

Epilepsy in Dravet Syndrome—Current and Future Therapeutic Opportunities
 ,
, 
Abstract
1. Introduction and Syndrome Characteristics
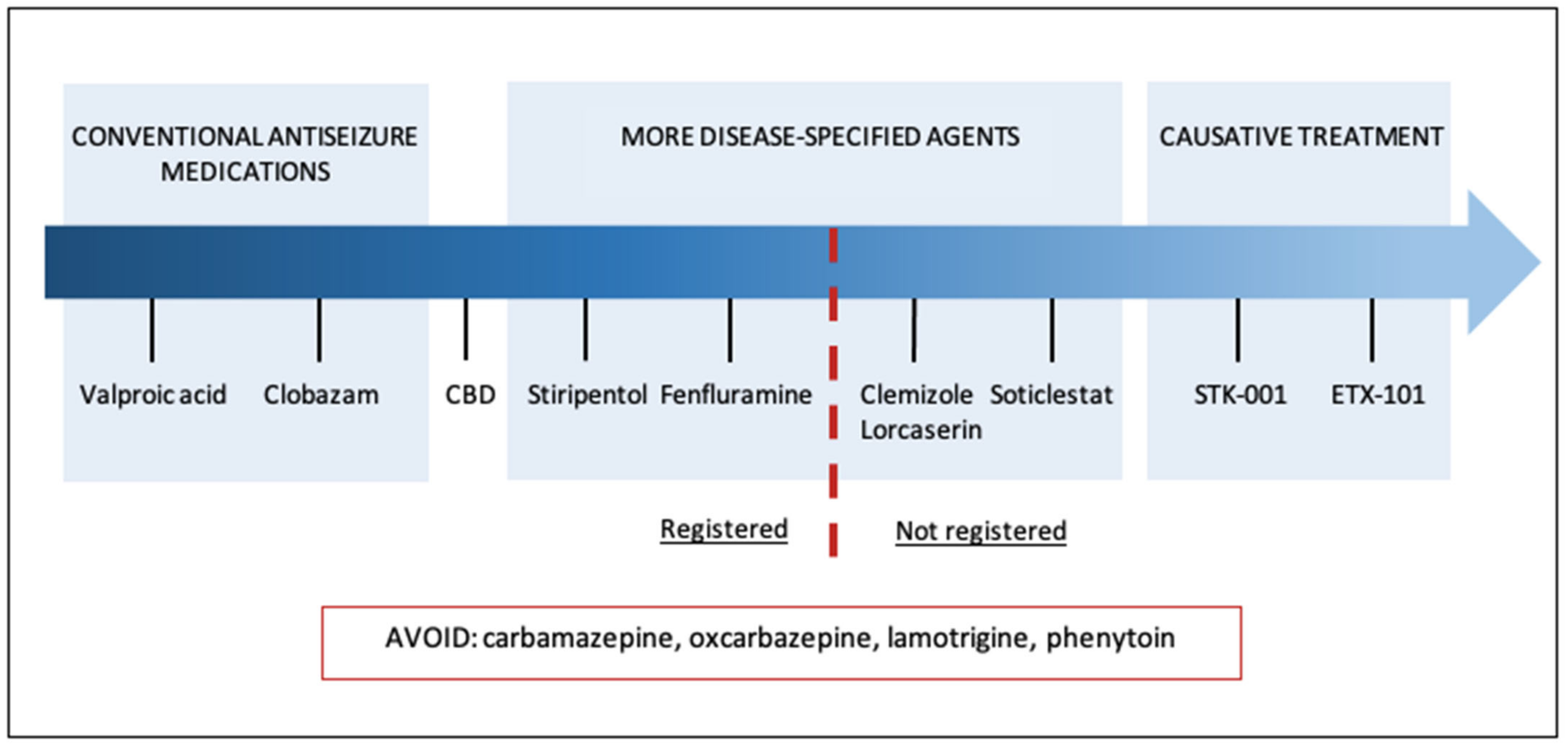
2. Current Treatment Strategies
2.1. Limitation of Provoking Factors
2.2. Contraindicated Medications
2.3. Acute Treatment
2.4. Chronic Management
2.4.1. Stiripentol (STP)
2.4.2. Fenfluramine (FFA)
2.4.3. Cannabidiol (CBD)
3. Novel Therapies
3.1. Modulators of Serotonin Signaling
3.1.1. Clemizole (EPX-100)
3.1.2. Lorcaserin (EPX-200)
3.1.3. Trazodone (EPX-300)
3.2. Soticlestat (TAK-935/OV935)
3.3. Gene Therapy
3.3.1. STK-001 (Antisense Oligonucleotides)
3.3.2. ETX101
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Symonds, J.; Zuberi, S.M.; Stewart, K.; McLellan, A.; O’Regan, M.; MacLeod, S.; Jollands, A.; Joss, S.; Kirkpatrick, M.; Brunklaus, A.; et al. Incidence and phenotypes of childhood-onset genetic epilepsies: A prospective population-based national cohort. Brain 2019, 142, 2303–2318. [Google Scholar] [CrossRef] [PubMed]
- Brunklaus, A.; Ellis, R.; Reavey, E.; Forbes, G.H.; Zuberi, S.M. Prognostic, clinical and demographic features in SCN1A mutation-positive Dravet syndrome. Brain 2012, 135 Pt 8, 2329–2336. [Google Scholar] [CrossRef]
- Bayat, A.; Hjalgrim, H.; Møller, R.S. The incidence of SCN1A-related Dravet syndrome in Denmark is 1:22,000: A population-based study from 2004 to 2009. Epilepsia 2015, 56, e36–e39. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.W.; Sullivan, J.; McDaniel, S.S.; Meisler, M.H.; Walsh, E.M.; Li, S.X.; Kuzniewicz, M.W. Incidence of Dravet Syndrome in a US Population. Pediatrics 2015, 136, e1310–e1315. [Google Scholar] [CrossRef] [PubMed]
- Dravet, C. The core Dravet syndrome phenotype. Epilepsia 2011, 52, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Wirrell, E.C.; Laux, L.; Donner, E.; Jette, N.; Knupp, K.; Meskis, M.A.; Miller, I.; Sullivan, J.; Welborn, M.; Berg, A.T. Optimizing the Diagnosis and Management of Dravet Syndrome: Recommendations from a North American Consensus Panel. Pediatr. Neurol. 2017, 68, 18–34.e3. [Google Scholar] [CrossRef]
- Catarino, C.B.; Liu, J.Y.; Liagkouras, I.; Gibbons, V.S.; Labrum, R.W.; Ellis, R.; Woodward, C.; Davis, M.B.; Smith, S.J.; Cross, J.H.; et al. Dravet syndrome as epileptic encephalopathy: Evidence from long-term course and neuropathology. Brain 2011, 134, 2982–3010. [Google Scholar] [CrossRef]
- Takayama, R.; Fujiwara, T.; Shigematsu, H.; Imai, K.; Takahashi, Y.; Yamakawa, K.; Inoue, Y. Long-term course of Dravet syndrome: A study from an epilepsy center in Japan. Epilepsia 2014, 55, 528–538. [Google Scholar] [CrossRef]
- Ouss, L.; Leunen, D.; Laschet, J.; Chemaly, N.; Barcia, G.; Losito, E.M.; Aouidad, A.; Barrault, Z.; Desguerre, I.; Breuillard, D.; et al. Autism spectrum disorder and cognitive profile in children with Dravet syndrome: Delineation of a specific phenotype. Epilepsia Open 2018, 4, 40–53. [Google Scholar] [CrossRef]
- Cardenal-Munoz, E.; Auvin, S.; Villanueva, V.; Cross, H.J.; Zuberi, S.; Lagae, L.; Aibar, J.A. Guidance on Dravet syndrome from infant to adult care: Roadmap for treatemnet planning in Europe. Epilepsia Open 2022, 7, 11–26. [Google Scholar] [CrossRef]
- Steel, D.; Symonds, J.D.; Zuberi, S.M.; Brunklaus, A. Dravet syndrome and its mimics: Beyond SCN1A. Epilepsia 2017, 58, 1807–1816. [Google Scholar] [CrossRef] [PubMed]
- Mantegazza, M.; Gambardella, A.; Rusconi, R.; Schiavon, E.; Annesi, F.; Cassulini, R.R.; Labate, A.; Carrideo, S.; Chifari, R.; Canevini, M.P.; et al. Identification of an Na v 1.1 sodium channel (SCN1A) loss-of-function mutation associated with familial simple febrile seizures. Proc. Natl. Acad. Sci. USA 2005, 102, 18177–18182. [Google Scholar] [CrossRef] [PubMed]
- Escayg, A.; Macdonald, B.T.; Meisler, M.H.; Baulac, S.; Huberfeld, G.; An-Gourfinkel, I.; Brice, A.; LeGuern, E.; Moulard, B.; Chaigne, D.; et al. Mutations of SCN1A, encoding a neuronal sodium channel, in two families with GEFS+2. Nat. Genet. 2000, 24, 343–345. [Google Scholar] [CrossRef]
- Freilich, E.R.; Jones, J.M.; Gaillard, W.D.; Conry, J.A.; Tsuchida, T.N.; Reyes, C.; Dib-Hajj, S.; Waxman, S.G.; Meisler, M.H.; Pearl, P.L. Novel SCN1A Mutation in a Proband w Malignant Migrating Partial Seizures of Infancy. Arch. Neurol. 2011, 68, 665–671. [Google Scholar] [CrossRef] [PubMed]
- Brunklaus, A.; Pérez-Palma, E.; Ghanty, I.; Xinge, J.; Brilstra, E.; Ceulemans, B.; Chemaly, N.; de Lange, I.; Depienne, C.; Guerrini, R.; et al. Development and Validation of a Prediction Model for Early Diagnosis of SCN1A-Related Epilepsies. Neurology 2022, 98, e1163–e1174. [Google Scholar] [CrossRef]
- Lagae, L.; Brambilla, I.; Mingorance, A.; Gibson, E.; Battersby, A. Quality of life and comorbidities associated with Dravet syndrome severity: A multinational cohort survey. Dev. Med. Child Neurol. 2018, 60, 63–72. [Google Scholar] [CrossRef]
- O’Reilly, H.; Eltze, C.; Bennett, K.; Verhaert, K.; Webb, R.; Merrett, A.; Scott, R.C.; Whitney, A.; Cross, J.H.; de Haan, M. Cognitive outcomes following epilepsy in infancy: A longitudinal community-based study. Epilepsia 2018, 59, 2240–2248. [Google Scholar] [CrossRef]
- Shmuely, S.; Sisodiya, S.M.; Gunning, W.B.; Sander, J.W.; Thijs, R.D. Mortality in Dravet syndrome: A review. Epilepsy Behav. 2016, 64, 69–74. [Google Scholar] [CrossRef]
- Gataullina, S.; Dulac, O. From genotype to phenotype in Dravet disease. Seizure 2017, 44, 58–64. [Google Scholar] [CrossRef]
- Ceulemans, B. Overall management of patients with Dravet syndrome. Dev. Med. Child Neurol. 2011, 53, 19–23. [Google Scholar] [CrossRef]
- Genton, P.; Velizarova, R.; Dravet, C. Dravet syndrome: The long-term outcome. Epilepsia 2011, 52, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Tro-Baumann, B.; von Spiczak, S.; Lotte, J.; Bast, T.; Haberlandt, E.; Sassen, R.; Freund, A.; Leiz, S.; Stephani, U.; Boor, R.; et al. A retrospective study of the relation between vaccination and occurrence of seizures in Dravet syndrome. Epilepsia 2011, 52, 175–178. [Google Scholar] [CrossRef] [PubMed]
- Clayton, L.M.; Balestrini, S.; Cross, J.H.; Wilson, G.; Eldred, C.; Evans, H.; Koepp, M.J.; Sisodiya, S.M. The impact of SARS-CoV-2 vaccination in Dravet syndrome: A UK survey. Epilepsy Behav. 2021, 124, 108258. [Google Scholar] [CrossRef]
- Hood, V.; Berg, A.T.; Knupp, K.G.; Koh, S.; Laux, L.; Meskis, M.A.; Zulfiqar-Ali, Q.; Perry, M.S.; Scheffer, I.E.; Sullivan, J.; et al. COVID-19 vaccine in patients with Dravet syndrome: Observations and real-world experiences. Epilepsia 2022, 63, 1778–1786. [Google Scholar] [CrossRef] [PubMed]
- De Lange, I.M.; Gunning, B.; Sonsma, A.C.M.; van Gemert, L.; van Kempen, M.; Verbeek, N.E.; Nicolai, J.; Knoers, N.V.A.M.; Koeleman, B.P.C.; Brilstra, E.H. Influence of contraindicated medication use on cognitive outcome in Dravet syndrome and age at first afebrile seizure as a clinical predictor in SCN1A -related seizure phenotypes. Epilepsia 2018, 59, 1154–1165. [Google Scholar] [CrossRef]
- Guerrini, R.; Dravet, C.; Genton, P.; Belmonte, A.; Kaminska, A.; Dulac, O. Lamotrigine and Seizure Aggravation in Severe Myoclonic Epilepsy. Epilepsia 1998, 39, 508–512. [Google Scholar] [CrossRef]
- Mueller, A.; Boor, R.; Coppola, G.; Striano, P.; Dahlin, M.; von Stuelpnagel, C.; Lotte, J.; Staudt, M.; Kluger, G. Low long-term efficacy and tolerability of add-on rufinamide in patients with Dravet syndrome. Epilepsy Behav. 2011, 21, 282–284. [Google Scholar] [CrossRef]
- Snoeijen-Schouwenaars, F.; Veendrick, M.; van Mierlo, P.; van Erp, G.; de Louw, A.; Kleine, B.; Schelhaas, H.; Tan, I. Carbamazepine and oxcarbazepine in adult patients with Dravet syndrome: Friend or foe? Seizure 2015, 29, 114–118. [Google Scholar] [CrossRef]
- Fişgin, T.; Gurer, Y.; Tezic, T.; Senbil, N.; Zorlu, P.; Okuyaz, C.; Akgün, D. Effects of Intranasal Midazolam and Rectal Diazepam on Acute Convulsions in Children: Prospective Randomized Study. J. Child Neurol. 2002, 17, 123–126. [Google Scholar] [CrossRef]
- Bhattacharyya, M.; Kalra, V.; Gulati, S. Intranasal Midazolam vs Rectal Diazepam in Acute Childhood Seizures. Pediatr. Neurol. 2006, 34, 355–359. [Google Scholar] [CrossRef]
- French, J.A.; Wechsler, R.; Gelfand, M.A.; Pollard, J.R.; Vazquez, B.; Friedman, D.; Gong, L.H.; Kamemoto, E.; Isojarvi, J.; Cassella, J.V. Inhaled alprazolam rapidly suppresses epileptic activity in photosensitive participants. Epilepsia 2019, 60, 1602–1609. [Google Scholar] [CrossRef] [PubMed]
- Vaitkevicius, H.; Ramsay, R.E.; Swisher, C.B.; Husain, A.M.; Aimetti, A.; Gasior, M. Intravenous ganaxolone for the treatment of refractory status epilepticus: Results from an open-label, dose-finding, phase 2 trial. Epilepsia 2022, 63, 2381–2391. [Google Scholar] [CrossRef] [PubMed]
- Poisson, M.; Huguet, F.; Savattier, A.; Bakri-Logeais, F.; Narcisse, G. A new type of anticonvulsant, stiripentol. Pharmacological profile and neurochemical study. Arzneimittelforschung 1984, 34, 199–204. [Google Scholar]
- Quilichini, P.P.; Chiron, C.; Ben-Ari, Y.; Gozlan, H. Stiripentol, a Putative Antiepileptic Drug, Enhances the Duration of Opening of GABAA-Receptor Channels. Epilepsia 2006, 47, 704–716. [Google Scholar] [CrossRef] [PubMed]
- Tran, A.; Rey, E.; Pons, G.; Rousseau, M.; D’Athis, P.; Olive, G.; Mather, G.G.; Bishop, F.E.; Wurden, C.J.; Labroo, R.; et al. Influence of stiripentol on cytochrome P450-mediated metabolic pathways in humans: In vitro and in vivo comparison and calculation of in vivo inhibition constants. Clin. Pharmacol. Ther. 1997, 62, 490–504. [Google Scholar] [CrossRef] [PubMed]
- Giraud, C.; Treluyer, J.-M.; Rey, E.; Chiron, C.; Vincent, J.; Pons, G.; Tran, A. In vitro and in vivo inhibitory effect of stiripentol on clobazam metabolism. Drug Metab. Dispos. 2006, 34, 608–611. [Google Scholar] [CrossRef]
- Chiron, C. Stiripentol for the treatment of seizures associated with Dravet syndrome. Expert Rev. Neurother. 2019, 19, 301–310. [Google Scholar] [CrossRef]
- Chiron, C.; Marchand, M.C.; Tran, A.; Rey, E.; D’Athis, P.; Vincent, J.; Dulac, O.; Pons, G.; STICLO Study Group. Stiripentol in severe myoclonic epilepsy in infancy: A randomised placebo-controlled syndrome-dedicated trial. Lancet 2000, 356, 1638–1642. [Google Scholar] [CrossRef]
- Chiron, C.; Helias, M.; Kaminska, A.; Laroche, C.; de Toffol, B.; Dulac, O.; Nabbout, R.; An, I. Do children with Dravet syndrome continue to benefit from stiripentol for long through adulthood? Epilepsia 2018, 59, 1705–1717. [Google Scholar] [CrossRef]
- Inoue, Y.; Ohtsuka, Y. Long-term safety and efficacy of stiripentol for the treatment of Dravet syndrome: A multicenter, open-label study in Japan. Epilepsy Res. 2015, 113, 90–97. [Google Scholar] [CrossRef]
- Yamada, M.; Suzuki, K.; Matsui, D.; Inoue, Y.; Ohtsuka, Y. Long-term safety and effectiveness of stiripentol in patients with Dravet syndrome: Interim report of a post-marketing surveillance study in Japan. Epilepsy Res. 2020, 170, 106535. [Google Scholar] [CrossRef] [PubMed]
- Habermehl, L.; Mross, P.; Krause, K.; Immisch, I.; Chiru, D.; Zahnert, F.; Gorny, I.; Strzelczyk, A.; Rosenow, F.; Möller, L.; et al. Stiripentol in the treatment of adults with focal epilepsy- a retrospective analysis. Seizure 2021, 88, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Balestrini, S.; Sisodiya, S.M. Audit of use of stiripentol in adults with Dravet syndrome. Acta Neurol. Scand. 2016, 135, 73–79. [Google Scholar] [CrossRef]
- Abenhaim, L.; Moride, Y.; Brenot, F.; Rich, S.; Benichou, J.; Kurz, X.; Higenbottam, T.; Oakley, C.; Wouters, E.; Aubier, M.; et al. Appetite-Suppressant Drugs and the Risk of Primary Pulmonary Hypertension. N. Engl. J. Med. 1996, 335, 609–616. [Google Scholar] [CrossRef]
- Sourbron, J.; Smolders, I.; de Witte, P.; Lagae, L. Pharmacological Analysis of the Anti-epileptic Mechanisms of Fenfluramine in scn1a Mutant Zebrafish. Front. Pharmacol. 2017, 8, 191. [Google Scholar] [CrossRef]
- Griffin, A.; Hamling, K.R.; Knupp, K.; Hong, S.; Lee, L.P.; Baraban, S.C. Clemizole and modulators of serotonin signalling suppress seizures in Dravet syndrome. Brain 2017, 140, 669–683. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Kecskés, A.; Copmans, D.; Langlois, M.; Crawford, A.D.; Ceulemans, B.; Lagae, L.; de Witte, P.A.M.; Esguerra, C.V. Pharmacological Characterization of an Antisense Knockdown Zebrafish Model of Dravet Syndrome: Inhibition of Epileptic Seizures by the Serotonin Agonist Fenfluramine. PLoS ONE 2015, 10, e0125898. [Google Scholar] [CrossRef] [PubMed]
- Lagae, L.; Sullivan, J.; Knupp, K.; Laux, L.; Polster, T.; Nikanorova, M.; Devinsky, O.; Cross, J.H.; Guerrini, R.; Talwar, D.; et al. Fenfluramine hydrochloride for the treatment of seizures in Dravet syndrome: A randomised, double-blind, placebo-controlled trial. Lancet 2019, 394, 2243–2254. [Google Scholar] [CrossRef]
- Nabbout, R.; Mistry, A.; Zuberi, S.; Villeneuve, N.; Gil-Nagel, A.; Sanchez-Carpintero, R.; Stephani, U.; Laux, L.; Wirrell, E.; Knupp, K.; et al. Fenfluramine for Treatment-Resistant Seizures in Patients With Dravet Syndrome Receiving Stiripentol-Inclusive Regimens. JAMA Neurol. 2020, 77, 300–308. [Google Scholar] [CrossRef]
- Sullivan, J.; Specchio, N.; Devinsky, O.; Auvin, S.; Perry, M.S.; Strzelczyk, A.; Gil-Nagel, A.; Dai, D.; Galer, B.S.; Gammaitoni, A.R. Fenfluramine significantly reduces day-to-day seizure burden by increasing number of seizure-free days and time between seizures in patients with Dravet syndrome: A time-to-event analysis. Epilepsia 2022, 63, 130–138. [Google Scholar] [CrossRef]
- Specchio, N.; Pietrafusa, N.; Doccini, V.; Trivisano, M.; Darra, F.; Ragona, F.; Cossu, A.; Spolverato, S.; Battaglia, D.; Quintiliani, M.; et al. Efficacy and safety of Fenfluramine hydrochloride for the treatment of seizures in Dravet syndrome: A real-world study. Epilepsia 2020, 61, 2405–2414. [Google Scholar] [CrossRef] [PubMed]
- Strzelczyk, A.; Pringsheim, M.; Mayer, T.; Polster, T.; Klotz, K.A.; Muhle, H.; Alber, M.; Trollmann, R.; Spors, H.; Kluger, G.; et al. Efficacy, tolerability, and retention of fenfluramine for the treatment of seizures in patients with Dravet syndrome: Compassionate use program in Germany. Epilepsia 2021, 62, 2518–2527. [Google Scholar] [CrossRef] [PubMed]
- Devinsky, O.; Marsh, E.; Friedman, D.; Thiele, E.; Laux, L.; Sullivan, J.; Miller, I.; Flamini, R.; Wilfong, A.; Filloux, F.; et al. Cannabidiol in patients with treatment-resistant epilepsy: An open-label interventional trial. Lancet Neurol. 2016, 15, 270–278. [Google Scholar] [CrossRef]
- Devinsky, O.; Cross, J.H.; Laux, L.; Marsh, E.; Miller, I.; Nabbout, R.; Scheffer, I.E.; Thiele, E.A.; Wright, S. Trial of Cannabidiol for Drug-Resistant Seizures in the Dravet Syndrome. N. Engl. J. Med. 2017, 376, 2011–2020. [Google Scholar] [CrossRef]
- Miller, I.; Scheffer, I.E.; Gunning, B.; Sanchez-Carpintero, R.; Gil-Nagel, A.; Perry, M.S.; Saneto, R.P.; Checketts, D.; Dunayevich, E.; Knappertz, V.; et al. Dose-Ranging Effect of Adjunctive Oral Cannabidiol vs Placebo on Convulsive Seizure Frequency in Dravet Syndrome. JAMA Neurol. 2020, 77, 613–621, Erratum in: JAMA Neurol. 2020, 77, 655. [Google Scholar] [CrossRef]
- Geffrey, A.L.; Pollack, S.F.; Bruno, P.L.; Thiele, E.A. Drug-drug interaction between clobazam and cannabidiol in children with refractory epilepsy. Epilepsia 2015, 56, 1246–1251. [Google Scholar] [CrossRef] [PubMed]
- Iannone, L.F.; Arena, G.; Battaglia, D.; Bisulli, F.; Bonanni, P.; Boni, A.; Canevini, M.P.; Cantalupo, G.; Cesaroni, E.; Contin, M.; et al. Results from an Italian Expanded Access Program on Cannabidiol Treatment in Highly Refractory Dravet Syndrome and Lennox–Gastaut Syndrome. Front. Neurol. 2021, 12, 673135. [Google Scholar] [CrossRef]
- Laux, L.C.; Bebin, E.M.; Checketts, D.; Chez, M.; Flamini, R.; Marsh, E.D.; Miller, I.; Nichol, K.; Park, Y.; Segal, E.; et al. Long-term safety and efficacy of cannabidiol in children and adults with treatment resistant Lennox-Gastaut syndrome or Dravet syndrome: Expanded access program results. Epilepsy Res. 2019, 154, 13–20. [Google Scholar] [CrossRef]
- Scheffer, I.E.; Halford, J.J.; Miller, I.; Nabbout, R.; Sanchez-Carpintero, R.; Shiloh-Malawsky, Y.; Wong, M.; Zolnowska, M.; Checketts, D.; Dunayevich, E.; et al. Add-on cannabidiol in patients with Dravet syndrome: Results of a long-term open-label extension trial. Epilepsia 2021, 62, 2505–2517. [Google Scholar] [CrossRef]
- Park, Y.D.; Linder, D.F.; Pope, J.; Flamini, J.R.; Moretz, K.; Diamond, M.P.; Long, S.A. Long-term efficacy and safety of cannabidiol (CBD) in children with treatment-resistant epilepsy: Results from a state-based expanded access program. Epilepsy Behav. 2020, 112, 107474. [Google Scholar] [CrossRef]
- Cross, J.H.; Caraballo, R.H.; Nabbout, R.; Vigevano, F.; Guerrini, R.; Lagae, L. Dravet syndrome: Treatment options and management of prolonged seizures. Epilepsia 2019, 60, S39–S48. [Google Scholar] [CrossRef] [PubMed]
- Wheless, J.W.; Fulton, S.P.; Mudigoudar, B.D. Dravet Syndrome: A Review of Current Management. Pediatr. Neurol. 2020, 107, 28–40. [Google Scholar] [CrossRef] [PubMed]
- Wirrell, E.C.; Hood, V.; Knupp, K.G.; Meskis, M.A.; Nabbout, R.; Scheffer, I.E.; Wilmshurst, J.; Sullivan, J. International consensus on diagnosis and management of Dravet syndrome. Epilepsia 2022, 63, 1761–1777. [Google Scholar] [CrossRef] [PubMed]
- Kossoff, E.H.; Zupec-Kania, B.A.; Auvin, S.; Ballaban-Gil, K.R.; Christina Bergqvist, A.G.; Blackford, R.; Buchhalter, J.R.; Caraballo, R.H.; Cross, J.H.; Dahlin, M.G.; et al. Optimal clinical management of children receiving dietary therapies for epilepsy: Updated recommendations of the International Ketogenic Diet Study Group. Epilepsia Open 2018, 3, 175–192. [Google Scholar] [CrossRef]
- Baraban, S.C. Forebrain electrophysiological recording in larval zebrafish. J Vis Exp. 2013, 71, 50104. [Google Scholar] [CrossRef]
- Dinday, M.T.; Baraban, S.C. Large-Scale Phenotype-Based Antiepileptic Drug Screening in a Zebrafish Model of Dravet Syndrome. Eneuro 2015, 2, ENEURO.0068-15.2015. [Google Scholar] [CrossRef]
- Higgins, G.A.; Fletcher, P.J.; Shanahan, W.R. Lorcaserin: A review of its preclinical and clinical pharmacology and therapeutic potential. Pharmacol. Ther. 2020, 205, 107417. [Google Scholar] [CrossRef]
- Tolete, P.; Knupp, K.; Karlovich, M.; DeCarlo, E.; Bluvstein, J.; Conway, E.; Friedman, D.; Dugan, P.; Devinsky, O. Lorcaserin therapy for severe epilepsy of childhood onset. Neurology 2018, 91, 837–839. [Google Scholar] [CrossRef]
- Nishi, T.; Kondo, S.; Miyamoto, M.; Watanabe, S.; Hasegawa, S.; Kondo, S.; Yano, J.; Watanabe, E.; Ishi, T.; Yoshikawa, M.; et al. Soticlestat, a novel cholesterol 24-hydroxylase inhibitor shows a therapeutic potential for neural hyperexcitation in mice. Sci. Rep. 2020, 10, 17081. [Google Scholar] [CrossRef]
- Koike, T.; Constantinescu, C.C.; Ikeda, S.; Nishi, T.; Sunahara, E.; Miyamoto, M.; Cole, P.; Barret, O.; Alagille, D.; Papin, C.; et al. Preclinical characterization of [18F]T-008, a novel PET imaging radioligand for cholesterol 24-hydroxylase. Eur. J. Nucl. Med. 2022, 49, 1148–1156. [Google Scholar] [CrossRef]
- Hawkins, N.A.; Jurado, M.; Thaxton, T.T.; Duarte, S.E.; Barse, L.; Tatsukawa, T.; Yamakawa, K.; Nishi, T.; Kondo, S.; Miyamoto, M.; et al. Soticlestat, a novel cholesterol 24-hydroxylase inhibitor, reduces seizures and premature death in Dravet syndrome mice. Epilepsia 2021, 62, 2845–2857. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Chen, G.; Pich, E.M.; Affinito, J.; Cwik, M.; Faessel, H. Safety, tolerability, pharmacokinetics, pharmacodynamics, bioavailability and food effect of single doses of soticlestat in healthy subjects. Br. J. Clin. Pharmacol. 2021, 87, 4354–4365. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Chen, G.; Pich, E.M.; Affinito, J.; Cwik, M.; Faessel, H.M. Pharmacokinetics, pharmacodynamics and safety assessment of multiple doses of soticlestat in healthy volunteers. Br. J. Clin. Pharmacol. 2022, 88, 2899–2908. [Google Scholar] [CrossRef]
- Hahn, C.D.; Jiang, Y.; Villanueva, V.; Zolnowska, M.; Arkilo, D.; Hsiao, S.; Asgharnejad, M.; Dlugos, D. A phase 2, randomized, double-blind, placebo-controlled study to evaluate the efficacy and safety of soticlestat as adjunctive therapy in pediatric patients with Dravet syndrome or Lennox–Gastaut syndrome (ELEKTRA). Epilepsia 2022, 63, 2671–2683. [Google Scholar] [CrossRef]
- Lenk, G.M.; Jafar-Nejad, P.; Hill, S.F.; Huffman, L.D.; Smolen, C.E.; Wagnon, J.L.; Petit, H.; Yu, W.; Ziobro, J.; Bhatia, K.; et al. Scn8a Antisense Oligonucleotide Is Protective in Mouse Models of SCN8A Encephalopathy and Dravet Syndrome. Ann. Neurol. 2020, 87, 339–346. [Google Scholar] [CrossRef]
- Han, Z.; Chen, C.; Christiansen, A.; Ji, S.; Lin, Q.; Anumonwo, C.; Liu, C.; Leiser, S.C.; Meena; Aznarez, I.; et al. Antisense oligonucleotides increase Scn1a expression and reduce seizures and SUDEP incidence in a mouse model of Dravet syndrome. Sci. Transl. Med. 2020, 12, eaaz6100. [Google Scholar] [CrossRef] [PubMed]
- Mora-Jimenez, L.; Valencia, M.; Sanchez-Carpintero, R.; Tønnesen, J.; Fadila, S.; Rubinstein, M.; Gonzalez-Aparicio, M.; Bunuales, M.; Fernandez-Pierola, E.; Nicolas, M.J.; et al. Transfer of SCN1A to the brain of adolescent mouse model of Dravet syndrome improves epileptic, motor, and behavioral manifestations. Mol. Ther. Nucleic Acids 2021, 25, 585–602. [Google Scholar] [CrossRef]
- Niibori, Y.; Lee, S.J.; Minassian, B.A.; Hampson, D.R. Sexually Divergent Mortality and Partial Phenotypic Rescue After Gene Therapy in a Mouse Model of Dravet Syndrome. Hum. Gene Ther. 2020, 31, 339–351. [Google Scholar] [CrossRef]
- Colasante, G.; Lignani, G.; Brusco, S.; Di Berardino, C.; Carpenter, J.; Giannelli, S.G.; Valassina, N.; Bido, S.; Ricci, R.; Castoldi, V.; et al. dCas9-Based Scn1a Gene Activation Restores Inhibitory Interneuron Excitability and Attenuates Seizures in Dravet Syndrome Mice. Mol. Ther. 2020, 28, 235–253. [Google Scholar] [CrossRef]
- Lim, K.H.; Han, Z.; Jeon, H.Y.; Kach, J.; Jing, E.; Weyn-Vanhentenryck, S.; Downs, M.; Corrionero, A.; Oh, R.; Scharner, J.; et al. Antisense oligonucleotide modulation of non-productive alternative splicing upregulates gene expression. Nat. Commun. 2020, 11, 3501. [Google Scholar] [CrossRef]
- Meena, M.; Ticho, B.; Barriere, O.; Gosselin, N. A pharmacokinetic (PK) model for STK-001, an antisense oligonucleotide (ASO), based on data from non-human primates (NHP) enables dose selection in patients with Dravet syndrome (DS) [Abstract 3.264]. In Proceedings of the Annual Meeting of the American Epilepsy Society, Chicago, IL, USA, 3–7 December 2021. [Google Scholar]
- Tanenhaus, A.; Stowe, T.; Young, A.; McLaughlin, J.; Aeran, R.; Lin, I.W.; Li, J.; Hosur, R.; Chen, M.; Leedy, J.; et al. Cell-Selective Adeno-Associated Virus-Mediated SCN1A Gene Regulation Therapy Rescues Mortality and Seizure Phenotypes in a Dravet Syndrome Mouse Model and Is Well Tolerated in Nonhuman Primates. Hum. Gene Ther. 2022, 33, 579–597. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Drug | Dose | Adverse Events | Monitoring | Special Warnings | ||
|---|---|---|---|---|---|---|
| Starting | Target | Max | ||||
| STP | 10–15 mg/kg | Up to 50 mg/kg, average 25–30 mg/kg | 3000 mg/d or 50 mg/kg | Decreased appetite and weight Insomnia Somnolence | CBC Liver function Weight | Neutropenia and thrombocytopenia |
| FFA | 0.2 mg/kg | 0.7 mg/kg/without STP 0.4 mg/kg/with STP | 26 mg/d without STP 17 mg/d with STP | Decreased appetite Diarrhea Pyrexia | Echocardiography Weight | Valvulopathy |
| CBD | 5 mg/kg | 10 mg/kg | 20 mg/kg | Somnolence Decreased appetite Diarrhea | Liver function | Liver dysfunction, Somnolence |
| Trial | Compound | Route, Dose | Mechanism of Action | Phase | Population Age |
|---|---|---|---|---|---|
| ENDEAVOR (NCT05419492) | ETX-001 | Intracerebroventricular Dose-escalation design | Increase expression of endogenous SCN1A, specifically in GABAergic interneurons | I/II | 6–36 months |
| NCT04442295, (MONARCH) | STK-001 | Intrathecal injection | Increase production of SCN1A mRNA isoforms | II | 2–18 years |
| NCT04740476 (SWALLOWTAIL) | Single ascending doses (10 mg, 20 mg, 30 mg, and 45 mg). | ||||
| NCT04442295 (ADMIRAL) | Multiple ascending doses—(20 mg, 30 mg, and 45 mg). | ||||
| NCT04462770 Argus Trial | EPX-100 (clemizole) | Oral | H1 antagonist with HTR2A and/or HTR2B receptors affinity | II | 2 years and above |
| Highest-tolerated dose titration | |||||
| NCT04572243 (MOMENTUM 1) | EPX-200 (lorcaserin) | Oral | HTR2C activator | III | 2 years and above |
| Up to 10, 20 mg/day for participants weighing 10 to <20, 20 to <40 kg, respectively. | |||||
| SKYLINE (NCT04940624). | TAK-935/OV935 (soticlestat) | Oral | selective inhibitor of cholesterol 24-hydroxylase | III | 2–21 years |
| Participants weighing <45 kg: soticlestat, mini-tablets, at the dose of 40 mg to 200 mg; participants weighing ≥45 kg: up to 300 mg |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, C.; Pielas, M.; Jiao, F.; Mei, D.; Wang, X.; Kotulska, K.; Jozwiak, S. Epilepsy in Dravet Syndrome—Current and Future Therapeutic Opportunities. J. Clin. Med. 2023, 12, 2532. https://doi.org/10.3390/jcm12072532
Gao C, Pielas M, Jiao F, Mei D, Wang X, Kotulska K, Jozwiak S. Epilepsy in Dravet Syndrome—Current and Future Therapeutic Opportunities. Journal of Clinical Medicine. 2023; 12(7):2532. https://doi.org/10.3390/jcm12072532
Chicago/Turabian StyleGao, Chao, Mikolaj Pielas, Fuyong Jiao, Daoqi Mei, Xiaona Wang, Katarzyna Kotulska, and Sergiusz Jozwiak. 2023. "Epilepsy in Dravet Syndrome—Current and Future Therapeutic Opportunities" Journal of Clinical Medicine 12, no. 7: 2532. https://doi.org/10.3390/jcm12072532
APA StyleGao, C., Pielas, M., Jiao, F., Mei, D., Wang, X., Kotulska, K., & Jozwiak, S. (2023). Epilepsy in Dravet Syndrome—Current and Future Therapeutic Opportunities. Journal of Clinical Medicine, 12(7), 2532. https://doi.org/10.3390/jcm12072532