
Advances in CAR-T Cell Therapy in Head and Neck Squamous Cell Carcinoma
 ,
, 


Abstract
1. Background
2. Brief Introduction to CAR-T Cell in Cancer Treatment
3. Application of CAR-T Cell Therapy in Head and Neck Squamous Cell Carcinomas
3.1. Preclinical Research of CAR-T Cell Therapy against HNSCC
3.2. Clinical Trials of CAR-T Cell Therapy against HNSCC
3.3. Potential Targets for CAR-T Cell Therapy against HNSCC
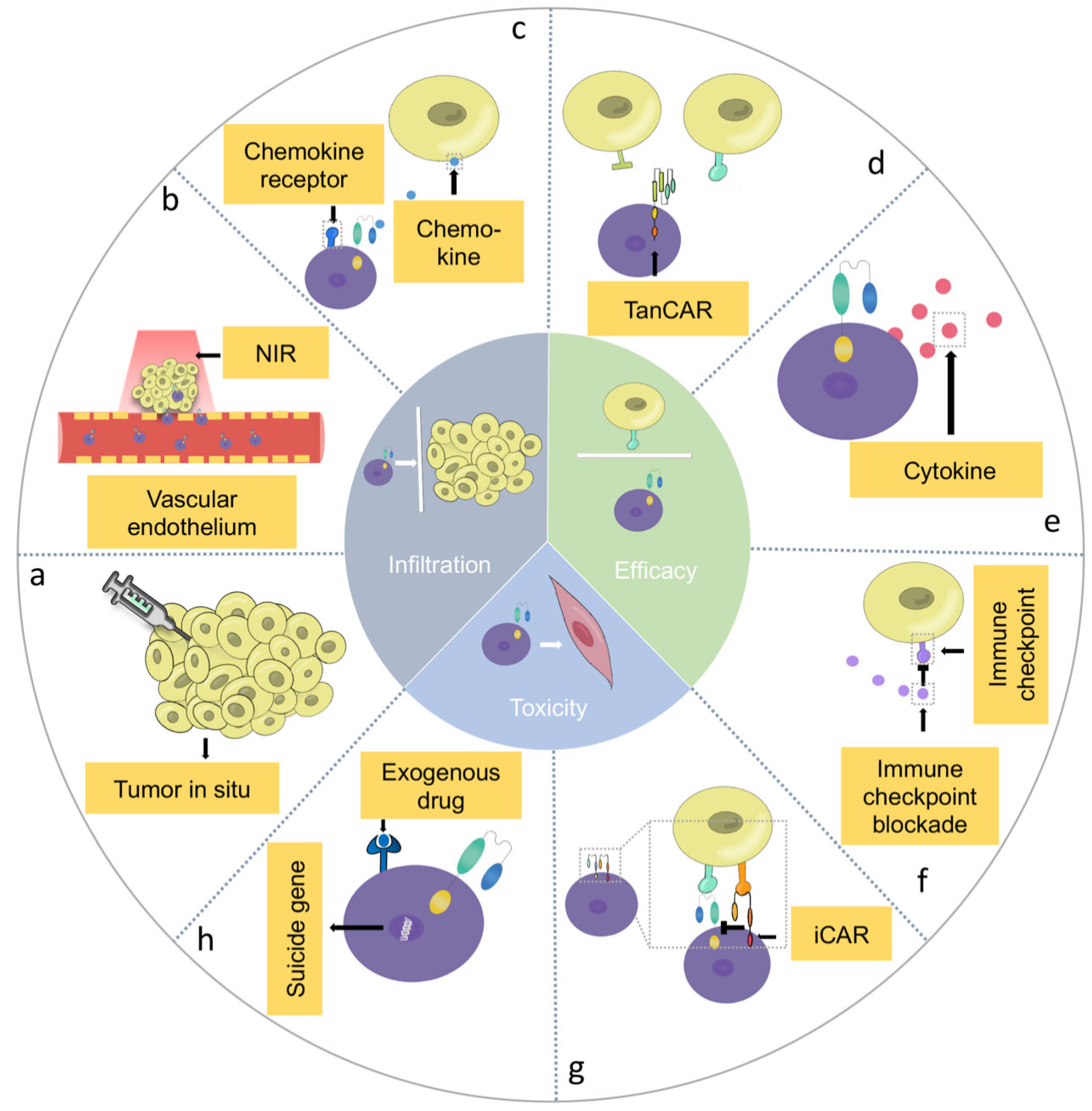
4. Barriers of CAR-T Cell Therapy in HNSCC and Potential Solutions
4.1. Infiltration Barriers of CAR-T Cells in HNSCC
4.1.1. Delivery Techniques
4.1.2. Combination Therapy
4.1.3. Genetically Engineered Multi-Functional CAR-T Cells
4.2. Efficacy Barriers of CAR-T Cells in HNSCC
4.2.1. Genetic Heterogeneity in HNSCC
4.2.2. Immunosuppressive Tumor Microenvironment in HNSCC
4.2.3. Immunogenicity of CAR-T Cells
4.3. The Safety of CAR-T Cells in HNSCC
4.3.1. On-Target, Off-Tumor” Toxicity in CAR-T Cell Therapy
4.3.2. Cytokine Release Syndrome in CAR-T Cell Therapy
5. Conclusions
Authors Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| HNSCC: | head and neck squamous cell carcinoma; |
| CAR-T: | chimeric antigen receptor T; |
| MHC: | non-major histocompatibility complex; |
| scFv: | single-chain variable fragment; |
| VL: | variable light chain; |
| VH: | variable heavy chain; |
| TRUCK: | T cells redirected for universal cytokine killing; |
| IL: | interleukin; |
| UniCAR-T: | universal CAR-T; |
| CRS: | cytokine release syndrome; |
| EGFR: | epidermal growth factor receptor; |
| GBM: | glioblastoma; |
| HER2: | human epidermal growth factor receptor 2; |
| GD2: | ganglioside D2; |
| FPA: | fibroblast activation protein; |
| MUC1: | mucin 1; |
| EpCAM: | epithelial cell adhesion molecule; |
| NKG2DL: | natural killer group 2 member D ligand; |
| LMP1: | latent membrane protein; |
| TME: | tumor microenvironment; |
| NIR: | near infrared |
| aPDL1: | anti-programmed death-ligand-1-blocking antibody; |
| PTT: | photothermal therapy; |
| CXCR: | C-X-C motif chemokine receptor; |
| HPSE: | heparinase; |
| TanCAR-T: | tandem CAR-T cells; |
| SUPRA: | split, universal and programmable; |
| OVs: | oncolytic viruses; |
| CAd: | binary oncolytic adenovirus; |
| PD-L1: | programmed cell death-ligand 1; |
| FHVH33: | humanized heavy-chain variable domain; |
| synNotch: | synthetic notch; |
| EphA2: | ephrin type A receptor 2; |
| iCAR-T: | inhibitory CAR-T; |
| iC9: | caspase-9; |
| CID: | chemical inducer of dimerization; |
| STAT3: | signal transducer and activator of transcription 3. |
References
- Johnson, D.E.; Burtness, B.; Leemans, C.R.; Lui VW, Y.; Bauman, J.E.; Grandis, J.R. Head and neck squamous cell carcinoma. Nat. Rev. Dis. Prim. 2020, 6, 92. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.; Jemal, A. Cancer statistics. CA A Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, A.K.; Engels, E.A.; Pfeiffer, R.M.; Hernandez, B.Y.; Xiao, W.; Kim, E.; Jiang, B.; Goodman, M.T.; Sibug-Saber, M.; Cozen, W.; et al. Human papillomavirus and rising oropharyngeal cancer incidence in the United States. J. Clin. Oncol. 2011, 29, 4294–4301. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, M.H.; McMillan, N.A.; Johnson, N.W. HPV-associated head and neck cancers in the Asia Pacific: A critical literature review & meta-analysis. Cancer Epidemiol. 2015, 39, 923–938. [Google Scholar]
- Tseng, Y.-H.; Chang, K.-W.; Yang, C.-C.; Liu, C.-J.; Kao, S.-Y.; Liu, T.-Y.; Lin, S.-C. Association between areca-stimulated vimentin expression and the progression of head and neck cancers. Head Neck 2011, 34, 245–253. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Osazuwa-Peters, N.; Simpson, M.C.; Zhao, L.; Boakye, E.A.; Olomukoro, S.I.; Deshields, T.; Loux, T.M.; Varvares, M.A.; Schootman, M. Suicide risk among cancer survivors: Head and neck versus other cancers. Cancer 2018, 124, 4072–4079. [Google Scholar] [CrossRef]
- Cramer, J.D.; Burtness, B.; Ferris, R.L. Immunotherapy for head and neck cancer: Recent advances and future directions. Oral Oncol. 2019, 99, 104460. [Google Scholar] [CrossRef]
- Migden, M.R.; Rischin, D.; Schmults, C.D.; Guminski, A.; Hauschild, A.; Lewis, K.D.; Chung, C.H.; Hernandez-Aya, L.F.; Lim, A.M.; Chang, A.L.S.; et al. PD-1 Blockade with Cemiplimab in Advanced Cutaneous Squamous-Cell Carcinoma. N. Engl. J. Med. 2018, 379, 341–351. [Google Scholar] [CrossRef]
- Yoshitake, Y.; Fukuma, D.; Yuno, A.; Hirayama, M.; Nakayama, H.; Tanaka, T.; Nagata, M.; Takamune, Y.; Kawahara, K.; Nakagawa, Y.; et al. Phase II clinical trial of multiple peptide vaccination for advanced head and neck cancer patients revealed induction of immune responses and improved OS. Clin. Cancer Res. 2015, 21, 312–321. [Google Scholar] [CrossRef]
- Gross, G.; Waks, T.; Eshhar, Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc. Natl. Acad. Sci. USA 1989, 86, 10024–10028. [Google Scholar] [CrossRef]
- Thanindratarn, P.; Dean, D.C.; Nelson, S.D.; Hornicek, F.J.; Duan, Z. Chimeric antigen receptor T (CAR-T) cell immunotherapy for sarcomas: From mechanisms to potential clinical applications. Cancer Treat. Rev. 2019, 82, 101934. [Google Scholar] [CrossRef]
- Eshhar, Z.; Waks, T.; Gross, G.; Schindler, D.G. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding do-mains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc. Natl. Acad. Sci. USA 1993, 90, 720–724. [Google Scholar] [CrossRef]
- Maher, J.; Brentjens, R.J.; Gunset, G.; Riviere, I.; Sadelain, M. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRζ/CD28 receptor. Nat. Biotechnol. 2002, 20, 70–75. [Google Scholar] [CrossRef]
- Sadelain, M.; Rivière, I.; Brentjens, R. Targeting tumours with genetically enhanced T lymphocytes. Nat. Rev. Cancer 2003, 3, 35–45. [Google Scholar] [CrossRef]
- Kershaw, M.H.; Westwood, J.A.; Parker, L.L.; Wang, G.; Eshhar, Z.; Mavroukakis, S.A.; White, D.E.; Wunderlich, J.R.; Canevari, S.; Rogers-Freezer, L.; et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin. Cancer Res. 2006, 12, 6106–6115. [Google Scholar] [CrossRef]
- Chmielewski, M.; Kopecky, C.; Hombach, A.A.; Abken, H. IL-12 Release by Engineered T Cells Expressing Chimeric Antigen Receptors Can Effectively Muster an Antigen-Independent Macrophage Response on Tumor Cells That Have Shut Down Tumor Antigen Expression Elimination of Antigen-Loss Tumor Cells. Cancer Res. 2011, 71, 5697–5706. [Google Scholar] [CrossRef]
- Feins, S.; Kong, W.; Williams, E.F.; Milone, M.C.; Fraietta, J.A. An introduction to chimeric antigen receptor (CAR) T-cell immunotherapy for human cancer. Am. J. Hematol. 2019, 94, S3–S9. [Google Scholar] [CrossRef]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T cell immunotherapy for human cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef]
- Jin, L.; Tao, H.; Karachi, A.; Long, Y.; Hou, A.Y.; Na, M.; Dyson, K.A.; Grippin, A.J.; Deleyrolle, L.P.; Zhang, W.; et al. CXCR1- or CXCR2-modified CAR T cells co-opt IL-8 for maximal antitumor efficacy in solid tumors. Nat. Commun. 2019, 10, 4016. [Google Scholar] [CrossRef]
- Kueberuwa, G.; Kalaitsidou, M.; Cheadle, E.; Hawkins, R.E.; Gilham, D.E. CD19 CAR T Cells Expressing IL-12 Eradicate Lymphoma in Fully Lymphoreplete Mice through Induction of Host Immunity. Mol. Ther. Oncolytics 2018, 8, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Ran, T.; Eichmüller, S.B.; Schmidt, P.; Schlander, M. Cost of decentralized CAR T-cell production in an academic nonprofit setting. Int. J. Cancer 2020, 147, 3438–3445. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Lin, Q.; Song, Y.; Liu, D. Universal CARs, universal T cells, and universal CAR T cells. J. Hematol. Oncol. 2018, 11, 132. [Google Scholar] [CrossRef]
- Wei, J.; Liu, Y.; Wang, C.; Zhang, Y.; Tong, C.; Dai, G.; Wang, W.; Rasko, J.E.J.; Melenhorst, J.J.; Qian, W.; et al. The model of cytokine release syndrome in CAR T-cell treatment for B-cell non-Hodgkin lymphoma. Signal Transduct. Target. Ther. 2020, 5, 134. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Yi, M.; Qin, S.; Wu, K. Next generation chimeric antigen receptor T cells: Safety strategies to overcome toxicity. Mol. Cancer 2019, 18, 125. [Google Scholar] [CrossRef]
- Feldmann, A.; Arndt, C.; Koristka, S.; Berndt, N.; Bergmann, R.; Bachmann, M.P. Conventional CARs versus modular CARs. Cancer Immunol. Immunother. 2019, 68, 1713–1719. [Google Scholar] [CrossRef]
- Feldman, L.; Brown, C.; Badie, B. Chimeric Antigen Receptor T-Cell Therapy: Updates in Glioblastoma Treatment. Neurosurgery 2021, 88, 1056–1064. [Google Scholar] [CrossRef]
- Guardiola, S.; Varese, M.; Sánchez-Navarro, M.; Giralt, E. A Third Shot at EGFR: New Opportunities in Cancer Therapy. Trends Pharmacol. Sci. 2019, 40, 941–955. [Google Scholar] [CrossRef]
- Feng, K.; Guo, Y.; Dai, H.; Wang, Y.; Li, X.; Jia, H.; Han, W. Chimeric antigen receptor-modified T cells for the immunotherapy of patients with EGFR-expressing advanced relapsed/refractory non-small cell lung cancer. Sci. China Life Sci. 2016, 59, 468–479. [Google Scholar] [CrossRef]
- Liu, Y.; Guo, Y.; Wu, Z.; Feng, K.; Tong, C.; Wang, Y.; Dai, H.; Shi, F.; Yang, Q.; Han, W. Anti-EGFR chimeric antigen receptor-modified T cells in metastatic pancreatic carcinoma: A phase I clinical trial. Cytotherapy 2020, 22, 573–580. [Google Scholar] [CrossRef]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017, 9, eaaa0984. [Google Scholar] [CrossRef]
- Bookman, M.A. Biological therapy of ovarian cancer: Current directions. Semin. Oncol. 1998, 25, 381–396. [Google Scholar]
- Choi, B.D.; Curry, W.T.; Carter, B.S.; Maus, M.V. Chimeric antigen receptor T-cell immunotherapy for glioblastoma: Practical insights for neurosurgeons. Neurosurg. Focus 2018, 44, E13. [Google Scholar] [CrossRef]
- Fusco, N.; Bosari, S. HER2 aberrations and heterogeneity in cancers of the digestive system: Implications for pathologists and gastroenterologists. World J. Gastroenterol. 2016, 22, 7926–7937. [Google Scholar] [CrossRef]
- Hu, X.; Huang, W.; Fan, M. Emerging therapies for breast cancer. J. Hematol. Oncol. 2017, 10, 98. [Google Scholar] [CrossRef]
- Ahmed, N.; Brawley, V.; Hegde, M.; Bielamowicz, K.; Kalra, M.; Landi, D.; Robertson, C.; Gray, T.L.; Diouf, O.; Wakefield, A.; et al. HER2-Specific Chimeric Antigen Receptor-Modified Virus-Specific T Cells for Progressive Glioblastoma: A Phase 1 Dose-Escalation Trial. JAMA Oncol. 2017, 3, 1094–1101. [Google Scholar] [CrossRef]
- Ahmed, N.; Brawley, V.S.; Hegde, M.; Robertson, C.; Ghazi, A.; Gerken, C.; Liu, E.; Dakhova, O.; Ashoori, A.; Corder, A.; et al. Human Epidermal Growth Factor Receptor 2 (HER2) -Specific Chimeric Antigen Receptor-Modified T Cells for the Immunotherapy of HER2-Positive Sarcoma. J. Clin. Oncol. 2015, 33, 1688–1696. [Google Scholar] [CrossRef]
- Feng, K.; Liu, Y.; Guo, Y.; Qiu, J.; Wu, Z.; Dai, H.; Yang, Q.; Wang, Y.; Han, W. Phase I study of chimeric antigen receptor modified T cells in treating HER2-positive advanced biliary tract cancers and pancreatic cancers. Protein Cell 2017, 9, 838–847. [Google Scholar] [CrossRef]
- Kawakami, M.; Leland, P.; Puri, R. Mutation and functional analysis of IL-13 receptors in human malignant glioma cells. Oncol. Res. Featur. Preclin. Clin. Cancer Ther. 2001, 12, 459–467. [Google Scholar] [CrossRef]
- Brown, C.E.; Badie, B.; Barish, M.E.; Weng, L.; Ostberg, J.R.; Chang, W.C.; Naranjo, A.; Starr, R.; Wagner, J.; Wright, C.; et al. Bioactivity and Safety of IL13Ralpha2-Redirected Chimeric Antigen Receptor CD8+ T Cells in Patients with Recurrent Glioblastoma. Clin. Cancer Res. 2015, 21, 4062–4072. [Google Scholar] [CrossRef]
- Brown, C.E.; Alizadeh, D.; Starr, R.; Weng, L.; Wagner, J.R.; Naranjo, A.; Ostberg, J.R.; Blanchard, M.S.; Kilpatrick, J.; Simpson, J.; et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N. Engl. J. Med. 2016, 375, 2561–2569. [Google Scholar] [CrossRef] [PubMed]
- Rosewell Shaw, A.; Porter, C.E.; Watanabe, N.; Tanoue, K.; Sikora, A.; Gottschalk, S.; Brenner, M.K.; Suzuki, M. Adenovirotherapy Delivering Cytokine and Checkpoint Inhibitor Augments CAR T Cells against Metastatic Head and Neck Cancer. Mol. Ther. 2017, 25, 2440–2451. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.-H.; Ding, Y.-M.; Guo, W.; Huang, J.-W.; Yang, Z.; Zhang, Y.; Chen, X.-H. The functional verification of EGFR-CAR T-cells targeted to hypopharyngeal squamous cell carcinoma. OncoTargets Ther. 2018, 11, 7053–7059. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.P.; Jin, L.; Bennett, K.B.; Wang, D.; Fredenburg, K.M.; Tseng, J.E.; Chang, L.-J.; Huang, J.; Chan, E.K. CD70 as a target for chimeric antigen receptor T cells in head and neck squamous cell carcinoma. Oral Oncol. 2018, 78, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Mei, Z.; Zhang, K.; Lam, A.K.; Huang, J.; Qiu, F.; Qiao, B.; Zhang, Y. MUC1 as a target for CAR-T therapy in head and neck squamous cell carinoma. Cancer Med. 2019, 9, 640–652. [Google Scholar] [CrossRef]
- Haist, C.; Schulte, E.; Bartels, N.; Bister, A.; Poschinski, Z.; Ibach, T.C.; Geipel, K.; Wiek, C.; Wagenmann, M.; Monzel, C.; et al. CD44v6-targeted CAR T-cells specifically eliminate CD44 isoform 6 expressing head/neck squamous cell carcinoma cells. Oral Oncol. 2021, 116, 105259. [Google Scholar] [CrossRef]
- Scribner, J.A.; Brown, J.G.; Son, T.; Chiechi, M.; Li, P.; Sharma, S.; Li, H.; De Costa, A.; Li, Y.; Chen, Y.; et al. Preclinical Development of MGC018, a Duocarmycin-based Antibody-drug Conjugate Targeting B7-H3 for Solid Cancer. Mol. Cancer Ther. 2020, 19, 2235–2244. [Google Scholar] [CrossRef]
- Arndt, C.; Loureiro, L.R.; Feldmann, A.; Jureczek, J.; Bergmann, R.; Máthé, D.; Hegedüs, N.; Berndt, N.; Koristka, S.; Mitwasi, N.; et al. UniCAR T cell immunotherapy enables efficient elimination of radioresistant cancer cells. Oncoimmunology 2020, 9, 1743036. [Google Scholar] [CrossRef]
- van Schalkwyk, M.C.; Papa, S.E.; Jeannon, J.P.; Urbano, T.G.; Spicer, J.F.; Maher, J. Design of a phase I clinical trial to evaluate intratumoral delivery of ErbB-targeted chimeric antigen receptor T-cells in locally advanced or recurrent head and neck cancer. Hum. Gene Ther. Clin. Dev. 2013, 24, 134–142. [Google Scholar] [CrossRef]
- Papa, S.; Adami, A.; Metoudi, M.; Achkova, D.; van Schalkwyk, M.; Pereira, A.P.; Bosshard-Carter, L.; Whilding, L.; van der Stegen, S.; Davies, D.; et al. A phase I trial of T4 CAR T-cell immunotherapy in head and neck squamous cancer (HNSCC). J. Clin. Oncol. 2018, 36, 3046. [Google Scholar] [CrossRef]
- Kakarla, S.; Chow, K.K.; Mata, M.; Shaffer, D.R.; Song, X.-T.; Wu, M.-F.; Liu, H.; Wang, L.L.; Rowley, D.R.; Pfizenmaier, K.; et al. Antitumor effects of chimeric receptor engineered human T cells directed to tumor stroma. Mol. Ther. 2013, 21, 1611–1620. [Google Scholar] [CrossRef]
- Schuberth, P.C.; Hagedorn, C.; Jensen, S.M.; Gulati, P.; Broek, M.V.D.; Mischo, A.; Soltermann, A.; Jüngel, A.; Belaunzaran, O.M.; Stahel, R.; et al. Treatment of malignant pleural mesothelioma by fibroblast activation protein-specific re-directed T cells. J. Transl. Med. 2013, 11, 187. [Google Scholar] [CrossRef]
- Gulati, P.; Rühl, J.; Kannan, A.; Pircher, M.; Schuberth, P.; Nytko, K.J.; Pruschy, M.; Sulser, S.; Haefner, M.; Jensen, S.; et al. Aberrant Lck Signal via CD28 Costimulation Augments Antigen-Specific Functionality and Tumor Control by Redirected T Cells with PD-1 Blockade in Humanized Mice. Clin. Cancer Res. 2018, 24, 3981–3993. [Google Scholar] [CrossRef]
- Pollock, N.I.; Wang, L.; Wallweber, G.; Gooding, W.E.; Huang, W.; Chenna, A.; Winslow, J.; Sen, M.; DeGrave, K.A.; Li, H.; et al. Increased Expression of HER2, HER3, and HER2:HER3 Heterodimers in HPV-Positive HNSCC Using a Novel Proximity-Based Assay: Implications for Targeted Therapies. Clin. Cancer Res. 2015, 21, 4597–4606. [Google Scholar] [CrossRef]
- Zuo, B.-L.; Yan, B.; Zheng, G.-X.; Xi, W.-J.; Zhang, X.; Yang, A.-G.; Jia, L.-T. Targeting and suppression of HER3-positive breast cancer by T lymphocytes expressing a heregulin chimeric antigen receptor. Cancer Immunol. Immunother. 2017, 67, 393–401. [Google Scholar] [CrossRef]
- Murad, J.M.; Baumeister, S.H.; Werner, L.; Daley, H.; Trébéden-Negre, H.; Reder, J.; Sentman, C.L.; Gilham, D.; Lehmann, F.; Snykers, S.; et al. Manufacturing development and clinical production of NKG2D chimeric antigen receptor-expressing T cells for autologous adoptive cell therapy. Cytotherapy 2018, 20, 952–963. [Google Scholar] [CrossRef]
- Pollock, N.I.; Grandis, J.R. HER2 as a therapeutic target in head and neck squamous cell carcinoma. Clin. Cancer Res. 2015, 21, 526–533. [Google Scholar] [CrossRef]
- Warren, E.A.; Liu, H.C.; Porter, C.E.; Liao, K.S.; Hegde, M.; Yu, W.; Castro, P.D.; Sandulache, V.; Ahmed, N.; Suzuki, M.; et al. Abstract 574: Overexpression of HER2 in head and neck cancer represents a potential target for T cell immunotherapy. Cancer Res. 2019, 79 (Suppl. 13), 574. [Google Scholar] [CrossRef]
- De Meulenaere, A. P5 Retrospective analysis on the expression of CD70 in squamous cell carcinoma of the head and neck (HNSCC) and its relation to patient outcome. Oral Oncol. 2015, 51, e43–e44. [Google Scholar] [CrossRef]
- Huang, T.-Q.; Bi, Y.-N.; Cui, Z.; Guan, J.-P.; Huang, Y.-C. MUC1 confers radioresistance in head and neck squamous cell carcinoma (HNSCC) cells. Bioengineered 2020, 11, 769–778. [Google Scholar] [CrossRef]
- Feng, R.; Chen, Y.; Liu, Y.; Zhou, Q.; Zhang, W. The role of B7-H3 in tumors and its potential in clinical application. International Int. Immunopharmacol. 2021, 101, 108153. [Google Scholar] [CrossRef] [PubMed]
- Fu, R.; Li, H.; Li, R.; McGrath, K.; Dotti, G.; Gu, Z. Delivery Techniques for Enhancing CAR T Cell Therapy against Solid Tumors. Adv. Funct. Mater. 2021, 31, 2009489. [Google Scholar] [CrossRef]
- Ye, B.; Stary, C.M.; Li, X.; Gao, Q.; Kang, C.; Xiong, X. Engineering chimeric antigen receptor-T cells for cancer treatment. Mol. Cancer 2018, 17, 32. [Google Scholar] [CrossRef] [PubMed]
- Zhang, E.; Gu, J.; Xu, H. Prospects for chimeric antigen receptor-modified T cell therapy for solid tumors. Mol. Cancer 2018, 17, 7. [Google Scholar] [CrossRef] [PubMed]
- Beatty, G.L.; O’Hara, M. Chimeric antigen receptor-modified T cells for the treatment of solid tumors: Defining the challenges and next steps. Pharmacol. Ther. 2016, 166, 30–39. [Google Scholar] [CrossRef]
- Craddock, J.A.; Lu, A.; Bear, A.; Pule, M.; Brenner, M.K.; Rooney, C.M.; Foster, A.E. Enhanced tumor trafficking of GD2 chimeric antigen receptor T cells by expression of the chemokine receptor CCR2b. J. Immunother. 2010, 33, 780–788. [Google Scholar] [CrossRef]
- Fucà, G.; Reppel, L.; Landoni, E.; Savoldo, B.; Dotti, G. Enhancing Chimeric Antigen Receptor T-Cell Efficacy in Solid Tumors. Clin. Cancer Res. 2020, 26, 2444–2451. [Google Scholar] [CrossRef]
- Cui, J.; Zhang, Q.; Song, Q.; Wang, H.; Dmitriev, P.; Sun, M.Y.; Cao, X.; Wang, Y.; Guo, L.; Indig, I.H.; et al. Targeting hypoxia downstream signaling protein, CAIX, for CAR T-cell therapy against glioblastoma. Neuro-Oncol. 2019, 21, 1436–1446. [Google Scholar] [CrossRef]
- Liu, H.; Xu, Y.; Xiang, J.; Long, L.; Green, S.; Yang, Z.; Zimdahl, B.; Lu, J.; Cheng, N.; Horan, L.H.; et al. Targeting Alpha-Fetoprotein (AFP)-MHC Complex with CAR T-Cell Therapy for Liver Cancer. Clin. Cancer Res. 2017, 23, 478–488. [Google Scholar] [CrossRef]
- Tchou, J.; Zhao, Y.; Levine, B.L.; Zhang, P.J.; Davis, M.M.; Melenhorst, J.J.; Kulikovskaya, I.; Brennan, A.L.; Liu, X.; Lacey, S.F.; et al. Safety and Efficacy of Intratumoral Injections of Chimeric Antigen Receptor (CAR) T Cells in Metastatic Breast Cancer. Cancer Immunol. Res. 2017, 5, 1152–1161. [Google Scholar] [CrossRef]
- Van der Stegen, S.J.; Davies, D.M.; Wilkie, S.; Foster, J.; Sosabowski, J.K.; Burnet, J.; Whilding, L.M.; Petrovic, R.M.; Ghaem-Maghami, S.; Mather, S.; et al. Preclinical in vivo modeling of cytokine release syndrome induced by ErbB-retargeted human T cells: Identifying a window of therapeutic opportunity? J. Immunol. 2013, 191, 4589–4598. [Google Scholar] [CrossRef]
- Stephan, S.B.; Taber, A.M.; Jileaeva, I.; Pegues, E.P.; Sentman, C.L.; Stephan, M.T. Biopolymer implants enhance the efficacy of adoptive T-cell therapy. Nat. Biotechnol. 2014, 33, 97–101. [Google Scholar] [CrossRef]
- Hu, Q.; Li, H.; Archibong, E.; Chen, Q.; Ruan, H.; Ahn, S.; Dukhovlinova, E.; Kang, Y.; Wen, D.; Dotti, G.; et al. Inhibition of post-surgery tumour recurrence via a hydrogel releasing CAR-T cells and anti-PDL1-conjugated platelets. Nat. Biomed. Eng. 2021, 5, 1038–1047. [Google Scholar] [CrossRef]
- Coon, M.E.; Stephan, S.B.; Gupta, V.; Kealey, C.P.; Stephan, M.T. Nitinol thin films functionalized with CAR-T cells for the treatment of solid tumours. Nat. Biomed. Eng. 2019, 4, 195–206. [Google Scholar] [CrossRef]
- Chen, Q.; Hu, Q.; Dukhovlinova, E.; Chen, G.; Ahn, S.; Wang, C.; Ogunnaike, E.A.; Ligler, F.S.; Dotti, G.; Gu, Z. Photothermal Therapy Promotes Tumor Infiltration and Antitumor Activity of CAR T Cells. Adv. Mater. 2019, 31, e1900192. [Google Scholar] [CrossRef]
- Masola, V.; Zaza, G.; Gambaro, G.; Franchi, M.; Onisto, M. Role of heparanase in tumor progression: Molecular aspects and therapeutic options. Semin. Cancer Biol. 2019, 62, 86–98. [Google Scholar] [CrossRef]
- Caruana, I.; Savoldo, B.; Hoyos, V.; Weber, G.; Liu, H.; Kim, E.S.; Ittmann, M.M.; Marchetti, D.; Dotti, G. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat. Med. 2015, 21, 524–529. [Google Scholar] [CrossRef]
- Ossche, V.V.D.; Zaryouh, H.; Vara-Messler, M.; Vignau, J.; Machiels, J.-P.; Wouters, A.; Schmitz, S.; Corbet, C. Microenvironment-driven intratumoral heterogeneity in head and neck cancers: Clinical challenges and opportunities for precision medicine. Drug Resist. Updat. 2022, 60, 100806. [Google Scholar]
- Hegde, M.; Mukherjee, M.; Grada, Z.; Pignata, A.; Landi, D.; Navai, S.A.; Wakefield, A.; Fousek, K.; Bielamowicz, K.; Chow, K.K.; et al. Tandem CAR T cells targeting HER2 and IL13Ralpha2 mitigate tumor antigen escape. J. Clin. Investig. 2016, 126, 3036–3052. [Google Scholar] [CrossRef]
- Cho, J.H.; Collins, J.J.; Wong, W.W. Universal Chimeric Antigen Receptors for Multiplexed and Logical Control of T Cell Responses. Cell 2018, 173, 1426–1438.e11. [Google Scholar] [CrossRef]
- Harrington, K.; Freeman, D.J.; Kelly, B.; Harper, J.; Soria, J.-C. Optimizing oncolytic virotherapy in cancer treatment. Nat. Rev. Drug Discov. 2019, 18, 689–706. [Google Scholar] [CrossRef] [PubMed]
- Park, A.K.; Fong, Y.; Kim, S.-I.; Yang, J.; Murad, J.P.; Lu, J.; Jeang, B.; Chang, W.-C.; Chen, N.G.; Thomas, S.H.; et al. Effective combination immunotherapy using oncolytic viruses to deliver CAR targets to solid tumors. Sci. Transl. Med. 2020, 12, eaaz1863. [Google Scholar] [CrossRef] [PubMed]
- Kagoya, Y.; Tanaka, S.; Guo, T.; Anczurowski, M.; Wang, C.-H.; Saso, K.; Butler, M.O.; Minden, M.D.; Hirano, N. A novel chimeric antigen receptor containing a JAK-STAT signaling domain mediates superior antitumor effects. Nat. Med. 2018, 24, 352–359. [Google Scholar] [CrossRef] [PubMed]
- Khouzam, R.A.; Brodaczewska, K.; Filipiak, A.; Zeinelabdin, N.A.; Buart, S.; Szczylik, C.; Kieda, C.; Chouaib, S. Tumor Hypoxia Regulates Immune Escape/Invasion: Influence on Angiogenesis and Potential Impact of Hypoxic Biomarkers on Cancer Therapies. Front. Immunol. 2021, 11, 613114. [Google Scholar] [CrossRef] [PubMed]
- Postow, M.A.; Callahan, M.K.; Wolchok, J.D. Immune Checkpoint Blockade in Cancer Therapy. J. Clin. Oncol. 2015, 33, 1974–1982. [Google Scholar] [CrossRef]
- Toor, S.M.; Nair, V.S.; Decock, J.; Elkord, E. Immune checkpoints in the tumor microenvironment. Semin. Cancer Biol. 2019, 65, 1–12. [Google Scholar] [CrossRef]
- Yeku, O.O.; Purdon, T.J.; Koneru, M.; Spriggs, D.; Brentjens, R.J. Armored CAR T cells enhance antitumor efficacy and overcome the tumor microenvironment. Sci. Rep. 2017, 7, 10541. [Google Scholar] [CrossRef]
- Adachi, K.; Kano, Y.; Nagai, T.; Okuyama, N.; Sakoda, Y.; Tamada, K. IL-7 and CCL19 expression in CAR-T cells improves immune cell infiltration and CAR-T cell survival in the tumor. Nat. Biotechnol. 2018, 36, 346–351. [Google Scholar] [CrossRef]
- Chmielewski, M.; Abken, H. CAR T Cells Releasing IL-18 Convert to T-Bet(high) FoxO1(low) Effectors that Exhibit Augmented Activity against Advanced Solid Tumors. Cell Rep. 2017, 21, 3205–3219. [Google Scholar] [CrossRef]
- Giuffrida, L.; Sek, K.; Henderson, M.A.; House, I.G.; Lai, J.; Chen, A.X.; Todd, K.L.; Petley, E.V.; Mardiana, S.; Todorovski, I.; et al. IL-15 Preconditioning Augments CAR T Cell Responses to Checkpoint Blockade for Improved Treatment of Solid Tumors. Mol. Ther. 2020, 28, 2379–2393. [Google Scholar] [CrossRef]
- Ma, X.; Shou, P.; Smith, C.; Chen, Y.; Du, H.; Sun, C.; Kren, N.P.; Michaud, D.; Ahn, S.; Vincent, B.; et al. Interleukin-23 engineering improves CAR T cell function in solid tumors. Nat. Biotechnol. 2020, 38, 448–459. [Google Scholar] [CrossRef]
- Huang, B.; Luo, L.; Wang, J.; He, B.; Feng, R.; Xian, N.; Zhang, Q.; Chen, L.; Huang, G. B7-H3 specific T cells with chimeric antigen receptor and decoy PD-1 receptors eradicate established solid human tumors in mouse models. Oncoimmunology 2019, 9, 1684127. [Google Scholar] [CrossRef]
- Zou, F.; Lu, L.; Liu, J.; Xia, B.; Zhang, W.; Hu, Q.; Liu, W.; Zhang, Y.; Lin, Y.; Jing, S.; et al. Engineered triple inhibitory receptor resistance improves anti-tumor CAR-T cell performance via CD56. Nat. Commun. 2019, 10, 4109. [Google Scholar] [CrossRef]
- Wagner, D.L.; Fritsche, E.; Pulsipher, M.A.; Ahmed, N.; Hamieh, M.; Hegde, M.; Ruella, M.; Savoldo, B.; Shah, N.N.; Turtle, C.J.; et al. Immunogenicity of CAR T cells in cancer therapy. Nat. Rev. Clin. Oncol. 2021, 18, 379–393. [Google Scholar] [CrossRef]
- Turtle, C.J.; Hanafi, L.-A.; Berger, C.; Gooley, T.A.; Cherian, S.; Hudecek, M.; Sommermeyer, D.; Melville, K.; Pender, B.; Budiarto, T.M.; et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J. Clin. Investig. 2016, 126, 2123–2138. [Google Scholar] [CrossRef]
- Hege, K.M.; Bergsland, E.K.; Fisher, G.A.; Nemunaitis, J.J.; Warren, R.S.; McArthur, J.G.; Lin, A.A.; Schlom, J.; June, C.H.; Sherwin, S.A. Safety, tumor trafficking and immunogenicity of chimeric antigen receptor (CAR)-T cells specific for TAG-72 in colorectal cancer. J. Immunother. Cancer 2017, 5, 22. [Google Scholar] [CrossRef]
- Lanitis, E.; Poussin, M.; Hagemann, I.; Coukos, G.; Sandaltzopoulos, R.; Scholler, N.; Powell, D.J. Redirected antitumor activity of primary human lymphocytes transduced with a fully human anti-mesothelin chimeric receptor. Mol. Ther. 2012, 20, 633–643. [Google Scholar] [CrossRef]
- Lam, N.; Trinklein, N.D.; Buelow, B.; Patterson, G.H.; Ojha, N.; Kochenderfer, J.N. Anti-BCMA chimeric antigen receptors with fully human heavy-chain-only antigen recognition domains. Nat. Commun. 2020, 11, 283. [Google Scholar] [CrossRef]
- Zhao, Z.; Chen, Y.; Francisco, N.M.; Zhang, Y.; Wu, M. The application of CAR-T cell therapy in hematological malignancies: Advantages and challenges. Acta Pharm. Sin. B 2018, 8, 539–551. [Google Scholar] [CrossRef]
- Bonifant, C.L.; Jackson, H.J.; Brentjens, R.J.; Curran, K.J. Toxicity and management in CAR T-cell therapy. Mol. Ther. Oncolytics 2016, 3, 16011. [Google Scholar] [CrossRef]
- Li, J.; Li, W.; Huang, K.; Zhang, Y.; Kupfer, G.; Zhao, Q. Chimeric antigen receptor T cell (CAR-T) immunotherapy for solid tumors: Lessons learned and strategies for moving forward. J. Hematol. Oncol. 2018, 11, 22. [Google Scholar] [CrossRef] [PubMed]
- Kosti, P.; Opzoomer, J.W.; Larios-Martinez, K.I.; Henley-Smith, R.; Scudamore, C.L.; Okesola, M.; Taher, M.Y.; Davies, D.M.; Muliaditan, T.; Larcombe-Young, D.; et al. Hypoxia-sensing CAR T cells provide safety and efficacy in treating solid tumors. Cell Rep. Med. 2021, 2, 100227. [Google Scholar] [CrossRef] [PubMed]
- Choe, J.H.; Watchmaker, P.B.; Simic, M.S.; Gilbert, R.D.; Li, A.W.; Krasnow, N.A.; Downey, K.M.; Yu, W.; Carrera, D.A.; Celli, A.; et al. SynNotch-CAR T cells overcome challenges of specificity, heterogeneity, and persistence in treating glioblastoma. Sci. Transl. Med. 2021, 13, eabe7378. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Han, X.; Bo, J.; Han, W. Target selection for CAR-T therapy. J. Hematol. Oncol. 2019, 12, 62. [Google Scholar] [CrossRef]
- Fedorov, V.D.; Themeli, M.; Sadelain, M. Sadelain, PD-1- and CTLA-4-based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Sci. Transl. Med. 2013, 5, 215ra172. [Google Scholar] [CrossRef]
- Morris, E.C.; Neelapu, S.S.; Giavridis, T.; Sadelain, M. Cytokine release syndrome and associated neurotoxicity in cancer immunotherapy. Nat. Rev. Immunol. 2021, 22, 85–96. [Google Scholar] [CrossRef]
- Lee, D.W.; Gardner, A.R.; Porter, D.L.; Louis, C.U.; Ahmed, N.; Jensen, M.C.; Grupp, S.A.; Mackall, C.L. Current concepts in the diagnosis and management of cytokine release syndrome. Blood 2014, 124, 188–195. [Google Scholar] [CrossRef]
- Huarte, E.; O’Connor, R.S.; Peel, M.T.; Nunez-Cruz, S.; Leferovich, J.; Juvekar, A.; Yang, Y.-O.; Truong, L.; Huang, T.; Naim, A.; et al. Itacitinib (INCB039110), a JAK1 Inhibitor, Reduces Cytokines Associated with Cytokine Release Syndrome Induced by CAR T-cell Therapy. Clin. Cancer Res. 2020, 26, 6299–6309. [Google Scholar] [CrossRef]
- Gargett, T.; Brown, M.P. The inducible caspase-9 suicide gene system as a safety switch to limit on-target, off-tumor toxicities of chimeric antigen receptor T cells. Front. Pharm. 2014, 5, 235. [Google Scholar] [CrossRef]
- Diaconu, I.; Ballard, B.; Zhang, M.; Chen, Y.; West, J.; Dotti, G.; Savoldo, B. Inducible Caspase-9 Selectively Modulates the Toxicities of CD19-Specific Chimeric Antigen Receptor-Modified T Cells. Mol. Ther. 2017, 25, 580–592. [Google Scholar] [CrossRef]
- Wei, Y.; Li, C.; Bian, H.; Qian, W.; Jin, K.; Xu, T.; Guo, X.; Lu, X.; Su, F. Targeting CDK7 suppresses super enhancer-linked inflammatory genes and alleviates CAR T cell-induced cytokine release syndrome. Mol. Cancer 2021, 20, 5. [Google Scholar] [CrossRef]
- Zou, S.; Tong, Q.; Liu, B.; Huang, W.; Tian, Y.; Fu, X. Targeting STAT3 in Cancer Immunotherapy. Mol. Cancer 2020, 19, 145. [Google Scholar] [CrossRef]
- Kotch, C.; Barrett, D.; Teachey, D.T. Teachey, Tocilizumab for the treatment of chimeric antigen receptor T cell-induced cytokine release syndrome. Expert Rev. Clin. Immunol. 2019, 15, 813–822. [Google Scholar] [CrossRef]
- Schoenfeld, J.D. Immunity in head and neck cancer. Cancer Immunol. Res. 2015, 3, 12–17. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Target | Introduction | Reference |
|---|---|---|
| Preclinical study | ||
| HER2 | CD28. CD3-ζ/CAd | [42] |
| EGFR | - | [43] |
| CD70 | 4-1BB.CD3-ζ | [44] |
| MUCI | 4-1BB.CD3-ζ/IL-12 | [45] |
| CD44v6 | CD28. CD3-ζ | [46] |
| B7-H3 | - | [47] |
| CD98 + EGFR | UniCAR-T | [48] |
| Clinical trial | ||
| ERBb2/HER2 | NCT01818323 NCT03740256 | [49,50] |
| EpCAM | NCT02915445 | |
| NKG2DL | NCT04107142 | |
| LMP1 | NCT02980315 | |
| Potential target | ||
| FAP | Overexpression of HNSCC cells compared to adjacent tissue controls (p < 0.0005); FAP-targeting CAR-T cells have been used in treating other solid tumors | [44,51,52,53] |
| HER3 | Overexpression of HPV-positive HNSCC cells compared to HPV-negative HNSCC cells (p = 0.0007); HER3-targeting CAR-T cells have been used in treating mice bearing breast tumor cells | [54,55] |
| NKGD2 | Overexpression of MICA and MICB in HNSCC cells compared to adjacent tissue controls (p < 0.0005); NKGD2-targeting CAR-T cells for treatment of acute myeloid leukemia and multiple myeloma have been used in clinical trial | [44,56] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, H.-Q.; Fu, R.; Man, Q.-W.; Yang, G.; Liu, B.; Bu, L.-L. Advances in CAR-T Cell Therapy in Head and Neck Squamous Cell Carcinoma. J. Clin. Med. 2023, 12, 2173. https://doi.org/10.3390/jcm12062173
Wang H-Q, Fu R, Man Q-W, Yang G, Liu B, Bu L-L. Advances in CAR-T Cell Therapy in Head and Neck Squamous Cell Carcinoma. Journal of Clinical Medicine. 2023; 12(6):2173. https://doi.org/10.3390/jcm12062173
Chicago/Turabian StyleWang, Han-Qi, Ruxing Fu, Qi-Wen Man, Guang Yang, Bing Liu, and Lin-Lin Bu. 2023. "Advances in CAR-T Cell Therapy in Head and Neck Squamous Cell Carcinoma" Journal of Clinical Medicine 12, no. 6: 2173. https://doi.org/10.3390/jcm12062173
APA StyleWang, H.-Q., Fu, R., Man, Q.-W., Yang, G., Liu, B., & Bu, L.-L. (2023). Advances in CAR-T Cell Therapy in Head and Neck Squamous Cell Carcinoma. Journal of Clinical Medicine, 12(6), 2173. https://doi.org/10.3390/jcm12062173