
The Sodium-Glucose Co-Transporter 2 (SGLT2) Inhibitor Empagliflozin Reverses Hyperglycemia-Induced Monocyte and Endothelial Dysfunction Primarily through Glucose Transport-Independent but Redox-Dependent Mechanisms
 , , , ,
, , , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Primary Human Monocytes and Primary Endothelial Cells Express SGLT-2
2.2. CD14++CD16− Monocytes Exposed to Hyperglycemic Conditions Do Not Exhibit an Enhanced Transmigration Phenotype
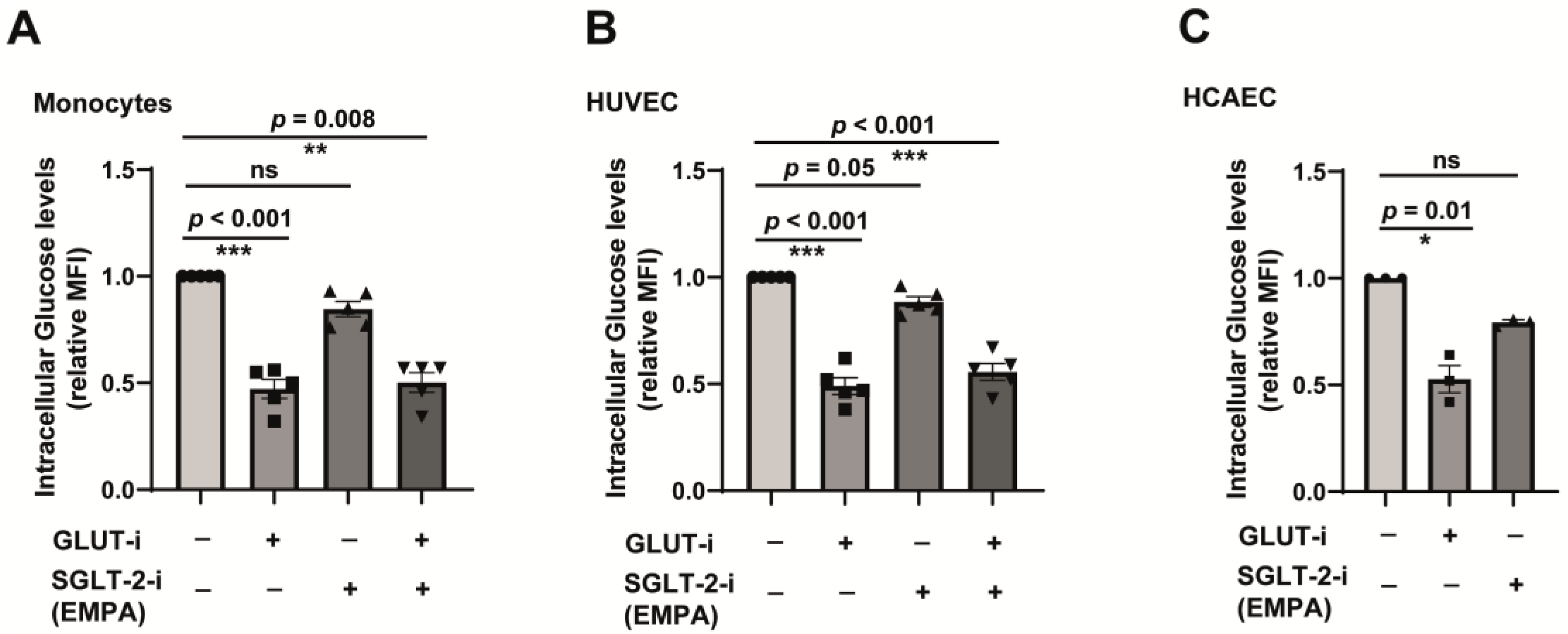
2.3. SGLT-2 Is Weakly Involved in the Glucose Transport in Both Monocytes and Endothelial Cells
2.4. Hyperglycemia-Induced Monocyte Dysfunction Is Oxidative Stress-Dependent, and Empagliflozin Alleviates ROS Accumulation and Reverses Monocyte Dysfunction
2.5. Oxidative Stress-Dependent Impairment of VEGFR-2 Contributes to Endothelial Dysfunction in Hyperglycemia, and Empagliflozin Restores VEGFR-2 to Alleviate Endothelial Dysfunction
2.6. A General Antioxidant Improves Cell Function, Whereas Induction of Oxidative Stress Reverses the Beneficial Effects of Empagliflozin
3. Discussion
4. Materials and Methods
4.1. Monocyte Isolation from Clinical Cohorts and Healthy Individuals
4.2. Human Umbilical Vein Endothelial Cells Isolation and Ethics
4.3. Human Fetoplacental Endothelial Cell Isolation from Clinical Cohorts and Healthy Individuals
4.4. Monocyte, HUVEC, HCAEC and HPEC Culture
4.5. Reagents
4.6. RNA Isolation and qPCR
4.7. Glucose Uptake Assay
4.8. Monocyte Chemotaxis and Chemokinesis Assay
4.9. Chemotaxis Assay of Endothelial Cells
4.10. Intracellular Reactive Oxygen Species Detection
4.11. Detection of the Surface Expression of VEGFR-2
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zinman, B.; Lachin, J.M.; Inzucchi, S.E. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N. Engl. J. Med. 2016, 374, 1094. [Google Scholar] [CrossRef] [PubMed]
- Prevalence of small vessel and large vessel disease in diabetic patients from 14 centres. The World Health Organisation Multinational Study of Vascular Disease in Diabetics. Diabetes Drafting Group. Diabetologia 1985, 28, 615–640. [CrossRef] [PubMed]
- Aronson, D.; Edelman, E.R. Coronary artery disease and diabetes mellitus. Cardiol. Clin. 2014, 32, 439–455. [Google Scholar] [CrossRef]
- Giacco, F.; Brownlee, M. Oxidative stress and diabetic complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef] [PubMed]
- Suryavanshi, S.V.; Kulkarni, Y.A. NF-kappabeta: A Potential Target in the Management of Vascular Complications of Diabetes. Front. Pharmacol. 2017, 8, 798. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Yamamoto, H. RAGE-Mediated Inflammation, Type 2 Diabetes, and Diabetic Vascular Complication. Front. Endocrinol. 2013, 4, 105. [Google Scholar] [CrossRef]
- Tchaikovski, V.; Olieslagers, S.; Bohmer, F.D.; Waltenberger, J. Diabetes mellitus activates signal transduction pathways resulting in vascular endothelial growth factor resistance of human monocytes. Circulation 2009, 120, 150–159. [Google Scholar] [CrossRef]
- Gao, X.; Zhang, H.; Schmidt, A.M.; Zhang, C. AGE/RAGE produces endothelial dysfunction in coronary arterioles in type 2 diabetic mice. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H491–H498. [Google Scholar] [CrossRef]
- Coughlan, M.T.; Thorburn, D.R.; Penfold, S.A.; Laskowski, A.; Harcourt, B.E.; Sourris, K.C.; Tan, A.L.; Fukami, K.; Thallas-Bonke, V.; Nawroth, P.P.; et al. RAGE-induced cytosolic ROS promote mitochondrial superoxide generation in diabetes. J. Am. Soc. Nephrol. 2009, 20, 742–752. [Google Scholar] [CrossRef]
- Wautier, M.P.; Chappey, O.; Corda, S.; Stern, D.M.; Schmidt, A.M.; Wautier, J.L. Activation of NADPH oxidase by AGE links oxidant stress to altered gene expression via RAGE. Am. J. Physiol. Endocrinol. Metab. 2001, 280, E685–E694. [Google Scholar] [CrossRef]
- Rieg, T.; Masuda, T.; Gerasimova, M.; Mayoux, E.; Platt, K.; Powell, D.R.; Thomson, S.C.; Koepsell, H.; Vallon, V. Increase in SGLT1-mediated transport explains renal glucose reabsorption during genetic and pharmacological SGLT2 inhibition in euglycemia. Am. J. Physiol. Ren. Physiol. 2014, 306, F188–F193. [Google Scholar] [CrossRef]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef]
- Packer, M.; Anker, S.D.; Butler, J.; Filippatos, G.; Pocock, S.J.; Carson, P.; Januzzi, J.; Verma, S.; Tsutsui, H.; Brueckmann, M.; et al. Cardiovascular and Renal Outcomes with Empagliflozin in Heart Failure. N. Engl. J. Med. 2020, 383, 1413–1424. [Google Scholar] [CrossRef]
- Zannad, F.; Ferreira, J.P.; Pocock, S.J.; Anker, S.D.; Butler, J.; Filippatos, G.; Brueckmann, M.; Ofstad, A.P.; Pfarr, E.; Jamal, W.; et al. SGLT2 inhibitors in patients with heart failure with reduced ejection fraction: A meta-analysis of the EMPEROR-Reduced and DAPA-HF trials. Lancet 2020, 396, 819–829. [Google Scholar] [CrossRef]
- Fitchett, D.; Zinman, B.; Wanner, C.; Lachin, J.M.; Hantel, S.; Salsali, A.; Johansen, O.E.; Woerle, H.J.; Broedl, U.C.; Inzucchi, S.E.; et al. Heart failure outcomes with empagliflozin in patients with type 2 diabetes at high cardiovascular risk: Results of the EMPA-REG OUTCOME(R) trial. Eur. Heart J. 2016, 37, 1526–1534. [Google Scholar] [CrossRef]
- Han, J.H.; Oh, T.J.; Lee, G.; Maeng, H.J.; Lee, D.H.; Kim, K.M.; Choi, S.H.; Jang, H.C.; Lee, H.S.; Park, K.S.; et al. The beneficial effects of empagliflozin, an SGLT2 inhibitor, on atherosclerosis in ApoE (-/-) mice fed a western diet. Diabetologia 2017, 60, 364–376. [Google Scholar] [CrossRef]
- Byrne, N.J.; Parajuli, N.; Levasseur, J.L.; Boisvenue, J.; Beker, D.L.; Masson, G.; Fedak, P.W.M.; Verma, S.; Dyck, J.R.B. Empagliflozin Prevents Worsening of Cardiac Function in an Experimental Model of Pressure Overload-Induced Heart Failure. JACC Basic Transl. Sci. 2017, 2, 347–354. [Google Scholar] [CrossRef]
- Park, S.H.; Farooq, M.A.; Gaertner, S.; Bruckert, C.; Qureshi, A.W.; Lee, H.H.; Benrahla, D.; Pollet, B.; Stephan, D.; Ohlmann, P.; et al. Empagliflozin improved systolic blood pressure, endothelial dysfunction and heart remodeling in the metabolic syndrome ZSF1 rat. Cardiovasc. Diabetol. 2020, 19, 19. [Google Scholar] [CrossRef]
- Andreadou, I.; Efentakis, P.; Balafas, E.; Togliatto, G.; Davos, C.H.; Varela, A.; Dimitriou, C.A.; Nikolaou, P.E.; Maratou, E.; Lambadiari, V.; et al. Empagliflozin Limits Myocardial Infarction in Vivo and Cell Death in Vitro: Role of STAT3, Mitochondria, and Redox Aspects. Front. Physiol. 2017, 8, 1077. [Google Scholar] [CrossRef]
- Patel, D.K.; Strong, J. The Pleiotropic Effects of Sodium-Glucose Cotransporter-2 Inhibitors: Beyond the Glycemic Benefit. Diabetes Ther. 2019, 10, 1771–1792. [Google Scholar] [CrossRef]
- Satoh, H. Pleiotropic effects of SGLT2 inhibitors beyond the effect on glycemic control. Diabetol. Int. 2018, 9, 212–214. [Google Scholar] [CrossRef] [PubMed]
- Mone, P.; Varzideh, F.; Jankauskas, S.S.; Pansini, A.; Lombardi, A.; Frullone, S.; Santulli, G. SGLT2 Inhibition via Empagliflozin Improves Endothelial Function and Reduces Mitochondrial Oxidative Stress: Insights From Frail Hypertensive and Diabetic Patients. Hypertension 2022, 79, 1633–1643. [Google Scholar] [CrossRef] [PubMed]
- Uthman, L.; Homayr, A.; Juni, R.P.; Spin, E.L.; Kerindongo, R.; Boomsma, M.; Hollmann, M.W.; Preckel, B.; Koolwijk, P.; van Hinsbergh, V.W.M.; et al. Empagliflozin and Dapagliflozin Reduce ROS Generation and Restore NO Bioavailability in Tumor Necrosis Factor alpha-Stimulated Human Coronary Arterial Endothelial Cells. Cell Physiol. Biochem. 2019, 53, 865–886. [Google Scholar] [CrossRef]
- Hemling, P.; Zibrova, D.; Strutz, J.; Sohrabi, Y.; Desoye, G.; Schulten, H.; Findeisen, H.; Heller, R.; Godfrey, R.; Waltenberger, J. Hyperglycemia-induced endothelial dysfunction is alleviated by thioredoxin mimetic peptides through the restoration of VEGFR-2-induced responses and improved cell survival. Int. J. Cardiol. 2020, 308, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Avogaro, A.; Fadini, G.P.; Gallo, A.; Pagnin, E.; de Kreutzenberg, S. Endothelial dysfunction in type 2 diabetes mellitus. Nutr. Metab. Cardiovasc. Dis. 2006, 16 (Suppl. S1), S39–S45. [Google Scholar] [CrossRef]
- Waltenberger, J.; Lange, J.; Kranz, A. Vascular endothelial growth factor-A-induced chemotaxis of monocytes is attenuated in patients with diabetes mellitus: A potential predictor for the individual capacity to develop collaterals. Circulation 2000, 102, 185–190. [Google Scholar] [CrossRef]
- Oelze, M.; Kroller-Schon, S.; Welschof, P.; Jansen, T.; Hausding, M.; Mikhed, Y.; Stamm, P.; Mader, M.; Zinssius, E.; Agdauletova, S.; et al. The sodium-glucose co-transporter 2 inhibitor empagliflozin improves diabetes-induced vascular dysfunction in the streptozotocin diabetes rat model by interfering with oxidative stress and glucotoxicity. PLoS ONE 2014, 9, e112394. [Google Scholar] [CrossRef]
- Steven, S.; Oelze, M.; Hanf, A.; Kroller-Schon, S.; Kashani, F.; Roohani, S.; Welschof, P.; Kopp, M.; Godtel-Armbrust, U.; Xia, N.; et al. The SGLT2 inhibitor empagliflozin improves the primary diabetic complications in ZDF rats. Redox Biol. 2017, 13, 370–385. [Google Scholar] [CrossRef]
- Werner, G.S.; Richartz, B.M.; Heinke, S.; Ferrari, M.; Figulla, H.R. Impaired acute collateral recruitment as a possible mechanism for increased cardiac adverse events in patients with diabetes mellitus. Eur. Heart J. 2003, 24, 1134–1142. [Google Scholar] [CrossRef]
- Waltenberger, J. Impaired collateral vessel development in diabetes: Potential cellular mechanisms and therapeutic implications. Cardiovasc. Res. 2001, 49, 554–560. [Google Scholar] [CrossRef]
- Warren, C.M.; Ziyad, S.; Briot, A.; Der, A.; Iruela-Arispe, M.L. A ligand-independent VEGFR2 signaling pathway limits angiogenic responses in diabetes. Sci. Signal. 2014, 7, ra1. [Google Scholar] [CrossRef]
- Sasso, F.C.; Torella, D.; Carbonara, O.; Ellison, G.M.; Torella, M.; Scardone, M.; Marra, C.; Nasti, R.; Marfella, R.; Cozzolino, D.; et al. Increased vascular endothelial growth factor expression but impaired vascular endothelial growth factor receptor signaling in the myocardium of type 2 diabetic patients with chronic coronary heart disease. J. Am. Coll. Cardiol. 2005, 46, 827–834. [Google Scholar] [CrossRef]
- Dorenkamp, M.; Muller, J.P.; Shanmuganathan, K.S.; Schulten, H.; Muller, N.; Loffler, I.; Muller, U.A.; Wolf, G.; Bohmer, F.D.; Godfrey, R.; et al. Hyperglycaemia-induced methylglyoxal accumulation potentiates VEGF resistance of diabetic monocytes through the aberrant activation of tyrosine phosphatase SHP-2/SRC kinase signalling axis. Sci. Rep. 2018, 8, 14684. [Google Scholar] [CrossRef]
- Fadini, G.P.; Albiero, M.; Bonora, B.M.; Avogaro, A. Angiogenic Abnormalities in Diabetes Mellitus: Mechanistic and Clinical Aspects. J. Clin. Endocrinol. Metab. 2019, 104, 5431–5444. [Google Scholar] [CrossRef]
- Ruiter, M.S.; van Golde, J.M.; Schaper, N.C.; Stehouwer, C.D.; Huijberts, M.S. Diabetes impairs arteriogenesis in the peripheral circulation: Review of molecular mechanisms. Clin. Sci. 2010, 119, 225–238. [Google Scholar] [CrossRef]
- Waltenberger, J. VEGF resistance as a molecular basis to explain the angiogenesis paradox in diabetes mellitus. Biochem. Soc. Trans. 2009, 37, 1167–1170. [Google Scholar] [CrossRef]
- Waltenberger, J. Stress testing at the cellular and molecular level to unravel cellular dysfunction and growth factor signal transduction defects: What Molecular Cell Biology can learn from Cardiology. Thromb. Haemost. 2007, 98, 975–979. [Google Scholar] [CrossRef]
- Chen, J.; Williams, S.; Ho, S.; Loraine, H.; Hagan, D.; Whaley, J.M.; Feder, J.N. Quantitative PCR tissue expression profiling of the human SGLT2 gene and related family members. Diabetes Ther. 2010, 1, 57–92. [Google Scholar] [CrossRef]
- Rahmoune, H.; Thompson, P.W.; Ward, J.M.; Smith, C.D.; Hong, G.; Brown, J. Glucose transporters in human renal proximal tubular cells isolated from the urine of patients with non-insulin-dependent diabetes. Diabetes 2005, 54, 3427–3434. [Google Scholar] [CrossRef]
- Dimitriadis, G.; Maratou, E.; Boutati, E.; Psarra, K.; Papasteriades, C.; Raptis, S.A. Evaluation of glucose transport and its regulation by insulin in human monocytes using flow cytometry. Cytom. A 2005, 64, 27–33. [Google Scholar] [CrossRef]
- Tumova, S.; Kerimi, A.; Porter, K.E.; Williamson, G. Transendothelial glucose transport is not restricted by extracellular hyperglycaemia. Vascul. Pharmacol. 2016, 87, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Dludla, P.V.; Dias, S.C.; Obonye, N.; Johnson, R.; Louw, J.; Nkambule, B.B. A Systematic Review on the Protective Effect of N-Acetyl Cysteine Against Diabetes-Associated Cardiovascular Complications. Am. J. Cardiovasc. Drugs 2018, 18, 283–298. [Google Scholar] [CrossRef] [PubMed]
- Juni, R.P.; Al-Shama, R.; Kuster, D.W.D.; van der Velden, J.; Hamer, H.M.; Vervloet, M.G.; Eringa, E.C.; Koolwijk, P.; van Hinsbergh, V.W.M. Empagliflozin restores chronic kidney disease-induced impairment of endothelial regulation of cardiomyocyte relaxation and contraction. Kidney Int. 2021, 99, 1088–1101. [Google Scholar] [CrossRef] [PubMed]
- Sukhanov, S.; Higashi, Y.; Yoshida, T.; Mummidi, S.; Aroor, A.R.; Jeffrey Russell, J.; Bender, S.B.; DeMarco, V.G.; Chandrasekar, B. The SGLT2 inhibitor Empagliflozin attenuates interleukin-17A-induced human aortic smooth muscle cell proliferation and migration by targeting TRAF3IP2/ROS/NLRP3/Caspase-1-dependent IL-1beta and IL-18 secretion. Cell Signal 2021, 77, 109825. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Belcastro, E.; Hasan, H.; Matsushita, K.; Marchandot, B.; Abbas, M.; Toti, F.; Auger, C.; Jesel, L.; Ohlmann, P.; et al. Angiotensin II-induced upregulation of SGLT1 and 2 contributes to human microparticle-stimulated endothelial senescence and dysfunction: Protective effect of gliflozins. Cardiovasc. Diabetol. 2021, 20, 65. [Google Scholar] [CrossRef]
- Albertoni Borghese, M.F.; Majowicz, M.P.; Ortiz, M.C.; Passalacqua Mdel, R.; Sterin Speziale, N.B.; Vidal, N.A. Expression and activity of SGLT2 in diabetes induced by streptozotocin: Relationship with the lipid environment. Nephron. Physiol. 2009, 112, p45–p52. [Google Scholar] [CrossRef]
- Umino, H.; Hasegawa, K.; Minakuchi, H.; Muraoka, H.; Kawaguchi, T.; Kanda, T.; Tokuyama, H.; Wakino, S.; Itoh, H. High Basolateral Glucose Increases Sodium-Glucose Cotransporter 2 and Reduces Sirtuin-1 in Renal Tubules through Glucose Transporter-2 Detection. Sci. Rep. 2018, 8, 6791. [Google Scholar] [CrossRef]
- Katsurada, K.; Nandi, S.S.; Sharma, N.M.; Patel, K.P. Enhanced Expression and Function of Renal SGLT2 (Sodium-Glucose Cotransporter 2) in Heart Failure: Role of Renal Nerves. Circ. Heart Fail. 2021, 14, e008365. [Google Scholar] [CrossRef]
- Malide, D.; Davies-Hill, T.M.; Levine, M.; Simpson, I.A. Distinct localization of GLUT-1, -3, and -5 in human monocyte-derived macrophages: Effects of cell activation. Am. J. Physiol. 1998, 274, E516–E526. [Google Scholar] [CrossRef]
- Huang, Y.; Lei, L.; Liu, D.; Jovin, I.; Russell, R.; Johnson, R.S.; Di Lorenzo, A.; Giordano, F.J. Normal glucose uptake in the brain and heart requires an endothelial cell-specific HIF-1alpha-dependent function. Proc. Natl. Acad. Sci. USA 2012, 109, 17478–17483. [Google Scholar] [CrossRef]
- Ganbaatar, B.; Fukuda, D.; Shinohara, M.; Yagi, S.; Kusunose, K.; Yamada, H.; Soeki, T.; Hirata, K.I.; Sata, M. Empagliflozin ameliorates endothelial dysfunction and suppresses atherogenesis in diabetic apolipoprotein E-deficient mice. Eur. J. Pharmacol. 2020, 875, 173040. [Google Scholar] [CrossRef]
- Mucka, S.; Miodonska, M.; Jakubiak, G.K.; Starzak, M.; Cieslar, G.; Stanek, A. Endothelial Function Assessment by Flow-Mediated Dilation Method: A Valuable Tool in the Evaluation of the Cardiovascular System. Int. J. Environ. Res. Public Health 2022, 19, 11242. [Google Scholar] [CrossRef]
- Mone, P.; Lombardi, A.; Kansakar, U.; Varzideh, F.; Jankauskas, S.S.; Pansini, A.; Marzocco, S.; De Gennaro, S.; Famiglietti, M.; Macina, G.; et al. Empagliflozin Improves the MicroRNA Signature of Endothelial Dysfunction in Patients with Heart Failure with Preserved Ejection Fraction and Diabetes. J. Pharmacol. Exp. Ther. 2023, 384, 116–122. [Google Scholar] [CrossRef]
- Cornuault, L.; Rouault, P.; Duplaa, C.; Couffinhal, T.; Renault, M.A. Endothelial Dysfunction in Heart Failure With Preserved Ejection Fraction: What are the Experimental Proofs? Front. Physiol. 2022, 13, 906272. [Google Scholar] [CrossRef]
- Miyosawa, K.; Iwata, H.; Minami-Takano, A.; Hayashi, H.; Tabuchi, H.; Sekita, G.; Kadoguchi, T.; Ishii, K.; Nozaki, Y.; Funamizu, T.; et al. Enhanced monocyte migratory activity in the pathogenesis of structural remodeling in atrial fibrillation. PLoS ONE 2020, 15, e0240540. [Google Scholar] [CrossRef]
- Das, N.A.; Carpenter, A.J.; Belenchia, A.; Aroor, A.R.; Noda, M.; Siebenlist, U.; Chandrasekar, B.; DeMarco, V.G. Empagliflozin reduces high glucose-induced oxidative stress and miR-21-dependent TRAF3IP2 induction and RECK suppression, and inhibits human renal proximal tubular epithelial cell migration and epithelial-to-mesenchymal transition. Cell Signal 2020, 68, 109506. [Google Scholar] [CrossRef]
- Spengler, K.; Kryeziu, N.; Grosse, S.; Mosig, A.S.; Heller, R. VEGF Triggers Transient Induction of Autophagy in Endothelial Cells via AMPKalpha1. Cells 2020, 9, 687. [Google Scholar] [CrossRef]
- Strutz, J.; Cvitic, S.; Hackl, H.; Kashofer, K.; Appel, H.M.; Thuringer, A.; Desoye, G.; Koolwijk, P.; Hiden, U. Gestational diabetes alters microRNA signatures in human feto-placental endothelial cells depending on fetal sex. Clin. Sci. 2018, 132, 2437–2449. [Google Scholar] [CrossRef]
- Tchaikovski, V.; Tchaikovski, S.; Olieslagers, S.; Waltenberger, J. Monocyte dysfunction as a previously unrecognized pathophysiological mechanism in ApoE-/- mice contributing to impaired arteriogenesis. Int. J. Cardiol. 2015, 190, 214–216. [Google Scholar] [CrossRef]
- Godfrey, R.; Arora, D.; Bauer, R.; Stopp, S.; Muller, J.P.; Heinrich, T.; Bohmer, S.A.; Dagnell, M.; Schnetzke, U.; Scholl, S.; et al. Cell transformation by FLT3 ITD in acute myeloid leukemia involves oxidative inactivation of the tumor suppressor protein-tyrosine phosphatase DEP-1/PTPRJ. Blood 2012, 119, 4499–4511. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Semo, D.; Obergassel, J.; Dorenkamp, M.; Hemling, P.; Strutz, J.; Hiden, U.; Müller, N.; Müller, U.A.; Zulfikar, S.A.; Godfrey, R.; et al. The Sodium-Glucose Co-Transporter 2 (SGLT2) Inhibitor Empagliflozin Reverses Hyperglycemia-Induced Monocyte and Endothelial Dysfunction Primarily through Glucose Transport-Independent but Redox-Dependent Mechanisms. J. Clin. Med. 2023, 12, 1356. https://doi.org/10.3390/jcm12041356
Semo D, Obergassel J, Dorenkamp M, Hemling P, Strutz J, Hiden U, Müller N, Müller UA, Zulfikar SA, Godfrey R, et al. The Sodium-Glucose Co-Transporter 2 (SGLT2) Inhibitor Empagliflozin Reverses Hyperglycemia-Induced Monocyte and Endothelial Dysfunction Primarily through Glucose Transport-Independent but Redox-Dependent Mechanisms. Journal of Clinical Medicine. 2023; 12(4):1356. https://doi.org/10.3390/jcm12041356
Chicago/Turabian StyleSemo, Dilvin, Julius Obergassel, Marc Dorenkamp, Pia Hemling, Jasmin Strutz, Ursula Hiden, Nicolle Müller, Ulrich Alfons Müller, Sajan Ahmad Zulfikar, Rinesh Godfrey, and et al. 2023. "The Sodium-Glucose Co-Transporter 2 (SGLT2) Inhibitor Empagliflozin Reverses Hyperglycemia-Induced Monocyte and Endothelial Dysfunction Primarily through Glucose Transport-Independent but Redox-Dependent Mechanisms" Journal of Clinical Medicine 12, no. 4: 1356. https://doi.org/10.3390/jcm12041356
APA StyleSemo, D., Obergassel, J., Dorenkamp, M., Hemling, P., Strutz, J., Hiden, U., Müller, N., Müller, U. A., Zulfikar, S. A., Godfrey, R., & Waltenberger, J. (2023). The Sodium-Glucose Co-Transporter 2 (SGLT2) Inhibitor Empagliflozin Reverses Hyperglycemia-Induced Monocyte and Endothelial Dysfunction Primarily through Glucose Transport-Independent but Redox-Dependent Mechanisms. Journal of Clinical Medicine, 12(4), 1356. https://doi.org/10.3390/jcm12041356