
Congenital Stationary Night Blindness (CSNB)—Case Reports and Review of Current Knowledge

Abstract
1. Introduction
2. Types of CSNB Based on Electrophysiological Findings
- The Riggs type is characterized by selective dysfunction of rod receptors, (DA—dark adaptation, LA—light adaptation):
- DA 0.01 ERG—not detectable;
- DA 3.0 ERG—reduction in amplitude of a- and b-waves;
- LA 3.0 ERG—normal response [7];
3. Types of CSNB with Fundus Lesions
- Fundus albipunctatus (FAP) is a rare inherited retinal disorder characterized by the presence of small white or yellowish-white punctate lesions in the mid-periphery of the fundus, sparing the macula at the level of the retinal pigment epithelium. Using fundus autofluorescence (FAF), the accumulation of lipofuscin in the RPE of a patient with FAP can be observed. The disease manifests in early childhood and primarily affects rod photoreceptors [9].In ERG examination the following is observed:
- DA 0.01 ERG—absent, but after prolonged dark adaptation (120 min) there is full recovery of the b-wave;
- DA 3.0 ERG—reduction in the amplitude of a- and b-waves, with b-wave reduction possibly greater than a-wave reduction (negative ERG pattern);
- LA 30 Hz—reduction in interpeak values;
- LA 3.0 ERG—normal.
- Oguchi disease is a rare form of congenital stationary night blindness, characterized by the Mizuo–Nakamura phenomenon (a metallic sheen across the entire retina, which disappears after approximately 3 h of dark adaptation). The ERG is similar to the Riggs type—prolonged dark adaptation results in an improved rod response. Further research on the genes involved in phototransduction and light adaptation is needed to determine the pathogenesis of this rare disease [10].
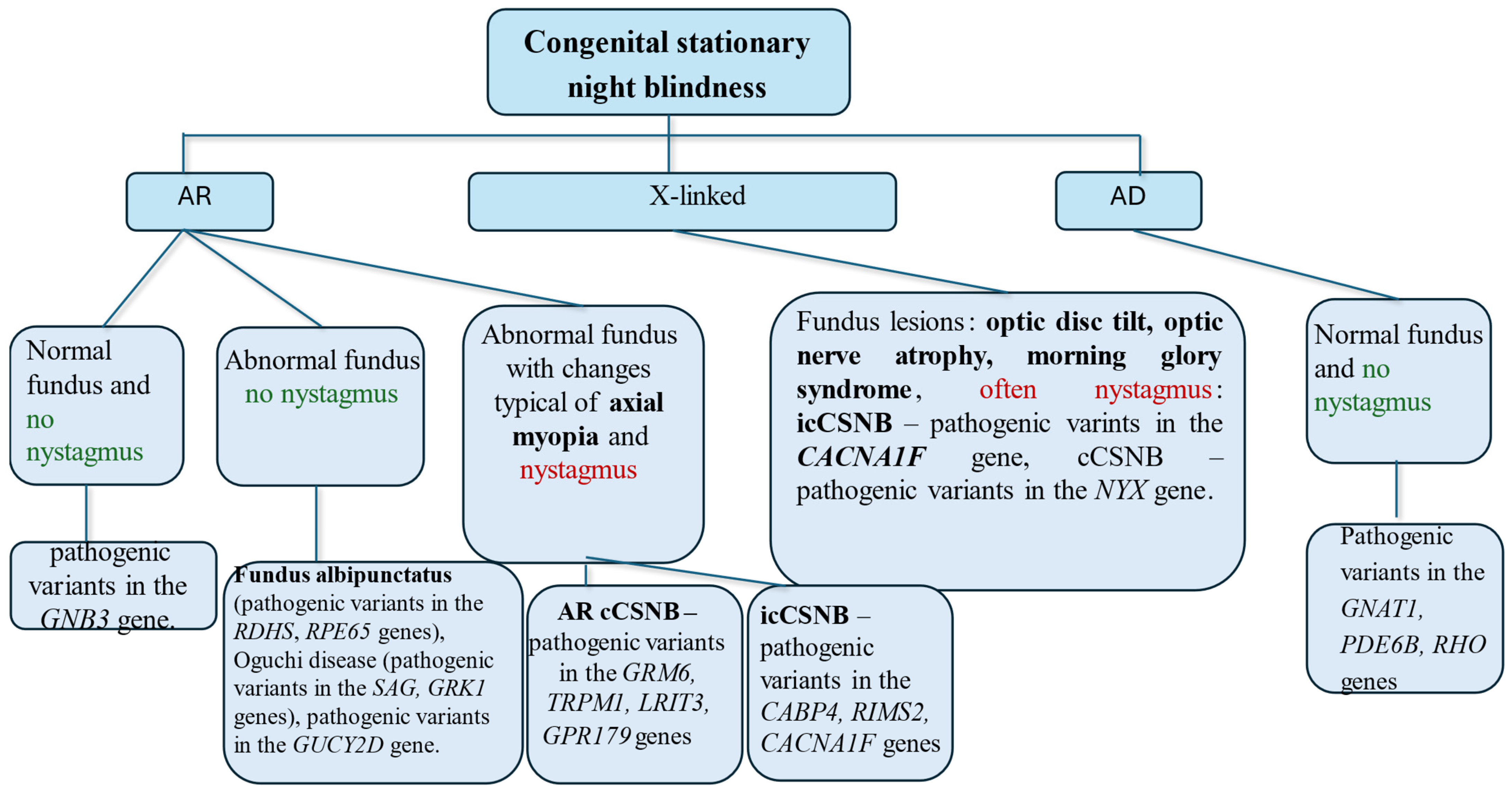
4. Characteristics of CSNB Based on Inheritance Patterns, Fundus Lesions, and Presence of Nystagmus
- A normal fundus and an absence of nystagmus suggest a pathogenic variant in the GNB3 gene;
- An abnormal fundus but no nystagmus suggests Fundus albipunctatus (pathogenic variants in the RDHS and RPE65 genes), Oguchi disease (pathogenic variants in the SAG and GRK1 genes), or mutations in the GUCY2D gene;
- An abnormal fundus with changes typical of axial myopia and nystagmus suggests the following:
- cCSNB—pathogenic variants in the GRM6, TRPM1, LRIT3, or GPR179 genes are suspected;
- icCSNB—pathogenic variants in the CABP4, RIMS2, or CACNA1F genes are suspected.
In X-linked inheritance:- icCSNB—pathogenic variants in the CACNA1F gene—nystagmus is common, and fundus changes are possible:
- cCSNB—mutations in the NYX gene.
In autosomal dominant inheritance:- Normal fundus and no nystagmus—pathogenic variants in the GNAT1, PDE6B, or RHO genes.
5. Case Reports and Review of Current Knowledge
5.1. Section 1 Cases Report
5.1.1. Case 1
5.1.2. Case 2
5.1.3. Case 3
5.2. Section 2 Review of Current Knowledge
Conditions with Electronegative ERG
6. Discussion
6.1. Electrophysiological Studies in CSNB Available in the Literature
6.1.1. Flicker ERG
6.1.2. MfERG
- Normal P1 wave amplitude with prolonged peak time in the foveal area (R1);
- Significant reduction in P1 wave amplitude in the parafoveal area (R2) [24].
- Normal amplitude with normal peak time in the first ring;
- Normal amplitude with prolonged peak time in the second ring;
- Reduction in amplitude in rings 3–5 and prolonged peak time in rings 2–6.
6.1.3. VEP
6.1.4. Electrooculography (EOG)
6.1.5. Pattern ERG
6.2. Combined Structural and Functional Exams Improve Diagnostics of CSNB
6.3. Genetics in CSNB
6.4. Monitoring
6.5. Treatment of Conditions Associated with CSNB
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Suppiej, A.; Marino, S.; Reffo, M.E.; Maritan, V.; Vitaliti, G.; Mailo, J.; Falsaperla, R. Early onset retinal dystrophies: Clinical clues to diagnosis for pediatricians. Ital. J. Pediatr. 2019, 45, 168. [Google Scholar] [CrossRef] [PubMed]
- Van Genderen, M.; Riemslag, F.; Jorritsma, F.; Hoeben, F.; Meire, F.; Stilma, J. The key role of electrophysiology in the diagnosis of visually impaired children. Acta Ophthalmol. Scand. 2006, 84, 799–806. [Google Scholar] [CrossRef]
- MacDonald, I.M.; Hoang, S.; Tuupanen, S. X-Linked Congenital Stationary Night Blindness; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J., Eds.; GeneReviews® Seattle, University of Washington: Seattle, WA, USA, 1993. Available online: http://www.ncbi.nlm.nih.gov/books/NBK1245/ (accessed on 28 April 2024).
- William, A.; Kohl, S.; Zeitz, C.; Willmann, G.; Zrenner, E.; Bartz-Schmidt, K.-U.; Gekeler, F.; Schatz, A. Macular sensitivity in patients with congenital stationary night-blindness. Br. J. Ophthalmol. 2018, 103, 1507–1510. [Google Scholar] [CrossRef] [PubMed]
- Zeitz, C.; Robson, A.G.; Audo, I. Congenital stationary night blindness: An analysis and update of genotype–phenotype correlations and pathogenic mechanisms. Prog. Retin. Eye Res. 2015, 45, 58–110. [Google Scholar] [CrossRef]
- Schubert, G.; Bornschein, H. Beitrag zur Analyse des menschlichen Elektroretinogramms. Ophthalmologica 1952, 123, 396–413. [Google Scholar] [CrossRef] [PubMed]
- Miyake, Y.; Yagasaki, K.; Horiguchi, M.; Kawase, Y. On- and off-responses in photopic electroretinogram in complete and incomplete types of congenital stationary night blindness. Jpn. J. Ophthalmol. 1987, 31, 81–87. [Google Scholar]
- Tsang, S.H.; Sharma, T. Congenital Stationary Night Blindness. In Atlas of Inherited Retinal Diseases; Tsang, S.H., Sharma, T., Eds.; Springer International Publishing: Berlin/Heidelberg, Germany, 2018; pp. 61–64. Available online: http://link.springer.com/10.1007/978-3-319-95046-4_13 (accessed on 28 April 2024).
- Tan, W.D.; Odom, J.V.; Leys, M. Fundus Albipunctatus Associated with Biallelic LRAT Gene Mutation: A Case Report with Long-Term Follow-Up. J. Clin. Med. 2023, 12, 6960. [Google Scholar] [CrossRef]
- Dai, Y.; Sun, T. Oguchi’s disease: Two cases and literature review. J. Int. Med. Res. 2021, 49, 03000605211019921. [Google Scholar] [CrossRef]
- Almutairi, F.; Almeshari, N.; Ahmad, K.; Magliyah, M.S.; Schatz, P. Congenital stationary night blindness: An update and review of the disease spectrum in Saudi Arabia. Acta Ophthalmol. 2020, 99, 581–591. [Google Scholar] [CrossRef]
- Pasutto, F.; Ekici, A.; Reis, A.; Kremers, J.; Huchzermeyer, C. Novel truncating mutation in CACNA1F in a young male patient diagnosed with optic atrophy. Ophthalmic Genet. 2018, 39, 741–748. [Google Scholar] [CrossRef]
- Du, M.; Li, Y.; Zheng, P.; Zhong, L.; Zhao, W.; Zhang, Y.; Gu, H.; Li, X.; Liu, Z. Identification of a novel CACNA1F mutation in a Chinese family with CORDX3. Mol. Genet. Genom. Med. 2022, 10, e2060. [Google Scholar] [CrossRef] [PubMed]
- Leahy, K.E.; Wright, T.; Grudzinska Pechhacker, M.K.; Audo, I.; Tumber, A.; Tavares, E.; MacDonald, H.; Locke, J.; VandenHoven, C.; Zeitz, C.; et al. Optic Atrophy and Inner Retinal Thinning in CACNA1F-Related Congenital Stationary Night Blindness. Genes 2021, 12, 330. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Ito, S.; Piao, C.-H.; Terasaki, H.; Miyake, Y. Retinal and Optic Disc Atrophy Associated with a CACNA1F Mutation in a Japanese Family. Arch. Ophthalmol. 2003, 121, 1028–1033. [Google Scholar] [CrossRef]
- Sakti, D.H.; Ali, H.; Korsakova, M.; Saakova, N.; Mustafic, N.; Fraser, C.L.; Jamieson, R.V.; Cornish, E.E.; Grigg, J.R. Electronegative electroretinogram in the modern multimodal imaging era. Clin. Exp. Ophthalmol. 2022, 50, 429–440. [Google Scholar] [CrossRef]
- Complete and Incomplete Types of CSNB. Electrodiagnosis of Retinal Diseases; Springer: Tokyo, Japan, 2006; pp. 90–113. Available online: https://link.springer.com/10.1007/4-431-30306-5_15 (accessed on 23 May 2024).
- Tremblay, F.; Parkinson, J. Gradient of deficit in cone responses in the incomplete form of congenital stationary night blindness revealed by multifocal electroretinography. Doc. Ophthalmol. 2007, 116, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Akula, J.D.; Ambrosio, L.; Howard, F.I.; Hansen, R.M.; Fulton, A.B. Extracting the ON and OFF contributions to the full-field photopic flash electroretinogram using summed growth curves. Exp. Eye Res. 2019, 189, 107827. [Google Scholar] [CrossRef]
- Quigley, M.; Roy, M.-S.; Barsoum-Homsy, M.; Chevrette, L.; Jacob, J.-L.; Milot, J. On- and off-responses in the photopic electroretinogram in complete-type congenital stationary night blindness. Doc. Ophthalmol. 1996, 92, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Kolb, H.; Fernandez, E.; Nelson, R. The Organization of the Retina and Visual System; University of Utah Health Sciences Center: Salt Lake City, UT, USA, 1995. Available online: http://www.ncbi.nlm.nih.gov/books/NBK11530/ (accessed on 24 September 2024).
- Miyake, Y.; Horiguchi, M.; Ota, I.; Shiroyama, N. Characteristic Erg Flicker Anomaly in Incomplete Congenital Stationary Night Blindness. Investig. Ophthalmol. Vis. Sci. 1987, 28, 1816–1823. [Google Scholar]
- Hoffmann, M.B.; Bach, M.; Kondo, M.; Li, S.; Walker, S.; Holopigian, K.; Viswanathan, S.; Robson, A.G. ISCEV standard for clinical multifocal electroretinography (mfERG) (2021 update). Doc. Ophthalmol. 2021, 142, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Kondo, M.; Miyake, Y.; Kondo, N.; Tanikawa, A.; Suzuki, S.; Horiguchi, M.; Terasaki, H. Multifocal ERG findings in complete type congenital stationary night blindness. Investig. Ophthalmol. Vis. Sci. 2001, 42, 1342–1348. [Google Scholar]
- Barnes, C.S.; Alexander, K.R.; Fishman, G.A. A distinctive form of congenital stationary night blindness with cone ON-pathway dysfunction. Ophthalmology 2002, 109, 575–583. [Google Scholar] [CrossRef] [PubMed]
- International Society for Clinical Electrophysiology of Vision; Odom, J.V.; Bach, M.; Brigell, M.; Holder, G.E.; McCulloch, D.L.; Mizota, A.; Tormene, A.P. ISCEV standard for clinical visual evoked potentials: (2016 update). Doc. Ophthalmol. 2016, 133, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Onoe, S.; Asamizu, N.; Mori, T.; Yoshimura, Y.; Tazawa, Y. Incomplete congenital stationary night blindness: Electroretinogram c-wave and electrooculogram light rise. Doc. Ophthalmol. 1988, 70, 67–75. [Google Scholar] [CrossRef]
- Weston, P.; Taranath, D.; Liebelt, J.; Smith, N. A clinical and electrophysiological case study of a child with a novel frame shift mutation in the CACNA1F and missense variation of RIMS1 genes. Doc. Ophthalmol. 2022, 145, 163–174. [Google Scholar] [CrossRef]
- Ung, T.; Allen, L.E.; Moore, A.T.; Trump, D.; Yates, J.; Bradshaw, K. Is Optic Nerve Fibre Mis-Routing a Feature of Congenital Stationary Night Blindness? Doc Ophthalmol. 2005, 111, 169–178. [Google Scholar] [CrossRef]
- Zeitz, C.; Gross, A.K.; Leifert, D.; Kloeckener-Gruissem, B.; McAlear, S.D.; Lemke, J.; Neidhardt, J.; Berger, W. Identification and Functional Characterization of a Novel Rhodopsin Mutation Associated with Autosomal Dominant CSNB. Investig. Opthalmology Vis. Sci. 2008, 49, 4105–4114. [Google Scholar] [CrossRef]
- Sergouniotis, P.I.; Robson, A.G.; Li, Z.; Devery, S.; Holder, G.E.; Moore, A.T.; Webster, A.R. A phenotypic study of congenital stationary night blindness (CSNB) associated with mutations in the GRM6 gene. Acta Ophthalmol. 2011, 90, e192–e197. [Google Scholar] [CrossRef] [PubMed]
- Malaichamy, S.; Sen, P.; Sachidanandam, R.; Arokiasamy, T.; Lancelot, M.E.; Audo, I.; Zeitz, C.; Soumittra, N. Molecular profiling of complete congenital stationary night blindness: A pilot study on an Indian cohort. Mol. Vis. 2014, 20, 341–351. [Google Scholar] [PubMed]
- Ivanova, M.E.; Zolnikova, I.V.; Gorgisheli, K.V.; Atarshchikov, D.S.; Ghosh, P.; Barh, D. Novel frameshift mutation in NYX gene in a Russian family with complete congenital stationary night blindness. Ophthalmic Genet. 2019, 40, 558–563. [Google Scholar] [CrossRef]
- Hoda, J.C.; Zaghetto, F.; Koschak, A.; Striessnig, J. Congenital Stationary Night Blindness Type 2 Mutations S229P, G369D, L1068P, and W1440X Alter Channel Gating or Functional Expression of Cav1.4 L-type Ca2+ Channels. J. Neurosci. 2005, 25, 252–259. [Google Scholar] [PubMed]
- Stockner, T.; Koschak, A. What can naturally occurring mutations tell us about Cav1.x channel function? Biochim. Biophys. Acta-Biomembranes 2013, 1828, 1598–1607. [Google Scholar] [CrossRef] [PubMed]
- Boycott, K.M.; Pearce, W.G.; Bech-Hansen, N.T. Clinical variability among patients with incomplete X-linked congenital stationary night blindness and a founder mutation in CACNA1F. Can. J. Ophthalmol. 2000, 35, 204–213. [Google Scholar] [CrossRef]
- Wutz, K.; Sauer, C.; Zrenner, E.; Lorenz, B.; Alitalo, T.; Broghammer, M.; Hergersberg, M.; de La Chapelle, A.; Weber, B.H.F.; Wissinger, B.; et al. Thirty distinct CACNA1F mutations in 33 families with incomplete type of XLCSNB and Cacna1f expression profiling in mouse retina. Eur. J. Hum. Genet. 2002, 10, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Abdelkader, E.; AlHilali, S.; Neuhaus, C.; Bergmann, C.; AlMurshed, T.; Schatz, P. Congenital stationary night blindness associated with morning glory disc malformation: A novel hemizygous mutation in CACNA1F. Ophthalmic Genet. 2018, 39, 659–661. [Google Scholar] [CrossRef]
- Hendriks, M.; Verhoeven, V.J.M.; Buitendijk, G.H.S.; Polling, J.R.; Meester-Smoor, M.A.; Hofman, A.; van Huet, R.A.; Klevering, B.J.; Bax, N.M.; Lambertus, S.; et al. Development of Refractive Errors—What Can We Learn from Inherited Retinal Dystrophies? Am. J. Ophthalmol. 2017, 182, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Al-Hujaili, H.; Taskintuna, I.; Neuhaus, C.; Bergmann, C.; Schatz, P. Long-term follow-up of retinal function and structure in TRPM1-associated complete congenital stationary night blindness. Mol. Vis. 2019, 25, 851–858. [Google Scholar]
- Kurata, K.; Hosono, K.; Hotta, Y. Long-Term Clinical Course in a Patient with Complete Congenital Stationary Night Blindness. Case Rep. Ophthalmol. 2017, 8, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Miyadera, K.; Santana, E.; Roszak, K.; Iffrig, S.; Visel, M.; Iwabe, S.; Boyd, R.F.; Bartoe, J.T.; Sato, Y.; Gray, A.; et al. Targeting ON-bipolar cells by AAV gene therapy stably reverses LRIT3 -congenital stationary night blindness. Proc. Natl. Acad. Sci. USA 2022, 119, e2117038119. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| cCSNB | icCSNB | |
|---|---|---|
| DA 0.01 ERG | None | Reduced b-wave amplitude (subnormal) |
| DA 3.0 ERG | Negative ERG type: b-wave amplitude is smaller than the a-wave amplitude | Negative ERG type Analogous to cCSNB |
| LA 30 Hz | Normal | Splitting of the upper peak; reduction in interpeak values |
| LA 3.0 ERG | Broadened a-wave, normal amplitude | Subnormal response (reduced b-wave amplitude) |
| ON-OFF ERG | Abnormal response from ON bipolar cells; normal response from OFF bipolar cells | Abnormal ON and OFF response |
| ERG | cCSNB | icCSNB | Normal |
|---|---|---|---|
| DA 0.01 |  |  | 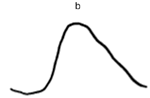 |
| DA 3.0 |  |  | 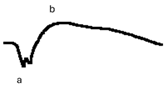 |
| LA 30 Hz | 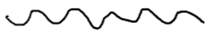 |  |  |
| LA 3.0 |  | 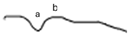 |  |
| ON-OFF response | 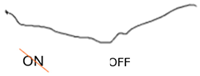 | 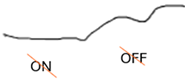 | 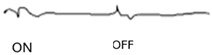 |
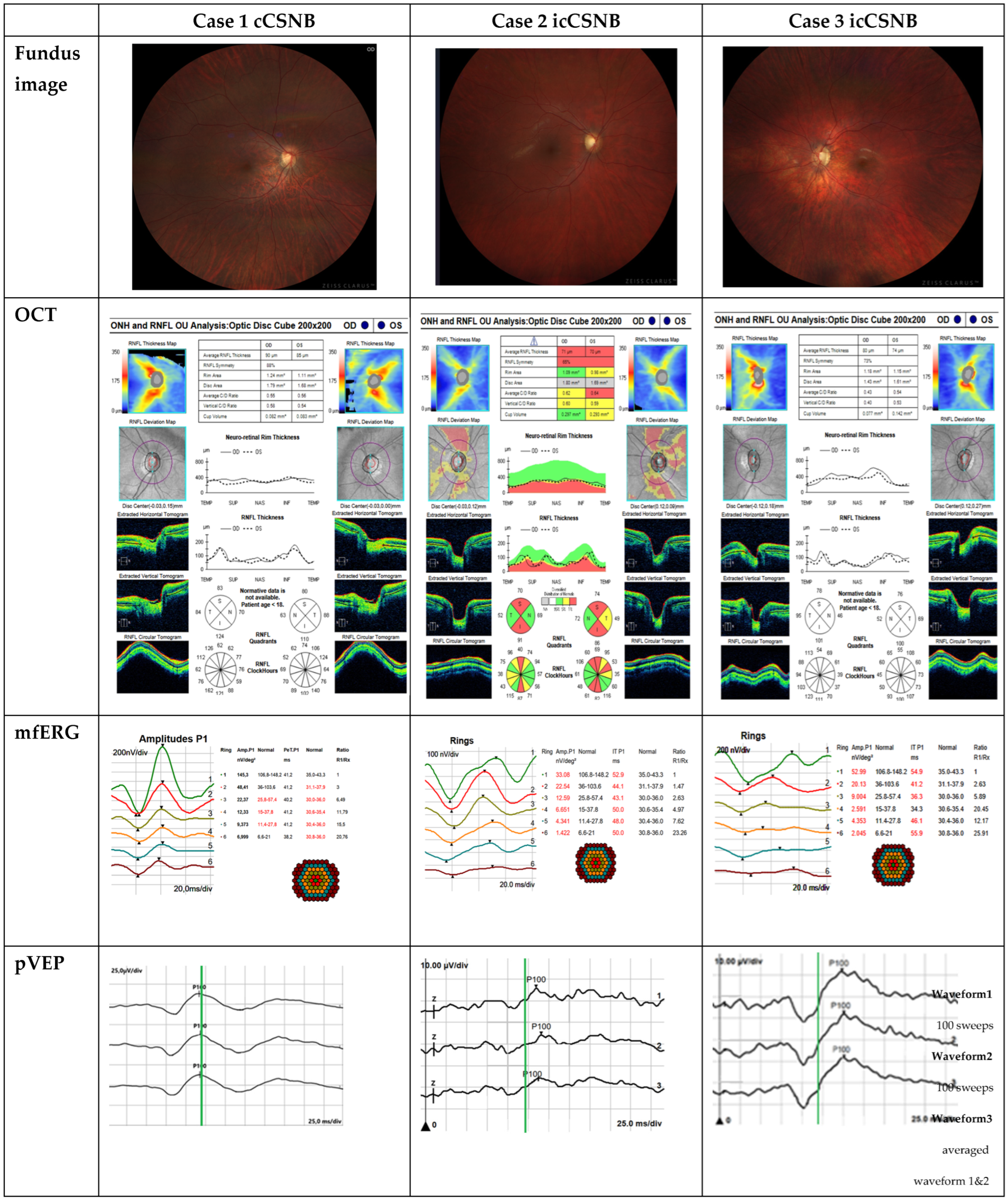
| Case 1 cCSNB | Case 2 icCSNB | Case 3 icCSNB | |
|---|---|---|---|
| BCVA RE [Snellen Chart] | 0.9 | 0.5 | 0.5 |
| BCVA LE [Snellen Chart] | 1.0 | 0.5 | 0.5 |
| Refractive error RE [D] | −2.75 | −3.5 | −3.0 |
| Refractive error LE [D] | −3.75 | −2.5 | −3.0 |
| Fundus examination | changes indicative of axial myopia | optic disc cupping, slight temporal pallor in both eyes | optic disc pink, pale, temporally cupped, vertically oval, peripapillary pigment clumping |
| OCT | sectoral RNFL thinning | sectoral thinning in the superior and inferior regions; reduced GCC thickness in both eyes | thinning RNFL; reduced GCC thickness in both eyes |
| Farnsworth Test | normal | normal | normal |
| Kinetic perimetry | normal | normal | normal |
| Intraocular pressure | normal | normal | normal |
| ON-OFF-response ERG | defect in ON | defects in both ON and OFF | defects in both ON and OFF |
| mfERG | dysfunction 5′–30′ from central point R2-R6 reduction in P1 density response associated with an increase in the implicit time of the P1 wave | dysfunction of the cone system’s bioelectrical function across the entire analyzed area including the macular region | dysfunction of the cone system’s bioelectrical function across the entire analyzed area including the macular region |
| pVEP | normal | prolonged P100 wave latency | prolonged P100 wave latency |
| Genetic testing | GPR179 gene, pathogenic variant ENST00000342292 homozygous | CACNA1F gene, NM_005183.4: c2576+1G>A, a pathogenic variant inherited in an X-linked manner | CACNA1F gene, NM_005183.4: c2576+1G>A, a pathogenic variant inherited in an X-linked manner |
| ERG | cCSNB | Normal |
|---|---|---|
| DA 0.01 | 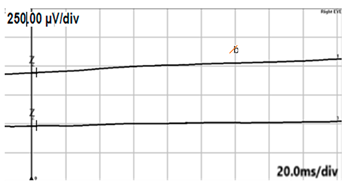 | 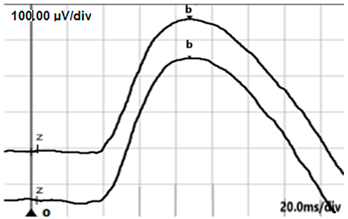 |
| DA 3.0 | 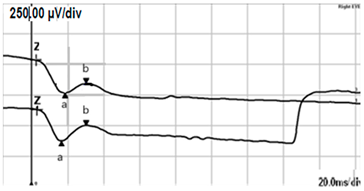 | 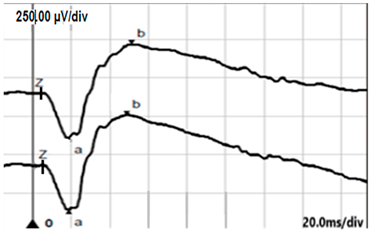 |
| LA 30 Hz |  |  |
| LA 3.0 | 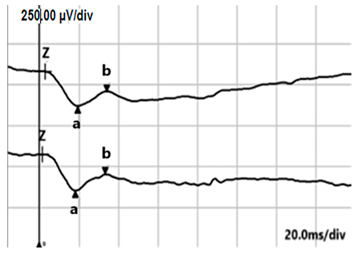 | 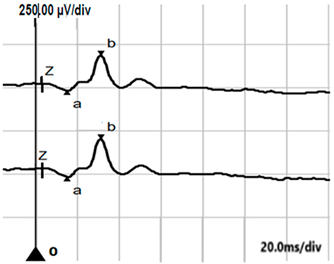 |
| ON-OFF response |  |  |
| ERG | icCSNB(1) | Normal |
|---|---|---|
| DA 0.01 | 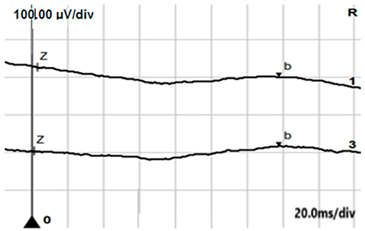 | 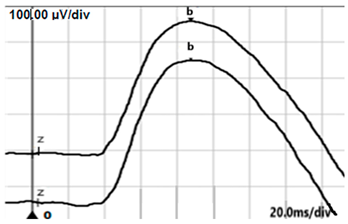 |
| DA 3.0 | 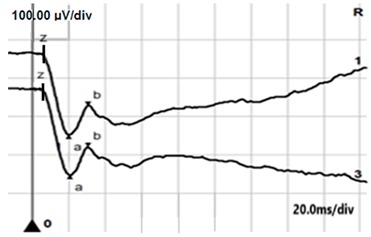 | 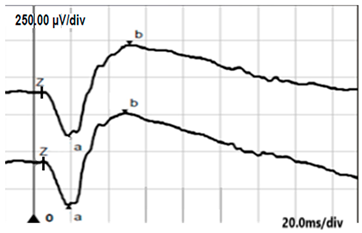 |
| LA 30 Hz | 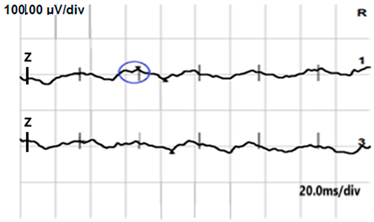 | 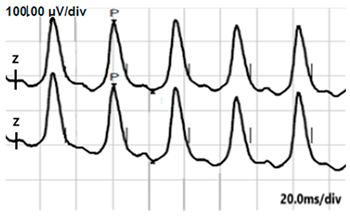 |
| LA 3.0 | 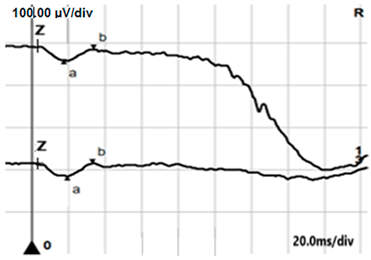 | 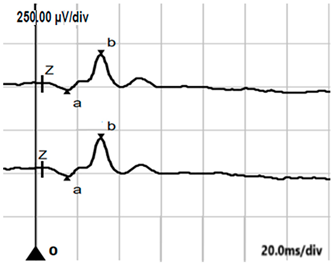 |
| ON-OFF response |  |  |
| ERG | icCSNB(2) | Normal |
|---|---|---|
| DA 0.01 | 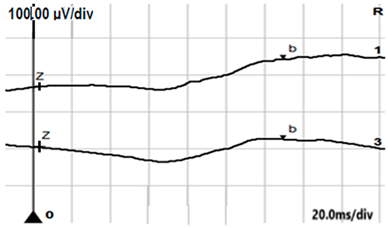 | 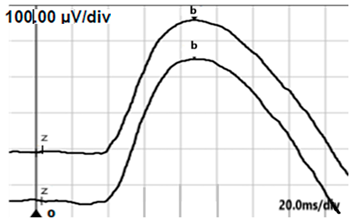 |
| DA 3.0 | 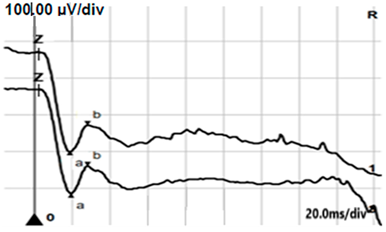 | 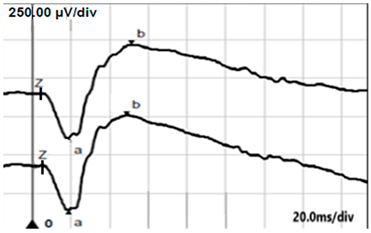 |
| LA 30 Hz | 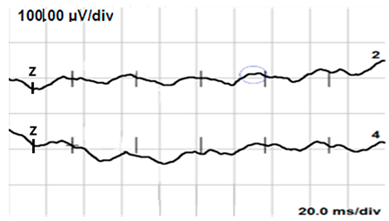 |  |
| LA 3.0 | 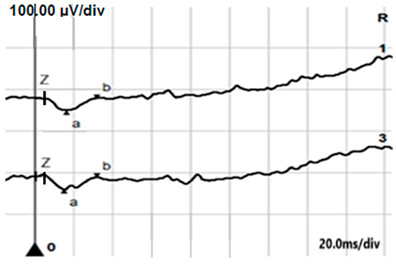 | 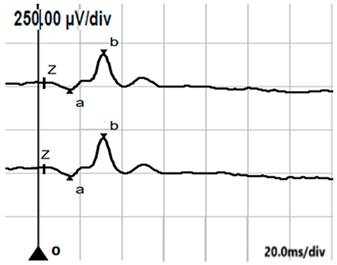 |
| ON-OFF response | 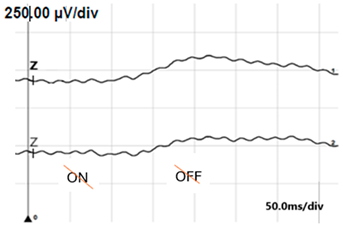 | 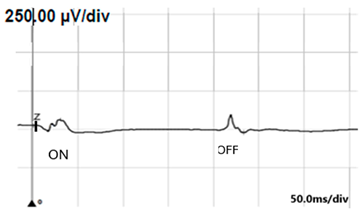 |
| Unilateral | Unilateral/Bilateral Asymmetrically | Bilateral | |
|---|---|---|---|
| Ischemia (e.g., post-CRAO) | Autoimmune retinopathy (+MAR, CAR) | Vitamin A deficiency—acquired night blindness | |
| Siderosis | Birdshot chorioretinopathy | Photoreceptor dystrophies | |
| Occlusive vasculitis | Retinoschisis | ||
| Batten disease (juvenile neuronal ceroid lipofuscinosis). | |||
| Vigabatrin | Methanol | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Durajczyk, M.; Lubiński, W. Congenital Stationary Night Blindness (CSNB)—Case Reports and Review of Current Knowledge. J. Clin. Med. 2025, 14, 1238. https://doi.org/10.3390/jcm14041238
Durajczyk M, Lubiński W. Congenital Stationary Night Blindness (CSNB)—Case Reports and Review of Current Knowledge. Journal of Clinical Medicine. 2025; 14(4):1238. https://doi.org/10.3390/jcm14041238
Chicago/Turabian StyleDurajczyk, Magdalena, and Wojciech Lubiński. 2025. "Congenital Stationary Night Blindness (CSNB)—Case Reports and Review of Current Knowledge" Journal of Clinical Medicine 14, no. 4: 1238. https://doi.org/10.3390/jcm14041238
APA StyleDurajczyk, M., & Lubiński, W. (2025). Congenital Stationary Night Blindness (CSNB)—Case Reports and Review of Current Knowledge. Journal of Clinical Medicine, 14(4), 1238. https://doi.org/10.3390/jcm14041238