
Premature Pubarche: Time to Revise the Diagnostic Approach?
 , , , , ,
, , , , ,  and
and
Abstract
1. Introduction
2. Materials and Methods
Statistical Analysis
3. Results
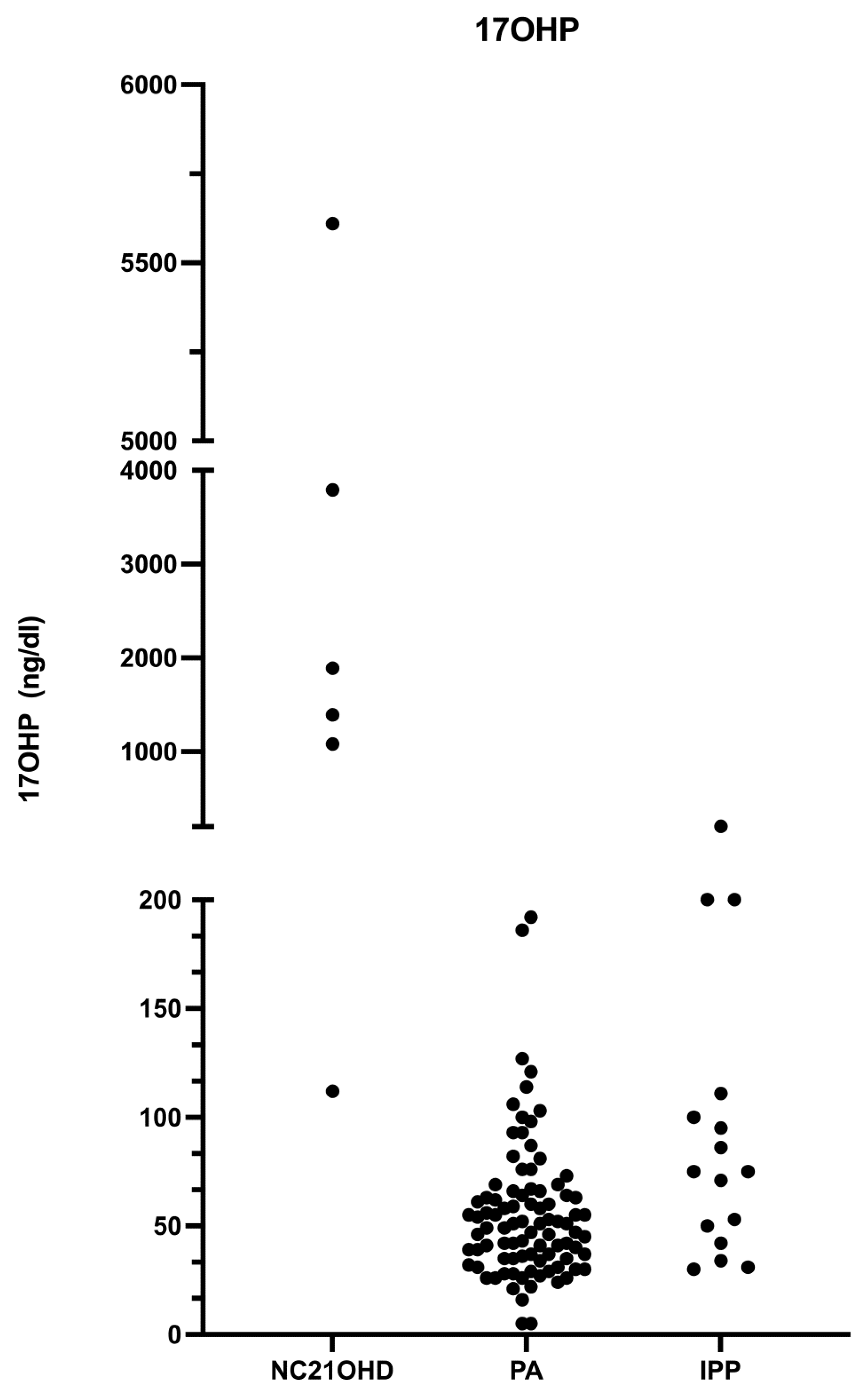
3.1. Adrenal Steroid Evaluation
3.2. Anthropometric Parameters and Bone Age
3.3. Puberty
3.4. NC21OHD Patients
4. Statistical Correlations
5. Discussion
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rosenfield, R.L. Normal and Premature Adrenarche. Endocr. Rev. 2021, 42, 783–814. [Google Scholar] [CrossRef] [PubMed]
- Vottero, A.; Capelletti, M.; Giuliodori, S.; Viani, I.; Ziveri, M.; Neri, T.M.; Bernasconi, S.; Ghizzoni, L. Decreased Androgen Receptor Gene Methylation in Premature Pubarche: A Novel Pathogenetic Mechanism? J. Clin. Endocrinol. Metab. 2006, 91, 968–972. [Google Scholar] [CrossRef]
- Sopher, A.B.; Jean, A.M.; Zwany, S.K.; Winston, D.M.; Pomeranz, C.B.; Bell, J.J.; McMahon, D.J.; Hassoun, A.; Fennoy, I.; Oberfield, S.E. Bone Age Advancement in Prepubertal Children with Obesity and Premature Adrenarche: Possible Potentiating Factors. Obesity (Silver Spring) 2011, 19, 1259–1264. [Google Scholar] [CrossRef] [PubMed]
- DeSalvo, D.J.; Mehra, R.; Vaidyanathan, P.; Kaplowitz, P.B. In Children with Premature Adrenarche, Bone Age Advancement by 2 or More Years Is Common and Generally Benign. J. Pediatr. Endocrinol. Metab. 2013, 26, 215–221. [Google Scholar] [CrossRef]
- Voutilainen, R.; Jääskeläinen, J. Premature Adrenarche: Etiology, Clinical Findings, and Consequences. J. Steroid Biochem. Mol. Biol. 2015, 145, 226–236. [Google Scholar] [CrossRef]
- Liimatta, J.; Utriainen, P.; Voutilainen, R.; Jääskeläinen, J. Girls with a History of Premature Adrenarche Have Advanced Growth and Pubertal Development at the Age of 12 Years. Front. Endocrinol. (Lausanne) 2017, 8, 291. [Google Scholar] [CrossRef]
- New, M.I. Extensive Clinical Experience: Nonclassical 21-Hydroxylase Deficiency. J. Clin. Endocrinol. Metab. 2006, 91, 4205–4214. [Google Scholar] [CrossRef]
- Nordenström, A.; Falhammar, H. MANAGEMENT OF ENDOCRINE DISEASE: Diagnosis and Management of the Patient with Non-Classic CAH Due to 21-Hydroxylase Deficiency. Eur. J. Endocrinol. 2019, 180, R127–R145. [Google Scholar] [CrossRef]
- Wasniewska, M.G.; Morabito, L.A.; Baronio, F.; Einaudi, S.; Salerno, M.; Bizzarri, C.; Russo, G.; Chiarito, M.; Grandone, A.; Guazzarotti, L.; et al. Growth Trajectory and Adult Height in Children with Nonclassical Congenital Adrenal Hyperplasia. Horm. Res. Paediatr. 2020, 93, 173–181. [Google Scholar] [CrossRef] [PubMed]
- New, M.I.; Lorenzen, F.; Lerner, A.J.; Kohn, B.; Oberfield, S.E.; Pollack, M.S.; Dupont, B.; Stoner, E.; Levy, D.J.; Pang, S.; et al. Genotyping Steroid 21-Hydroxylase Deficiency: Hormonal Reference Data. J. Clin. Endocrinol. Metab. 1983, 57, 320–326. [Google Scholar] [CrossRef] [PubMed]
- Bachega, T.A.; Billerbeck, A.E.; Marcondes, J.A.; Madureira, G.; Arnhold, I.J.; Mendonca, B.B. Influence of Different Genotypes on 17-Hydroxyprogesterone Levels in Patients with Nonclassical Congenital Adrenal Hyperplasia Due to 21-Hydroxylase Deficiency. Clin. Endocrinol. (Oxf.) 2000, 52, 601–607. [Google Scholar] [CrossRef] [PubMed]
- Dewailly, D.; Vantyghem-Haudiquet, M.C.; Sainsard, C.; Buvat, J.; Cappoen, J.P.; Ardaens, K.; Racadot, A.; Lefebvre, J.; Fossati, P. Clinical and Biological Phenotypes in Late-Onset 21-Hydroxylase Deficiency. J. Clin. Endocrinol. Metab. 1986, 63, 418–423. [Google Scholar] [CrossRef] [PubMed]
- Ibáñez, L.; Bonnin, M.R.; Zampolli, M.; Prat, N.; Alia, P.J.; Navarro, M.A. Usefulness of an ACTH Test in the Diagnosis of Nonclassical 21-Hydroxylase Deficiency among Children Presenting with Premature Pubarche. Horm. Res. 1995, 44, 51–56. [Google Scholar] [CrossRef]
- Török, D.; Halász, Z.; Garami, M.; Homoki, J.; Fekete, G.; Sólyom, J. Limited Value of Serum Steroid Measurements in Identification of Mild Form of 21-Hydroxylase Deficiency. Exp. Clin. Endocrinol. Diabetes 2003, 111, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Armengaud, J.-B.; Charkaluk, M.-L.; Trivin, C.; Tardy, V.; Bréart, G.; Brauner, R.; Chalumeau, M. Precocious Pubarche: Distinguishing Late-Onset Congenital Adrenal Hyperplasia from Premature Adrenarche. J. Clin. Endocrinol. Metab. 2009, 94, 2835–2840. [Google Scholar] [CrossRef] [PubMed]
- Cacciari, E.; Milani, S.; Balsamo, A.; Dammacco, F.; De Luca, F.; Chiarelli, F.; Pasquino, A.M.; Tonini, G.; Vanelli, M. Italian Cross-Sectional Growth Charts for Height, Weight and BMI (6-20 y). Eur. J. Clin. Nutr. 2002, 56, 171–180. [Google Scholar] [CrossRef]
- Speiser, P.W.; Arlt, W.; Auchus, R.J.; Baskin, L.S.; Conway, G.S.; Merke, D.P.; Meyer-Bahlburg, H.F.L.; Miller, W.L.; Murad, M.H.; Oberfield, S.E.; et al. Congenital Adrenal Hyperplasia Due to Steroid 21-Hydroxylase Deficiency: An Endocrine Society Clinical Practice Guideline. J. Clin. Endocrinol. Metab. 2018, 103, 4043–4088. [Google Scholar] [CrossRef]
- Claahsen - van der Grinten, H.L.; Speiser, P.W.; Ahmed, S.F.; Arlt, W.; Auchus, R.J.; Falhammar, H.; Flück, C.E.; Guasti, L.; Huebner, A.; Kortmann, B.B.M.; et al. Congenital Adrenal Hyperplasia—Current Insights in Pathophysiology, Diagnostics, and Management. Endocr. Rev. 2021, 43, 91–159. [Google Scholar] [CrossRef]
- Greulich, W.; Pyle, S. Radiographic Atlas of Skeletal Development Ofthe Hand and Wrist; Stanford University Press: Stanford, CA, USA, 1959. [Google Scholar]
- Savas Erdeve, S.; Berberoglu, M.; Yurur-Kutlay, N.; Siklar, Z.; Hacihamdioglu, B.; Tukun, A.; Ocal, G. Characteristics and Prevalence of Non-Classical Congenital Adrenal Hyperplasia with a V2811 Mutation in Patients with Premature Pubarche. J. Pediatr. Endocrinol. Metab. 2011, 24, 965–970. [Google Scholar] [CrossRef]
- Grandone, A.; Marzuillo, P.; Luongo, C.; Toraldo, R.; Mariani, M.; Miraglia Del Giudice, E.; Perrone, L. Basal Levels of 17-Hydroxyprogesterone Can Distinguish Children with Isolated Precocious Pubarche. Pediatr. Res. 2018, 84, 533–536. [Google Scholar] [CrossRef]
- Leite, M.V.; Mendonça, B.B.; Arnhold, I.J.; Estefan, V.; Nunes, C.; Nicolau, W.; Bloise, W. Identification of Nonclassical 21-Hydroxylase Deficiency in Girls with Precocious Pubarche. J. Endocrinol. Invest. 1991, 14, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Bizzarri, C.; Crea, F.; Marini, R.; Benevento, D.; Porzio, O.; Ravà, L.; Cappa, M. Clinical Features Suggestive of Non-Classical 21-Hydroxylase Deficiency in Children Presenting with Precocious Pubarche. J. Pediatr. Endocrinol. Metab. 2012, 25, 1059–1064. [Google Scholar] [CrossRef] [PubMed]
- Neeman, B.; Bello, R.; Lazar, L.; Phillip, M.; de Vries, L. Central Precocious Puberty as a Presenting Sign of Nonclassical Congenital Adrenal Hyperplasia: Clinical Characteristics. J. Clin. Endocrinol. Metab. 2019, 104, 2695–2700. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Males (n.24) | Females (n.87) | ||
|---|---|---|---|
| Gestational age (wks) | 38 (3.5) | 39 (2.5) | NS |
| Neonatal weight (gr) | 2829 (375) | 3140 (643) | NS |
| Chronological age (yrs) | 8.9 (1.5) | 7.9 (1.0) | p < 0.01 |
| Bone age (yrs) | 10.8 (1.7) | 9.7 (1.2) | p < 0.01 |
| Delta BA-CA (yrs) | 2.1 (0.6) | 2.0 (0.6) | NS |
| Height SDS | 0.9 (0.9) | 0.9 (1.0) | NS |
| BMI SDS | 0.4 (1.0) | 0.7 (0.8) | NS |
| Pubarche onset (yrs) | 7.6 (1.6) | 6.7 (1.0) | p < 0.05 |
| Group 1 IPP (n = 15) | Group 2 PA (n = 90) | Group 3 NC21OHD (n = 6) | PA vs. IPP | NC21OHD vs. IPP | NC21OHD vs. PA | |
|---|---|---|---|---|---|---|
| Baseline 17-OHP (ng/dL) | 89.6 (61.6) | 55.3 (31.9) | 2312 (2022) | NS | p < 0.05 | p < 0.05 |
| Stimulated 17-OHP (ng/dL) | 331.2 (239.6) | 236.1 (104.3) | 4923 (1604) | NS | p < 0.05 | p < 0.05 |
| Baseline Δ4A (ng/dL) | 33.4 (19.6) | 56.9 (42.0) | 374 (145) | NS | p < 0.05 | p < 0.05 |
| DHEA-S (mcg/dL) | 31.1 (8.9) | 103.3 (51.3) | 229 (64) | NS | p < 0.05 | p < 0.05 |
| Testosterone (ng/mL) | 0.06 (0.04) | 0.2 (0.2) | 0.7 (0.3) | p < 0.05 | p < 0.05 | p < 0.05 |
| Height SDS | 0.7 (0.7) | 1 (0.9) | 0.6 (1.4) | NS | NS | NS |
| Weight SDS | 0.7 (0.6) | 0.8 (0.8) | 0.5 (1) | NS | NS | NS |
| BMI SDS | 0.3 (0.6) | 0.7 (09) | 0.3 (1.4) | NS | NS | NS |
| BA-CA (yrs) | 2.3 (0.9) | 1.9 (0.6) | 2.6 (0.8) | NS | NS | NS |
| Age at symptoms | 6.3 (1.3) | 7.0 (1.2) | 6.9 (0.7) | NS | NS | NS |
| Age at test | 7.2 (1.1) | 8.3 (1.2) | 8.9 (0.9) | p < 0.05 | p < 0.05 | NS |
| Groups (BA-CA) | A | B | C | B vs. A | C vs. A | C vs. B |
|---|---|---|---|---|---|---|
| (1 < BA-CA < 2) | (2 ≤ BA-CA < 3) | (3 ≤ BA-CA) | ||||
| n = 64 | n = 37 | n = 10 | ||||
| Baseline 17-OHP (ng/dL) | 58.2 (42.9) | 172 (398) | 1008 (1993) | NS | p < 0.05 | p < 0.05 |
| Baseline Δ4A(ng/dL) | 53.3 (47.8) | 77.8 (77) | 157 (212) | NS | p < 0.05 | p < 0.05 |
| DHEA-S (mcg/dL) | 93.4 (51.3) | 108.4 (72.6) | 115.3 (84.4) | NS | p < 0.05 | p < 0.05 |
| Testosterone (ng/mL) | 0.16 (0.2) | 0.20 (0.2) | 0.33 (0.4) | NS | p < 0.05 | NS |
| No. of pts with NC21OHD | 1 | 3 | 2 | - | - | - |
| Pt | Sex | CA yrs at ACTH Test | Height SDS | BMI SDS | BA-CA (yrs) | Early Puberty | Baseline Testosterone (ng/mL) | Baseline 17-OHP (ng/dL) | Stimulated 17-OHP (ng/dL) | Baseline Δ4A (ng/dL) | Baseline DHEA (ng/mL) | Baseline DHEA-S (mcg/dL) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | F | 9.3 | −1.2 | 1.3 | 1.4 | Yes | 0.5 | 112.0 | 3270 | 291.0 | 10.8 | 262.0 |
| 2 | F | 9.4 | −0.4 | 1.5 | 2.9 | Yes | 0.7 | 1080.0 | 3380 | 319.0 | 15.3 | 255.0 |
| 3 | M | 10.3 | 0.3 | −1.3 | 2.1 | Yes | 0.6 | 1390.0 | 5340 | 214.0 | 6.0 | 268.0 |
| 4 | F | 7.7 | 0.6 | −1.0 | 2.5 | No | 0.5 | 1890.0 | 7410 | 323.0 | 4.9 | 102.0 |
| 5 | F | 8.7 | 1.6 | −0.6 | 3.4 | No | 1.2 | 3790.0 | 4250 | 605.0 | 17.2 | 260.0 |
| 6 | F | 8.4 | 2.7 | 1.7 | 3.2 | Yes | 0.9 | 5610.0 | 5890 | 494.0 | 18.0 | 228.0 |
| Baseline 17-OHP (ng/dL) | Stimulated 17 OHP (ng/dL) | Δ4A (ng/dL) | Testosterone (ng/mL) | DHEA-S (ng/dL) | BA-CA (Years) | ||
|---|---|---|---|---|---|---|---|
| Cut-off | 111 | 200 | 3271 | 214 | 0.4 | 228 | 2 |
| Sensitivity (%) | 100 | 83.3 | 100 | 100 | 100 | 83.3 | 83.3 |
| Specificity (%) | 92.4 | 97.1 | 100 | 100 | 94.3 | 96.2 | 65.7 |
| PPV (%) | 42.9 | 62.5 | 100 | 100 | 50 | 55.6 | 12.2 |
| NPV (%) | 100 | 99 | 100 | 100 | 100 | 99 | 98.6 |
| PLR | 13.1 | 29.2 | - | - | 17.5 | 21.9 | 2.4 |
| NLR | 0 | 0.17 | - | - | 0 | 0.2 | 0.3 |
| AUC | 98.7 | - | 100 | 100 | 97.8 | 93.7 | 74.3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baronio, F.; Marzatico, A.; De Iasio, R.; Ortolano, R.; Fanolla, A.; Radetti, G.; Balsamo, A.; Pession, A.; Cassio, A. Premature Pubarche: Time to Revise the Diagnostic Approach? J. Clin. Med. 2023, 12, 2187. https://doi.org/10.3390/jcm12062187
Baronio F, Marzatico A, De Iasio R, Ortolano R, Fanolla A, Radetti G, Balsamo A, Pession A, Cassio A. Premature Pubarche: Time to Revise the Diagnostic Approach? Journal of Clinical Medicine. 2023; 12(6):2187. https://doi.org/10.3390/jcm12062187
Chicago/Turabian StyleBaronio, Federico, Alice Marzatico, Rosaria De Iasio, Rita Ortolano, Antonio Fanolla, Giorgio Radetti, Antonio Balsamo, Andrea Pession, and Alessandra Cassio. 2023. "Premature Pubarche: Time to Revise the Diagnostic Approach?" Journal of Clinical Medicine 12, no. 6: 2187. https://doi.org/10.3390/jcm12062187
APA StyleBaronio, F., Marzatico, A., De Iasio, R., Ortolano, R., Fanolla, A., Radetti, G., Balsamo, A., Pession, A., & Cassio, A. (2023). Premature Pubarche: Time to Revise the Diagnostic Approach? Journal of Clinical Medicine, 12(6), 2187. https://doi.org/10.3390/jcm12062187