
Blood-Induced Arthropathy: A Major Disabling Complication of Haemophilia


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
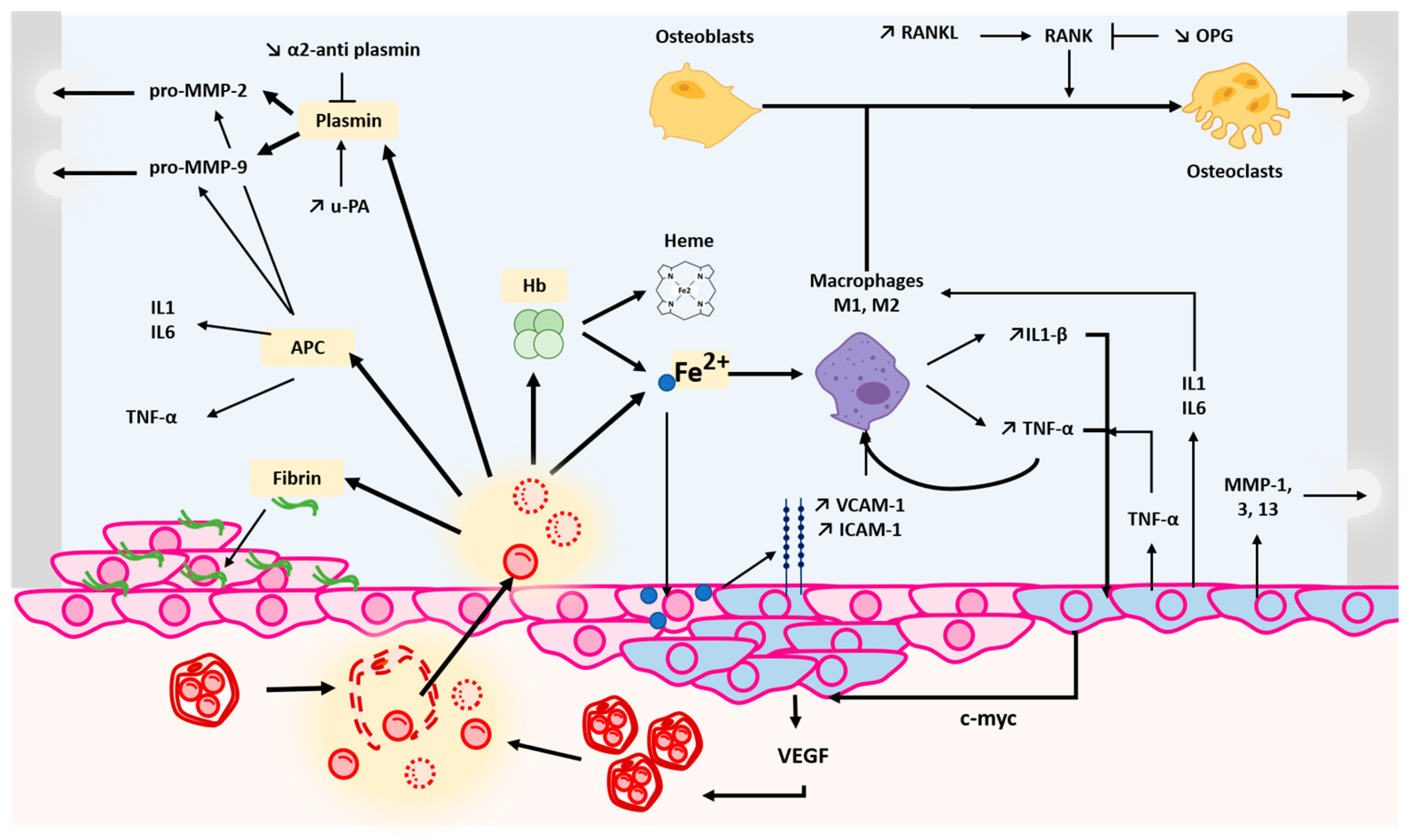
2. Pathophysiological Mechanisms of Haemophilic Arthropathy
2.1. Joint Bleeding
2.2. Effects of Blood Breakdown Products on Joint Health
2.2.1. Iron Deposition
2.2.2. Fibrinolysis Proteins
2.2.3. Activated Protein C
2.3. Synovial Inflammation
2.4. Consequences of Recurrent Bleeding Episodes
2.4.1. Pannus Formation
2.4.2. Angiogenesis Induction
2.4.3. Cartilage and Bone Degradation
3. Risk Factors for Haemophilic Arthropathy
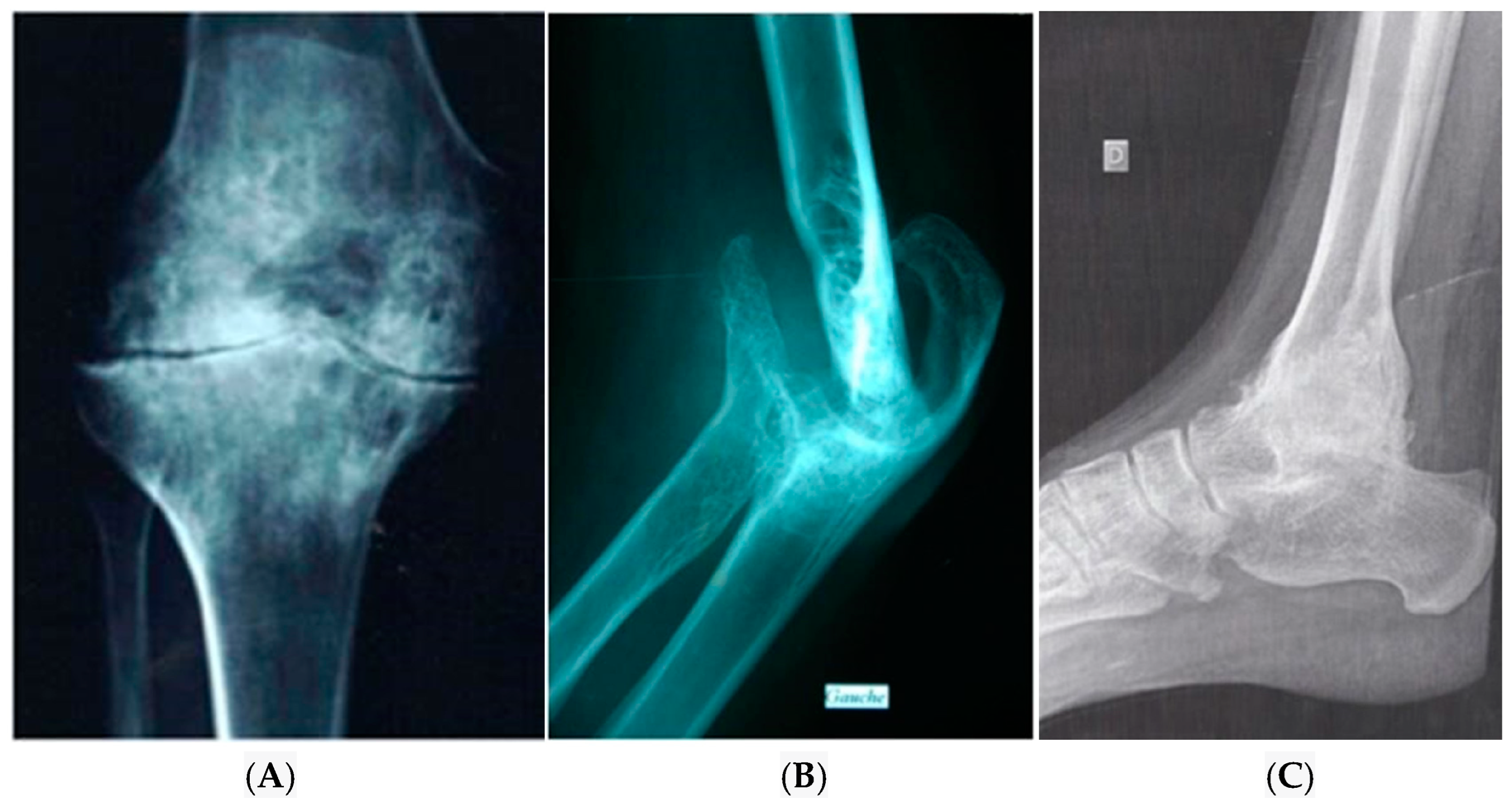
4. Diagnosis of Haemophilic Arthropathy
5. Treatment of Haemophilic Arthropathy
6. Prevention of Haemophilic Arthropathy: A Challenge for Today and Tomorrow
7. Management of Haemophilic Arthropathy with a Personalized Medicine Approach
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Drake, T.A.; Morrissey, J.H.; Edgington, T.S. Selective cellular expression of tissue factor in human tissues. Implications for disorders of hemostasis and thrombosis. Am. J. Pathol. 1989, 134, 1087–1097. [Google Scholar] [PubMed]
- Brinkmann, T.; Kähnert, H.; Prohaska, W.; Nordfang, O.; Kleesiek, K. Synthesis of tissue factor pathway inhibitor in human synovial cells and chondrocytes makes joints the predilected site of bleeding in haemophiliacs. Eur. J. Clin. Chem. Clin. Biochem. 1994, 32, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Dargaud, Y.; Simpson, H.; Chevalier, Y.; Scoazec, J.Y.; Hot, A.; Guyen, O.; Baglin, T.; Négrier, C. The potential role of synovial thrombomodulin in the pathophysiology of joint bleeds in haemophilia. Haemophilia 2012, 18, 818–823. [Google Scholar] [CrossRef] [PubMed]
- Peyvandi, F.; Garagiola, I.; Biguzzi, E. Advances in the treatment of bleeding disorders. J. Thromb. Haemost. 2016, 14, 2095–2106. [Google Scholar] [CrossRef] [PubMed]
- Mannucci, P.M. Hemophilia therapy: The future has begun. Haematologica 2020, 105, 545–553. [Google Scholar] [CrossRef]
- Stephensen, D.; Tait, R.C.; Brodie, N.; Collins, P.; Cheal, R.; Keeling, D.; Melton, K.; Dolan, G.; Haye, H.; Hayman, E.; et al. Changing patterns of bleeding in patients with severe haemophilia A. Haemophilia 2009, 15, 1210–1214. [Google Scholar] [CrossRef]
- Manco-Johnson, M.J.; Soucie, J.M.; Gill, J.C. Joint Outcomes Committee of the Universal Data Collection USHTCN. Prophylaxis usage, bleeding rates, and joint outcomes of haemophilia, 1999 to 2010: A surveillance project. Blood 2017, 129, 2368–2374. [Google Scholar] [CrossRef]
- Valentino, L.A. Blood-induced joint disease: The pathophysiology of hemophilic arthropathy. J. Thromb. Haemost. 2010, 8, 1895–1902. [Google Scholar] [CrossRef]
- Lafeber, F.P.J.G.; Miossec, P.; Valentino, L.A. Physiopathology of haemophilic arthropathy. Haemophilia 2008, 14, 3–9. [Google Scholar] [CrossRef]
- Fischer, K.; Collins, P.; Björkman, S.; Blanchette, V.; Oh, M.; Fritsch, S.; Schroth, P.; Spotts, G.; Ewenstein, B. Trends in bleeding patterns during prophylaxis for severe haemophilia: Observations from a series of prospective clinical trials. Haemophilia 2011, 17, 433–438. [Google Scholar] [CrossRef]
- van Dijk, K.; Fischer, K.; van der Bom, J.G.; Grobbee, D.E.; van den Berg, H.M. Variability in clinical phenotype of severe haemophilia: The role of the first joint bleed. Haemophilia 2005, 11, 438–443. [Google Scholar] [CrossRef] [PubMed]
- Roosendaal, G.; Vianen, M.E.; Wenting, M.J.; van Rinsum, A.C.; van den Berg, H.M.; Lafeber, F.P.; Bijlsma, J.W. Iron deposits and catabolic properties of synovial tissue from patients with haemophilia. J. Bone Jt. Surg. Br. 1998, 80, 540–545. [Google Scholar] [CrossRef]
- Zhou, J.Y.; Wong, J.H.; Berman, Z.T.; Lombardi, A.F.; Chang, E.Y.; von Drygalski, A. bleeding with iron deposition and vascular remodeling in subchondral cysts: A newly discovered feature unique to hemophilic arthropathy. Haemophilia 2021, 27, e730–e738. [Google Scholar] [CrossRef] [PubMed]
- von Drygalski, A.; Barnes, R.F.W.; Jang, H.; Ma, Y.; Wong, J.H.; Berman, Z.; Du, J.; Chang, E.Y. Advanced magnetic resonance imaging of cartilage components in haemophilic joints reveals that cartilage hemosiderin correlates with joint deterioration. Haemophilia 2019, 25, 851–858. [Google Scholar] [CrossRef] [PubMed]
- Walker, R.J.; Dymock, I.W.; Ansell, I.D.; Hamilton, E.B.; Williams, R. Synovial biopsy in haemochromatosis arthropathy. histological findings and iron deposition in relation to total body iron overload. Ann. Rheum. Dis. 1972, 31, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Nieuwenhuizen, L.; Schutgens, R.E.G.; van Asbeck, B.S.; Wenting, M.J.; van Veghel, K.; Roosendaal, G.; Biesma, D.H.; Lafeber, F.P.J.G. Identification and expression of iron regulators in human synovium: Evidence for upregulation in haemophilic arthropathy compared to rheumatoid arthritis, osteoarthritis, and healthy controls. Haemophilia 2013, 19, e218–e227. [Google Scholar] [CrossRef]
- Hooiveld, M.J.J.; Roosendaal, G.; van den Berg, H.M.; Bijlsma, J.W.J.; Lafeber, F.P.J.G. Haemoglobin-derived iron-dependent hydroxyl radical formation in blood-induced joint damage: An in vitro study. Rheumatol. Oxf. Engl. 2003, 42, 784–790. [Google Scholar] [CrossRef] [PubMed]
- Nell-Duxneuner, V.; Axmann, R.; Husar-Memmer, E.; Dallos, T.; Datz, C.; Stadlmayr, A.; Aigner, E.; Englbrecht, M.; Schett, G.; Zwerina, J. VCAM-1 Serum levels are associated with arthropathy in hereditary haemochromatosis. Ann. Rheum. Dis. 2013, 72, 2006–2010. [Google Scholar] [CrossRef]
- Sen, D.; Chapla, A.; Walter, N.; Daniel, V.; Srivastava, A.; Jayandharan, G.R. Nuclear factor (NF)-κB and its associated pathways are major molecular regulators of blood-induced joint damage in a murine model of hemophilia. J. Thromb. Haemost. 2013, 11, 293–306. [Google Scholar] [CrossRef]
- Zhang, F.-J.; Luo, W.; Lei, G.-H. Role of HIF-1α and HIF-2α in osteoarthritis. Jt. Bone Spine 2015, 82, 144–147. [Google Scholar] [CrossRef]
- Saito, T.; Kawaguchi, H. HIF-2α as a possible therapeutic target of osteoarthritis. Osteoarthr. Cartil. 2010, 18, 1552–1556. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Kim, J.; Ryu, J.-H.; Oh, H.; Chun, C.-H.; Kim, B.J.; Min, B.H.; Chun, J.-S. Hypoxia-inducible factor-2alpha is a catabolic regulator of osteoarthritic cartilage destruction. Nat. Med. 2010, 16, 687–693. [Google Scholar] [CrossRef] [PubMed]
- Nieuwenhuizen, L.; Roosendaal, G.; Coeleveld, K.; Lubberts, E.; Biesma, D.H.; Lafeber, F.P.J.G.; Schutgens, R.E.G. Haemarthrosis stimulates the synovial fibrinolytic system in haemophilic mice. Thromb. Haemost. 2013, 110, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Baramova, E.N.; Bajou, K.; Remacle, A.; L’Hoir, C.; Krell, H.W.; Weidle, U.H.; Noel, A.; Foidart, J.M. Involvement of PA/Plasmin system in the processing of pro-MMP-9 and in the second step of pro-MMP-2 activation. FEBS Lett. 1997, 405, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Nieuwenhuizen, L.; Roosendaal, G.; Mastbergen, S.C.; Coeleveld, K.; Biesma, D.H.; Lafeber, F.P.J.G.; Schutgens, R.E.G. Antiplasmin, but not amiloride, prevents synovitis and cartilage damage following hemarthrosis in hemophilic mice. J. Thromb. Haemost. 2014, 12, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Pernaute, O.; Largo, R.; Calvo, E.; Alvarez-Soria, M.; Egido, J.; Herrero-Beaumont, G. A fibrin based model for rheumatoid synovitis. Ann. Rheum. Dis. 2003, 62, 1135–1138. [Google Scholar] [CrossRef] [PubMed]
- Esmon, C.T. The protein C pathway. Chest 2003, 124, 26S–32S. [Google Scholar] [CrossRef]
- Jackson, M.T.; Smith, M.M.; Smith, S.M.; Jackson, C.J.; Xue, M.; Little, C.B. Activation of cartilage matrix metalloproteinases by activated protein C. Arthritis Rheum. 2009, 60, 780–791. [Google Scholar] [CrossRef]
- Xue, M.; Dervish, S.; McKelvey, K.J.; March, L.; Wang, F.; Little, C.B.; Jackson, C.J. Activated protein C targets immune cells and rheumatoid synovial fibroblasts to prevent inflammatory arthritis in mice. Rheumatol. Oxf. Engl. 2019, 58, 1850–1860. [Google Scholar] [CrossRef]
- Magisetty, J.; Kondreddy, V.; Keshava, S.; Das, K.; Esmon, C.T.; Pendurthi, U.R.; Rao, L.V.M. Selective Inhibition of activated protein C anticoagulant activity protects against hemophilic arthropathy in mice. Blood 2022, 139, 2830–2841. [Google Scholar] [CrossRef]
- van Meegeren, M.E.R.; Roosendaal, G.; Coeleveld, K.; Nieuwenhuizen, L.; Mastbergen, S.C.; Lafeber, F.P.J.G. A Single intra-articular injection with IL-4 plus IL-10 ameliorates blood-induced cartilage Degeneration in haemophilic mice. Br. J. Haematol. 2013, 160, 515–520. [Google Scholar] [CrossRef] [PubMed]
- Nieuwenhuizen, L.; Schutgens, R.E.G.; Coeleveld, K.; Mastbergen, S.C.; Roosendaal, G.; Biesma, D.H.; Lafeber, F.P.J.G. Hemarthrosis in Hemophilic mice results in alterations in M1-M2 monocyte/macrophage polarization. Thromb. Res. 2014, 133, 390–395. [Google Scholar] [CrossRef]
- Hoots, W.K. Pathogenesis of Hemophilic arthropathy. Semin. Hematol. 2006, 43, S18–S22. [Google Scholar] [CrossRef] [PubMed]
- Mainardi, C.L.; Levine, P.H.; Werb, Z.; Harris, E.D. Proliferative synovitis in hemophilia: Biochemical and morphologic observations. Arthritis Rheum. 1978, 21, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Acharya, S.S.; Kaplan, R.N.; Macdonald, D.; Fabiyi, O.T.; DiMichele, D.; Lyden, D. Neoangiogenesis Contributes to the development of hemophilic synovitis. Blood 2011, 117, 2484–2493. [Google Scholar] [CrossRef] [PubMed]
- Zetterberg, E.; Palmblad, J.; Wallensten, R.; Morfini, M.; Melchiorre, D.; Holmström, M. Angiogenesis is Increased in advanced haemophilic joint disease and characterised by normal pericyte coverage. Eur. J. Haematol. 2014, 92, 256–262. [Google Scholar] [CrossRef] [PubMed]
- Bhat, V.; Olmer, M.; Joshi, S.; Durden, D.L.; Cramer, T.J.; Barnes, R.; Ball, S.T.; Hughes, T.H.; Silva, M.; Luck, J.V.; et al. Vascular remodeling underlies re-bleeding in hemophilic arthropathy. Am. J. Hematol. 2015, 90, 1027–1035. [Google Scholar] [CrossRef]
- Melchiorre, D.; Milia, A.F.; Linari, S.; Romano, E.; Benelli, G.; Manetti, M.; Guiducci, S.; Ceccarelli, C.; Innocenti, M.; Carulli, C.; et al. RANK-RANKL-OPG in hemophilic arthropathy: From clinical and imaging diagnosis to histopathology. J. Rheumatol. 2012, 39, 1678–1686. [Google Scholar] [CrossRef]
- Pettit, A.R.; Walsh, N.C.; Manning, C.; Goldring, S.R.; Gravallese, E.M. RANKL Protein is expressed at the pannus–bone interface at sites of articular bone erosion in rheumatoid arthritis. Rheumatology 2006, 45, 1068–1076. [Google Scholar] [CrossRef]
- Gooding, R.; Thachil, J.; Alamelu, J.; Motwani, J.; Chowdary, P. Asymptomatic joint bleeding and joint health in hemophilia: A review of variables, methods, and biomarkers. J. Blood Med. 2021, 12, 209–220. [Google Scholar] [CrossRef]
- Schmidt, D.E.; Michalopoulou, A.; Fischer, K.; Motwani, J.; Andersson, N.G.; Pergantou, H.; Ranta, S.; PedNet Study Group. Long-term joint outcomes in adolescents with moderate or severe haemophilia A. Haemophilia 2022, 28, 1054–1061. [Google Scholar] [CrossRef] [PubMed]
- Manco-Johnson, M.J.; Abshire, T.C.; Shapiro, A.D.; Riske, B.; Hacker, M.R.; Kilcoyne, R.; Ingram, J.D.; Manco-Johnson, M.L.; Funk, S.; Jacobson, L.; et al. Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia. N. Engl. J. Med. 2007, 357, 535–544. [Google Scholar] [CrossRef] [PubMed]
- Gringeri, A.; Lundin, B.; von Mackensen, S.; Mantovani, L.; Mannucci, P.M.; ESPRIT Study Group. A randomized clinical trial of prophylaxis in children with hemophilia A (the ESPRIT Study). J. Thromb. Haemost. 2011, 9, 700–710. [Google Scholar] [CrossRef] [PubMed]
- Manco-Johnson, M.J.; Sanders, J.; Ewing, N.; Rodriguez, N.; Tarantino, M.; Humphries, T.; TEEN/TWEN Study Group. Consequences of switching from prophylactic treatment to on-demand treatment in late teens and early adults with severe haemophilia A: The TEEN/TWEN study. Haemophilia 2013, 19, 727–735. [Google Scholar] [CrossRef] [PubMed]
- Pasca, S.; Zanon, E. Savings without changing: How to use the MyPKfit® device to improve treatment strategies in a cohort of patients with haemophilia A. Thromb. Res. 2019, 183, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.; Santagostino, E.; Dougall, A.; Kitchen, S.; Sutherland, M.; Pipe, S.W.; Carcao, M.; Mahlangu, J.; Ragni, M.V.; Windyga, J.; et al. WFH guidelines for the management of hemophilia, 3rd edition. Haemophilia 2020, 26, 1–158. [Google Scholar] [CrossRef]
- Dargaud, Y.; Negrier, C. Thrombin generation testing in haemophilia comprehensive care centres. Haemophilia 2010, 16, 223–230. [Google Scholar] [CrossRef]
- Dargaud, Y.; Negrier, C.; Rusen, L.; Windyga, J.; Georgiev, P.; Bichler, J.; Solomon, C.; Knaub, S.; Lissitchkov, T.; Klamroth, R. Individual thrombin generation and spontaneous bleeding rate during personalized prophylaxis with Nuwiq® (human-cl rhFVIII) in previously treated patients with severe haemophilia A. Haemophilia 2018, 24, 619–627. [Google Scholar] [CrossRef]
- Delavenne, X.; Ollier, E.; Lienhart, A.; Dargaud, Y. A new paradigm for personalized prophylaxis for patients with severe haemophilia A. Haemophilia 2020, 26, 228–235. [Google Scholar] [CrossRef]
- Morfini, M.; Haya, S.; Tagariello, G.; Pollmann, H.; Quintana, M.; Siegmund, B.; Stieltjes, N.; Dolan, G.; Tusell, J. European study on orthopaedic status of haemophilia patients with inhibitors. Haemophilia 2007, 13, 606–612. [Google Scholar] [CrossRef]
- Lannoy, N.; Hermans, C. Principles of genetic variations and molecular diseases: Applications in hemophilia A. Crit. Rev. Oncol. Hematol. 2016, 104, 1–8. [Google Scholar] [CrossRef] [PubMed]
- López-Jiménez, J.J.; Ortega-Cervantes, R.; Luna-Záizar, H.; Fletes-Rayas, A.L.; Beltrán-Miranda, C.P.; Troyo-Sanromán, R.; Soto-Padilla, J.; Tlacuilo-Parra, A.; Jaloma-Cruz, A.R. Genetic biomarkers related to hemarthrosis, inflammation, and cartilage structure in pediatric patients with hemophilic arthropathy. Mol. Genet. Genom. Med. 2019, 7, e979. [Google Scholar] [CrossRef] [PubMed]
- Gomperts, E.D.; Schwarz, J.; Donfield, S.M.; Lail, A.E.; Astermark, J.; Hoots, W.K.; Winkler, C.A.; Berntorp, E. The importance of genetic factors for the development of arthropathy: A longitudinal study of children and adolescents with haemophilia A. Thromb. Haemost. 2017, 117, 277–285. [Google Scholar] [CrossRef] [PubMed]
- De la Corte-Rodriguez, H.; Rodriguez-Merchan, E.C.; Alvarez-Roman, M.T.; Martin-Salces, M.; Rivas-Pollmar, I.; Jimenez-Yuste, V. Arthropathy in people with mild haemophilia: Exploring risk factors. Thromb. Res. 2022, 211, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Fibbi, G.; Pucci, M.; Serni, U.; Cerinic, M.M. Antisense targeting of the urokinase receptor blocks urokinase-dependent proliferation, chemoinvasion, and chemotaxis of human synovial cells and chondrocytes in vitro. Proc. Assoc. Am. Physicians 1998, 110, 340–350. [Google Scholar] [PubMed]
- Janbain, M.; Enjolras, N.; Bordet, J.C.; Bolbos, R.; Brevet, M.; Leissinger, C.; Dargaud, Y. Hemostatic effect of tranexamic acidcombined with factor VIII concentrate in prophylactic setting in severe hemophilia A: A preclinical study. J. Thromb. Haemost. 2020, 18, 584–592. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, S.; Makris, M. Haemophilia and ageing. Br. J. Haematol. 2019, 184, 712–720. [Google Scholar] [CrossRef]
- Buzzard, B.M. Sports and hemophilia: Antagonist or protagonist. Clin. Orthop. Relat. Res. 1996, 328, 25–30. [Google Scholar] [CrossRef]
- Khawaji, M.; Astermark, J.; Akesson, K.; Berntorp, E. Physical activity for prevention of osteoporosis in patients with severe haemophilia on long-term prophylaxis. Haemophilia 2010, 16, 495–501. [Google Scholar] [CrossRef]
- McGee, S.; Raffini, L.; Witmer, C. Organized sports participation and the association with injury in paediatric patients with haemophilia. Haemophilia 2015, 21, 538–542. [Google Scholar] [CrossRef]
- Gomis, M.; Querol, F.; Gallach, J.E.; González, L.M.; Aznar, J. A Exercise and sport in the treatment of haemophilic patients: A systematic review. Haemophilia 2009, 15, 43–54. [Google Scholar] [CrossRef] [PubMed]
- Fijnvandraat, K.; Cnossen, M.H.; Leebeek, F.W.; Peters, M. Diagnosis and management of haemophilia. BMJ 2012, 344, e2707. [Google Scholar] [CrossRef] [PubMed]
- Negrier, C.; Seuser, A.; Forsyth, A.; Lobet, S.; Llinas, A.; Rosas, M.; Heijnen, L. The benefits of exercise for patients with haemophilia and recommendations for safe and effective physical activity. Haemophilia 2013, 19, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Wallny, T.A.; Scholz, D.T.; Oldenburg, J.; Nicolay, C.; Ezziddin, S.; Pennekamp, P.H.; Stoffel-Wagner, B.; Kraft, C.N. Osteoporosis in haemophilia—An underestimated comorbidity? Haemophilia 2007, 13, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Petkovic, M.J.; Tran, H.A.; Ebeling, P.R.; Zengin, A. Osteoporosis management and falls prevention in patients with haemophilia: Review of haemophilia guidelines. Haemophilia 2022, 28, 388–396. [Google Scholar] [CrossRef] [PubMed]
- Linari, S.; Montorzi, G.; Bartolozzi, D.; Borderi, M.; Melchiorre, D.; Benelli, M.; Morfini, M. Hypovitaminosis D and osteopenia/osteoporosis in a haemophilia population: A study in HCV/HIV or HCV infected patients. Haemophilia 2013, 19, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Bagnolesi, P.; Campassi, C.; Cilotti, A.; Lencioni, R.; Napoli, V.; Bartolozzi, C. Hemophilic arthropathy: Echography and radiology. Radiol. Med. 1993, 85, 28–33. [Google Scholar]
- Pettersson, H.; Ahlberg, A.; Nilsson, I.M. A radiologic classification of hemophilic arthropathy. Clin. Orthop. Relat. Res. 1980, 149, 153–159. [Google Scholar] [CrossRef]
- Doria, A.S.; Lundin, B.; Kilcoyne, R.F.; Babyn, P.S.; Miller, S.; Nuss, R.; Rivard, G.; Stephens, D.; Pettersson, H. Reliability of progressive and additive MRI scoring systems for evaluation of haemophilic arthropathy in children: Expert MRI working group of the international prophylaxis study group. Haemophilia 2005, 11, 245–253. [Google Scholar] [CrossRef]
- Lundin, B.; Pettersson, H.; Ljung, R. A new magnetic resonance imaging scoring method for assessment of haemophilic arthropathy. Haemophilia 2004, 10, 383–389. [Google Scholar] [CrossRef]
- von Drygalski, A.; Moore, R.E.; Nguyen, S.; Barnes, R.F.W.; Volland, L.M.; Hughes, T.H.; Du, J.; Chang, E.Y. Advanced hemophilic arthropathy: Sensitivity of soft tissue discrimination with musculoskeletal ultrasound. J. Ultrasound Med. 2018, 37, 1945–1956. [Google Scholar] [CrossRef] [PubMed]
- Martinoli, C.; Della Casa Alberighi, O.; Di Minno, G.; Graziano, E.; Molinari, A.C.; Pasta, G.; Russo, G.; Santagostino, E.; Tagliaferri, A.; Tagliafico, A.; et al. Development and definition of a simplified scanning procedure and scoring method for Haemophilia Early Arthropathy Detection with Ultrasound (HEAD-US). Thromb. Haemost. 2013, 109, 1170–1179. [Google Scholar] [CrossRef] [PubMed]
- Plut, D.; Kotnik, B.F.; Zupan, I.P.; Kljucevsek, D.; Vidmar, G.; Snoj, Z.; Martinoli, C.; Salapura, V. Diagnostic accuracy of haemophilia early arthropathy detection with ultrasound (HEAD-US): A comparative magnetic resonance imaging (MRI) study. Radiol. Oncol. 2019, 53, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Hilliard, P.; Funk, S.; Zourikian, N.; Bergstrom, B.M.; Bradley, C.S.; McLimont, M.; Manco-Johnson, M.; Petrini, P.; van den Berg, M.; Feldman, B.M. Hemophilia joint health score reliability study. Haemophilia 2006, 12, 518–525. [Google Scholar] [CrossRef] [PubMed]
- van Bergen, E.D.P.; Mastbergen, S.C.; Lafeber, F.P.J.G.; Schutgens, R.E.G.; van Vulpen, L.F.D. Challenges in biomarker research in haemophilic arthropathy. Haemophilia 2021, 27, e547–e548. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Merchan, E.C. Synovitis in hemophilia: Preventing, detecting, and treating joint bleeds. Expert. Rev. Hematol. 2023, 16, 525–534. [Google Scholar] [CrossRef]
- Jiang, J.; Leong, N.L.; Khalique, U.; Phan, T.M.; Lyons, K.M.; Luck, J.V., Jr. Connective tissue growth factor (CTGF/CCN2) in haemophilic arthropathy and arthrofibrosis: A histological analysis. Haemophilia 2016, 22, e527–e536. [Google Scholar] [CrossRef]
- Martin, E.J.; Cooke, E.J.; Ceponis, A.; Barnes, R.F.; Moran, C.M.; Holle, S.; Hughes, T.H.; Moore, R.E.; von Drygalski, A. Efficacy and safety of point-of-care ultrasound-guided intra-articular corticosteroid joint injections in patients with haemophilic arthropathy. Haemophilia 2017, 23, 135–143. [Google Scholar] [CrossRef]
- Li, T.Y.; Wu, Y.T.; Chen, L.C.; Cheng, S.N.; Pan, R.Y.; Chen, Y. CAn exploratory comparison of single intra-articular injection of platelet-rich plasma vs hyaluronic acid in treatment of haemophilic arthropathy of the knee. Haemophilia 2019, 25, 484–492. [Google Scholar] [CrossRef]
- Landro, M.E.; Daffunchio, C.; Cambiaggi, G.; Galatro, G.; Caviglia, H. Platelet-rich plasma vs platelet-rich plasma plus hyaluronic acid for haemophilic knee arthropathy treatment. Acta Orthop. Belg. 2021, 87, 705–712. [Google Scholar] [CrossRef]
- Fenelon, C.; Murphy, E.P.; Fahey, E.J.; Murphy, R.P.; O’Connell, N.M.; Queally, J.M. Total knee arthroplasty in hemophilia: Survivorship and outcomes-a systematic review and meta-analysis. J. Arthroplast. 2022, 37, 581–592. [Google Scholar] [CrossRef] [PubMed]
- Pezzotti, G.; Zhu, W.; Terai, Y.; Marin, E.; Boschetto, F.; Kawamoto, K.; Itaka, K. Raman spectroscopic insight into osteoarthritic cartilage regeneration by mRNA therapeutics encoding cartilage-anabolic transcription factor Runx1. Mater. Today Bio 2022, 13, 100210. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Madry, H.; Cucchiarini, M. application of alginate hydrogels for next-generation articular cartilage regeneration. Int. J. Mol. Sci. 2022, 23, 1147. [Google Scholar] [CrossRef] [PubMed]
- Iorio, A.; Königs, C.; Reding, M.T.; Rotellini, D.; Skinner, M.W.; Mancuso, M.E.; Berntorp, E. Prophylaxis use of clotting factor replacement products in people with non-severe haemophilia: A review of the literature. Haemophilia 2022, 29, 33–44. [Google Scholar] [CrossRef]
- Wilkins, R.A.; Stephensen, D.; Siddle, H.; Scott, M.J.; Xiang, H.; Horn, E.; Palmer, B.; Chapman, G.J.; Richards, M.; Walwyn, R.; et al. Twelve-month prevalence of haemarthrosis and joint disease using the Haemophilia Joint Health score: Evaluation of the UK National Haemophilia Database and Haemtrack patient reported data: An observational study. BMJ Open 2022, 12, e052358. [Google Scholar] [CrossRef]
- Greco, T.; Polichetti, C.; Cannella, A.; La Vergata, V.; Maccauro, G.; Perisano, C. Ankle hemophilic arthropathy: Literature review. Am. J. Blood Res. 2021, 11, 206–216. [Google Scholar]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leuci, A.; Dargaud, Y. Blood-Induced Arthropathy: A Major Disabling Complication of Haemophilia. J. Clin. Med. 2024, 13, 225. https://doi.org/10.3390/jcm13010225
Leuci A, Dargaud Y. Blood-Induced Arthropathy: A Major Disabling Complication of Haemophilia. Journal of Clinical Medicine. 2024; 13(1):225. https://doi.org/10.3390/jcm13010225
Chicago/Turabian StyleLeuci, Alexandre, and Yesim Dargaud. 2024. "Blood-Induced Arthropathy: A Major Disabling Complication of Haemophilia" Journal of Clinical Medicine 13, no. 1: 225. https://doi.org/10.3390/jcm13010225
APA StyleLeuci, A., & Dargaud, Y. (2024). Blood-Induced Arthropathy: A Major Disabling Complication of Haemophilia. Journal of Clinical Medicine, 13(1), 225. https://doi.org/10.3390/jcm13010225