed]
- Marangon, M.; Vianelli, N.; Palandri, F.; Mazzucconi, M.G.; Santoro, C.; Barcellini, W.; Fattizzo, B.; Volpetti, S.; Lucchini, E.; Polverelli, N.; et al. Rituximab in immune thrombocytopenia: Gender, age and response as predictors of long-term response. Eur. J. Haematol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Reboursiere, E.; Fouques, H.; Maigne, G.; Johnson, H.; Chantepie, S.; Gac, A.C.; Reman, O.; Macro, M.; Benabed, K.; Troussard, X.; et al. Rituximab salvage therapy in adults with immune thrombocytopenia: Retrospective study on efficacy and safety profiles. Int. J. Hematol. 2016, 104, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.L.; Mahevas, M.; Lee, S.Y.; Stasi, R.; Cunningham-Rundles, S.; Godeau, B.; Kanter, J. Outcomes 5 years after response to rituximab therapy in children and adults with immune thrombocytopenia. Blood 2012, 119, 5989–5995. [Google Scholar] [CrossRef] [PubMed]
- Ghanima, W.; Khelif, A.; Waage, A.; Michel, M.; Tjonnfjord, G.E.; Romdhan, N.B.; Kahrs, J. Rituximab as second-line treatment for adult immune thrombocytopenia (the RITP trial): A multicentre, randomised, double-blind, placebo-controlled trial. Lancet 2015, 385, 1653–1661. [Google Scholar] [CrossRef]
- Erickson-Miller, C.L.; DeLorme, E.; Tian, S.S.; Hopson, C.B.; Stark, K.; Giampa, L.; Valoret, E.I.; Duffy, K.J.; Luengo, J.L.; Rosen, J.; et al. Discovery and characterization of a selective, nonpeptidyl thrombopoietin receptor agonist. Exp. Hematol. 2005, 33, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Cohn, C.S.; Bussel, J.B. Romiplostim: A second-generation thrombopoietin agonist. Drugs Today 2009, 45, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Newland, A.; Godeau, B.; Priego, V.; Viallard, J.F.; Lopez Fernandez, M.F.; Orejudos, A.; Eisen, M. Remission and platelet responses with romiplostim in primary immune thrombocytopenia: Final results from a phase 2 study. Br. J. Haematol. 2016, 172, 262–273. [Google Scholar] [CrossRef] [PubMed]
- McKenzie, C.G.; Guo, L.; Freedman, J.; Semple, J.W. Cellular immune dysfunction in immune thrombocytopenia (ITP). Br. J. Haematol. 2013, 163, 10–23. [Google Scholar] [CrossRef] [PubMed]
- Klinger, M.H.; Jelkmann, W. Subcellular localization of thrombopoietin in human blood platelets and its release upon thrombin stimulation. Br. J. Haematol. 2001, 115, 421–427. [Google Scholar] [CrossRef] [PubMed]
- Stasi, R.; Bosworth, J.; Rhodes, E.; Shannon, M.S.; Willis, F.; Gordon-Smith, E.C. Thrombopoietic agents. Blood Rev. 2010, 24, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Kapur, R.; Zufferey, A.; Boilard, E.; Semple, J.W. Nouvelle Cuisine: Platelets Served with Inflammation. J. Immunol. 2015, 194, 5579–5587. [Google Scholar] [CrossRef] [PubMed]
- Kapur, R.; Semple, J.W. The nonhemostatic immune functions of platelets. Semin. Hematol. 2016, 53, S2–S6. [Google Scholar] [CrossRef] [PubMed]
- Kapur, R.; Semple, J.W. Platelets as immune-sensing cells. Blood Adv. 2016, 1, 10–14. [Google Scholar] [CrossRef]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).