
Bilateral Sector Macular Dystrophy Associated with PRPH2 Variant c.623G>A (p.Gly208Asp)
 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Clinical Diagnosis
2.2. Molecular Genetic Testing
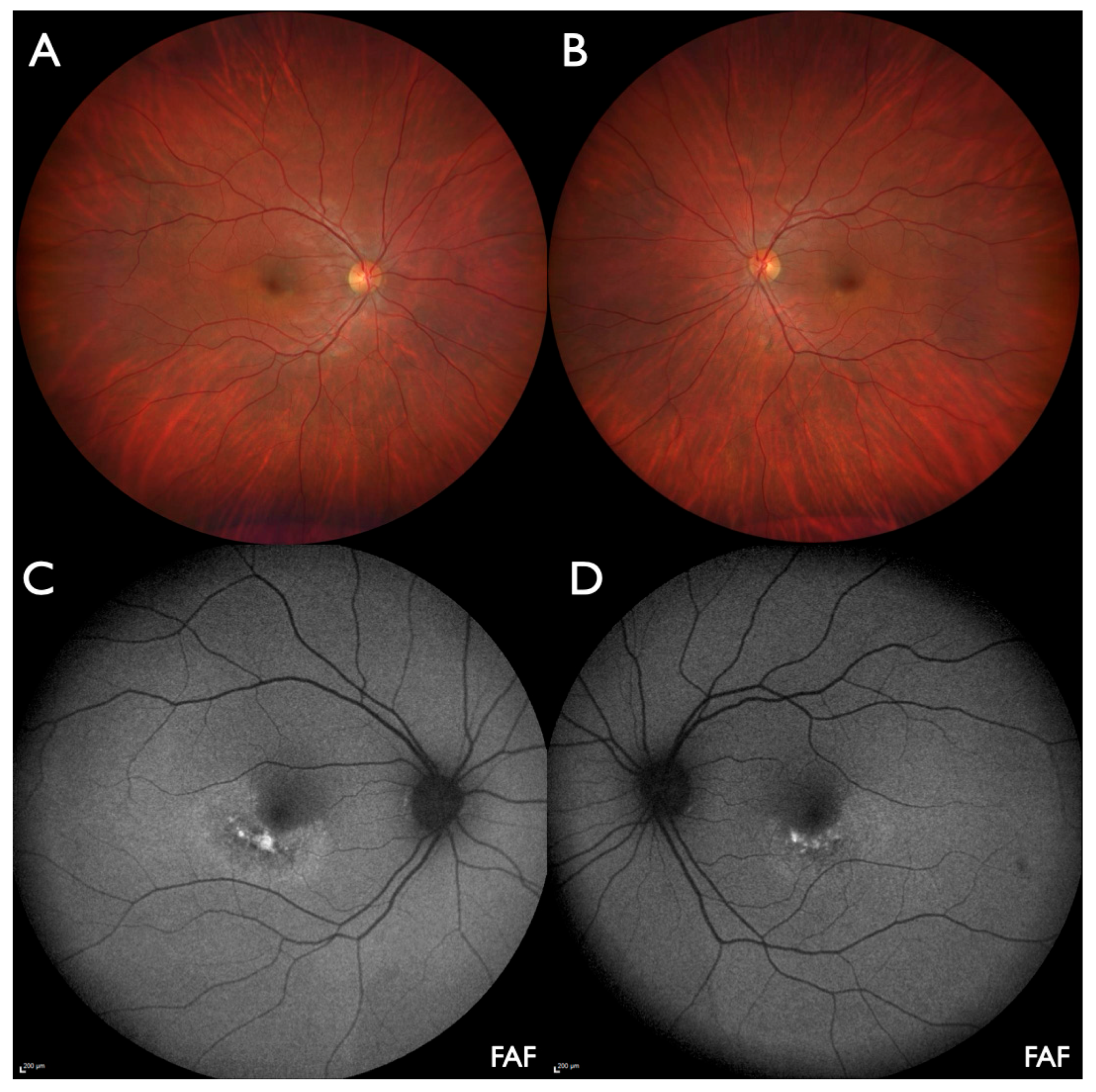
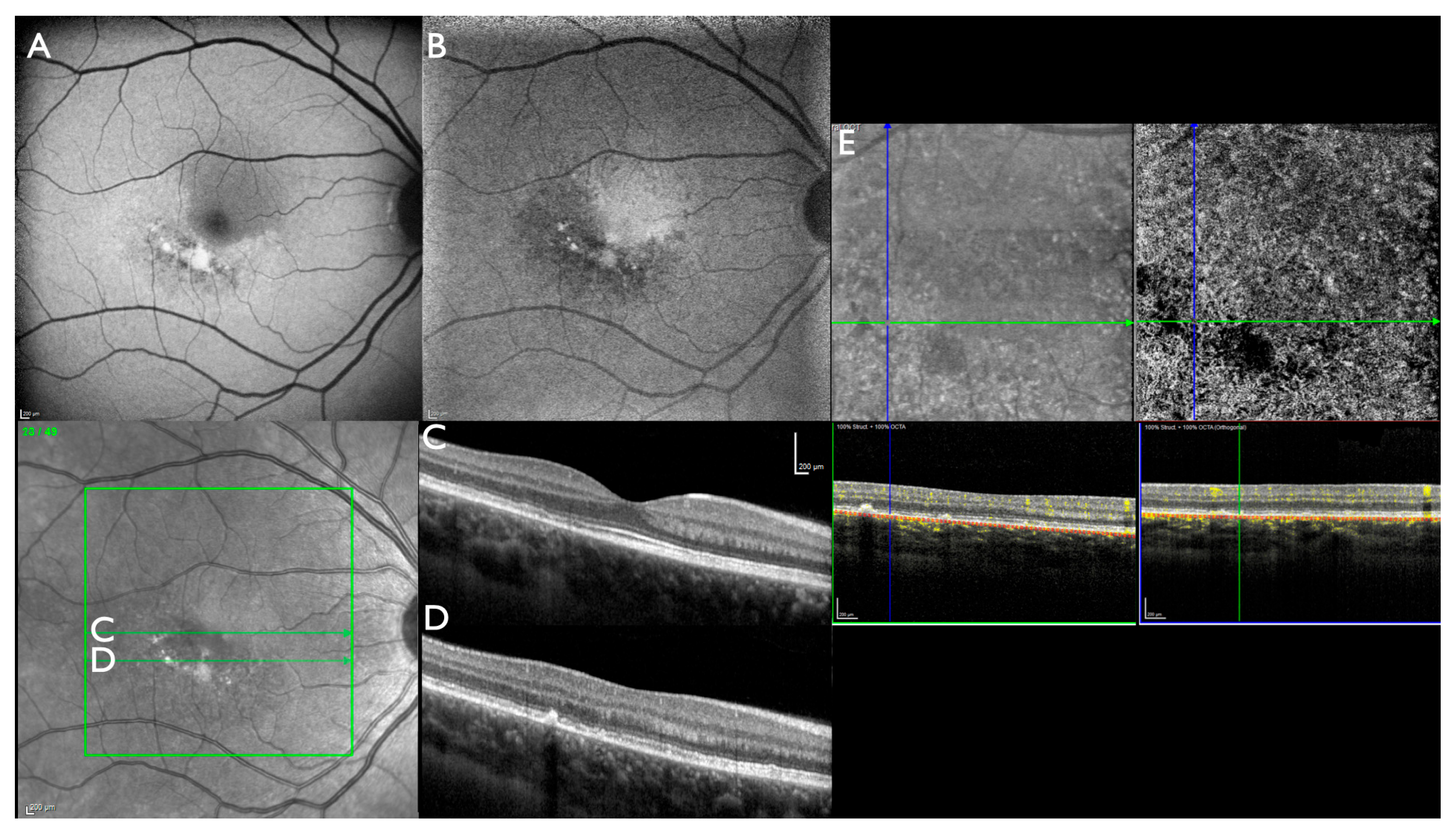
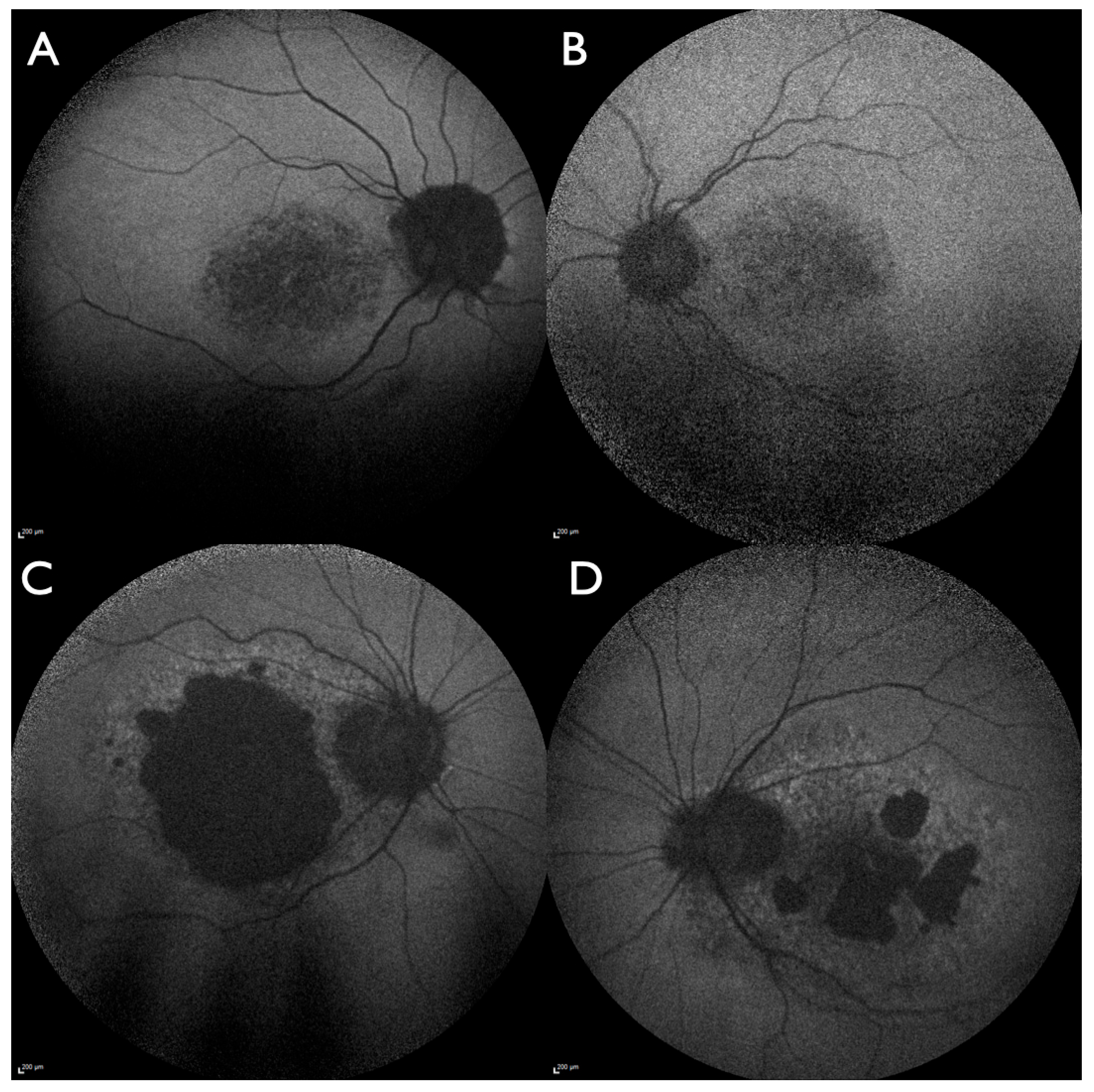
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| FAF | Multidisciplinary Digital Publishing Institute |
| IRD | inherited retinal dystrophies |
| NIA | near-infrared autofluorescence |
| OCT | optical coherence tomography |
| OCTA | OCT-angiography |
| PPE | pachychoroid pigment epitheliopathy |
| RPE | retinal pigment epithelium |
References
- Reeves, M.J.; Goetz, K.E.; Guan, B.; Ullah, E.; Blain, D.; Zein, W.M.; Tumminia, S.J.; Hufnagel, R.B. Genotype-phenotype associations in a large PRPH2-related retinopathy cohort. Hum. Mutat. 2020, 41, 1528–1539. [Google Scholar] [CrossRef] [PubMed]
- Peeters, M.; Khan, M.; Rooijakkers, A.; Mulders, T.; Haer-Wigman, L.; Boon, C.J.F.; Klaver, C.C.W.; van den Born, L.I.; Hoyng, C.B.; Cremers, F.P.M.; et al. PRPH2 mutation update: In silico assessment of 245 reported and 7 novel variants in patients with retinal disease. Hum. Mutat. 2021, 42, 1521–1547. [Google Scholar] [CrossRef] [PubMed]
- Heath Jeffery, R.C.; Thompson, J.A.; Lo, J.; Chelva, E.S.; Armstrong, S.; Pulido, J.S.; Procopio, R.; Vincent, A.L.; Bianco, L.; Battaglia Parodi, M.; et al. Retinal Dystrophies Associated With Peripherin-2: Genetic Spectrum and Novel Clinical Observations in 241 Patients. Investig. Ophthalmol. Vis. Sci. 2024, 65, 22. [Google Scholar] [CrossRef] [PubMed]
- AlAshwal, S.M.; Yassin, S.H.; Kalaw, F.G.P.; Borooah, S. Prph2-Associated Retinal Diseases: A Systematic Review of Phenotypic Findings. Am. J. Ophthalmol. 2025, 271, 7–30. [Google Scholar] [CrossRef]
- Antonelli, G.; Parravano, M.; Barbano, L.; Costanzo, E.; Bertelli, M.; Medori, M.C.; Parisi, V.; Ziccardi, L. Multimodal Study of PRPH2 Gene-Related Retinal Phenotypes. Diagnostics 2022, 12, 1851. [Google Scholar] [CrossRef]
- Soucy, M.; Kolesnikova, M.; Kim, A.H.; Tsang, S.H. Phenotypic variability in PRPH2 as demonstrated by a family with incomplete penetrance of autosomal dominant cone-rod dystrophy. Doc. Ophthalmol. 2023, 146, 267–272. [Google Scholar] [CrossRef]
- Fokkema, I.; Kroon, M.; Lopez Hernandez, J.A.; Asscheman, D.; Lugtenburg, I.; Hoogenboom, J.; den Dunnen, J.T. The LOVD3 platform: Efficient genome-wide sharing of genetic variants. Eur. J. Hum. Genet. 2021, 29, 1796–1803. [Google Scholar] [CrossRef]
- Saleh, M.S.H.; Kiel, C.; Kellner, S.; Weinitz, S.; Farmand, G.; Weber, B.H.F.; Lommatzsch, A.; Kellnr, U. Wide-field optical coherence tomography in ABCA4-associated inherited retinal dystrophies. J. Trans. Genet. Genom. 2021, 5, 250–264. [Google Scholar] [CrossRef]
- Deutsch, S.; Lommatzsch, A.; Weinitz, S.; Farmand, G.; Kellner, U. Optical coherence tomography angiography (OCT-A) in retinitis pigmentosa and macular dystrophy patients: A retrospective study. Graefes Arch. Clin. Exp. Ophthalmol. 2022, 260, 1923–1931. [Google Scholar] [CrossRef]
- Kiel, C.; Biasella, F.; Stohr, H.; Rating, P.; Spital, G.; Kellner, U.; Hufendiek, K.; Huchzermeyer, C.; Jaegle, H.; Ruether, K.; et al. 18-Years of single-centre DNA testing in over 7000 index cases with inherited retinal dystrophies and optic neuropathies. Sci. Rep. 2024, 14, 25529. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Trujillo Tiebas, M.J.; Gimenez Pardo, A.; Garcia Sandoval, B.; Ayuso Garcia, C. Phenotypic variation in a family affected by autosomal dominant retinal dystrophy caused by the Gly208Asp mutation in the RDS peripherin gene. Med. Clin. 2002, 118, 716. [Google Scholar] [CrossRef]
- Trujillo, M.J.; Bueno, J.; Osorio, A.; Sanz, R.; Garcia-Sandoval, B.; Ramos, C.; Ayuso, C. Three novel RDS-peripherin mutations (689delT, 857del17, G208D) in Spanish families affected with autosomal dominant retinal degenerations. Mutations in brief no. 147. Online. Hum. Mutat. 1998, 12, 70. [Google Scholar] [CrossRef]
- Martin-Merida, I.; Aguilera-Garcia, D.; Fernandez-San Jose, P.; Blanco-Kelly, F.; Zurita, O.; Almoguera, B.; Garcia-Sandoval, B.; Avila-Fernandez, A.; Arteche, A.; Minguez, P.; et al. Toward the Mutational Landscape of Autosomal Dominant Retinitis Pigmentosa: A Comprehensive Analysis of 258 Spanish Families. Investig. Ophthalmol. Vis. Sci. 2018, 59, 2345–2354. [Google Scholar] [CrossRef] [PubMed]
- Gamundi, M.J.; Hernan, I.; Muntanyola, M.; Trujillo, M.J.; Garcia-Sandoval, B.; Ayuso, C.; Baiget, M.; Carballo, M. High prevalence of mutations in peripherin/RDS in autosomal dominant macular dystrophies in a Spanish population. Mol. Vis. 2007, 13, 1031–1037. [Google Scholar]
- Garcia Bohorquez, B.; Aller, E.; Rodriguez Munoz, A.; Jaijo, T.; Garcia Garcia, G.; Millan, J.M. Updating the Genetic Landscape of Inherited Retinal Dystrophies. Front. Cell Dev. Biol. 2021, 9, 645600. [Google Scholar] [CrossRef] [PubMed]
- Palma, M.M.D.; Martin, D.; Salles, M.V.; Motta, F.L.T.; Abujamra, S.; Sallum, J.M.F. Retinal dystrophies and variants in PRPH2. Arq. Bras. Oftalmol. 2019, 82, 158–160. [Google Scholar] [CrossRef]
- Manes, G.; Guillaumie, T.; Vos, W.L.; Devos, A.; Audo, I.; Zeitz, C.; Marquette, V.; Zanlonghi, X.; Defoort-Dhellemmes, S.; Puech, B.; et al. High prevalence of PRPH2 in autosomal dominant retinitis pigmentosa in france and characterization of biochemical and clinical features. Am. J. Ophthalmol. 2015, 159, 302–314. [Google Scholar] [CrossRef]
- Weisschuh, N.; Obermaier, C.D.; Battke, F.; Bernd, A.; Kuehlewein, L.; Nasser, F.; Zobor, D.; Zrenner, E.; Weber, E.; Wissinger, B.; et al. Genetic architecture of inherited retinal degeneration in Germany: A large cohort study from a single diagnostic center over a 9-year period. Hum. Mutat. 2020, 41, 1514–1527. [Google Scholar] [CrossRef]
- Kohl, S.; Christ-Adler, M.; Apfelstedt-Sylla, E.; Kellner, U.; Eckstein, A.; Zrenner, E.; Wissinger, B. RDS/peripherin gene mutations are frequent causes of central retinal dystrophies. J. Med. Genet. 1997, 34, 620–626. [Google Scholar] [CrossRef]
- Birtel, J.; Eisenberger, T.; Gliem, M.; Muller, P.L.; Herrmann, P.; Betz, C.; Zahnleiter, D.; Neuhaus, C.; Lenzner, S.; Holz, F.G.; et al. Clinical and genetic characteristics of 251 consecutive patients with macular and cone/cone-rod dystrophy. Sci. Rep. 2018, 8, 4824. [Google Scholar] [CrossRef] [PubMed]
- Capalbo, A.; Valero, R.A.; Jimenez-Almazan, J.; Pardo, P.M.; Fabiani, M.; Jimenez, D.; Simon, C.; Rodriguez, J.M. Optimizing clinical exome design and parallel gene-testing for recessive genetic conditions in preconception carrier screening: Translational research genomic data from 14,125 exomes. PLoS Genet. 2019, 15, e1008409. [Google Scholar] [CrossRef]
- Panneman, D.M.; Hitti-Malin, R.J.; Holtes, L.K.; de Bruijn, S.E.; Reurink, J.; Boonen, E.G.M.; Khan, M.I.; Ali, M.; Andreasson, S.; De Baere, E.; et al. Cost-effective sequence analysis of 113 genes in 1,192 probands with retinitis pigmentosa and Leber congenital amaurosis. Front. Cell Dev. Biol. 2023, 11, 1112270. [Google Scholar] [CrossRef] [PubMed]
- Holtan, J.P.; Selmer, K.K.; Heimdal, K.R.; Bragadottir, R. Inherited retinal disease in Norway—A characterization of current clinical and genetic knowledge. Acta Ophthalmol. 2020, 98, 286–295. [Google Scholar] [CrossRef]
- Turro, E.; Astle, W.J.; Megy, K.; Graf, S.; Greene, D.; Shamardina, O.; Allen, H.L.; Sanchis-Juan, A.; Frontini, M.; Thys, C.; et al. Whole-genome sequencing of patients with rare diseases in a national health system. Nature 2020, 583, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Carss, K.J.; Arno, G.; Erwood, M.; Stephens, J.; Sanchis-Juan, A.; Hull, S.; Megy, K.; Grozeva, D.; Dewhurst, E.; Malka, S.; et al. Comprehensive Rare Variant Analysis via Whole-Genome Sequencing to Determine the Molecular Pathology of Inherited Retinal Disease. Am. J. Hum. Genet. 2017, 100, 75–90. [Google Scholar] [CrossRef]
- Duncan, J.L.; Talcott, K.E.; Ratnam, K.; Sundquist, S.M.; Lucero, A.S.; Day, S.; Zhang, Y.; Roorda, A. Cone structure in retinal degeneration associated with mutations in the peripherin/RDS gene. Invest. Ophthalmol. Vis. Sci. 2011, 52, 1557–1566. [Google Scholar] [CrossRef]
- Zampaglione, E.; Kinde, B.; Place, E.M.; Navarro-Gomez, D.; Maher, M.; Jamshidi, F.; Nassiri, S.; Mazzone, J.A.; Finn, C.; Schlegel, D.; et al. Copy-number variation contributes 9% of pathogenicity in the inherited retinal degenerations. Genet. Med. 2020, 22, 1079–1087. [Google Scholar] [CrossRef]
- Ibanez, M.B.t.; de Guimaraes, T.A.C.; Capasso, J.; Bello, N.; Levin, A.V. Stargardt misdiagnosis: How ocular genetics helps. Am. J. Med. Genet. A 2021, 185, 814–819. [Google Scholar] [CrossRef]
- Warrow, D.J.; Hoang, Q.V.; Freund, K.B. Pachychoroid pigment epitheliopathy. Retina 2013, 33, 1659–1672. [Google Scholar] [CrossRef]
- Pang, C.E.; Freund, K.B. Pachychoroid neovasculopathy. Retina 2015, 35, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Yagi, M.; Miyake, M.; Mori, Y.; Hosoda, Y.; Takahashi, A.; Muraoka, Y.; Ueda-Arakawa, N.; Miyata, M.; Yamashiro, K.; Tamura, H.; et al. Natural Course of Pachychoroid Pigment Epitheliopathy. Ophthalmol. Sci. 2022, 2, 100201. [Google Scholar] [CrossRef] [PubMed]
- Dansingani, K.K.; Balaratnasingam, C.; Naysan, J.; Freund, K.B. En Face Imaging of Pachychoroid Spectrum Disorders with Swept-Source Optical Coherence Tomography. Retina 2016, 36, 499–516. [Google Scholar] [CrossRef] [PubMed]
- Woof, W.A.; de Guimaraes, T.A.C.; Al-Khuzaei, S.; Daich Varela, M.; Sen, S.; Bagga, P.; Mendes, B.; Shah, M.; Burke, P.; Parry, D.; et al. Quantification of Fundus Autofluorescence Features in a Molecularly Characterized Cohort of >3500 Patients with Inherited Retinal Disease from the United Kingdom. Ophthalmol. Sci. 2025, 5, 100652. [Google Scholar] [CrossRef]
- Romano, F.; Cozzi, E.; Boon, C.J.; Staurenghi, G.; Salvetti, A.P. Multimodal Retinal Imaging Reveals New Pathogenic Insights in Central Areolar Choroidal Dystrophy: A Case Series. Retin. Cases Brief Rep. 2024, 18, 32–38. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kellner, S.; Weinitz, S.; Farmand, G.; Stöhr, H.; Weber, B.H.F.; Kellner, U. Bilateral Sector Macular Dystrophy Associated with PRPH2 Variant c.623G>A (p.Gly208Asp). J. Clin. Med. 2025, 14, 4893. https://doi.org/10.3390/jcm14144893
Kellner S, Weinitz S, Farmand G, Stöhr H, Weber BHF, Kellner U. Bilateral Sector Macular Dystrophy Associated with PRPH2 Variant c.623G>A (p.Gly208Asp). Journal of Clinical Medicine. 2025; 14(14):4893. https://doi.org/10.3390/jcm14144893
Chicago/Turabian StyleKellner, Simone, Silke Weinitz, Ghazaleh Farmand, Heidi Stöhr, Bernhard H. F. Weber, and Ulrich Kellner. 2025. "Bilateral Sector Macular Dystrophy Associated with PRPH2 Variant c.623G>A (p.Gly208Asp)" Journal of Clinical Medicine 14, no. 14: 4893. https://doi.org/10.3390/jcm14144893
APA StyleKellner, S., Weinitz, S., Farmand, G., Stöhr, H., Weber, B. H. F., & Kellner, U. (2025). Bilateral Sector Macular Dystrophy Associated with PRPH2 Variant c.623G>A (p.Gly208Asp). Journal of Clinical Medicine, 14(14), 4893. https://doi.org/10.3390/jcm14144893