
Anti-dsDNA B-Cell ELISpot as a Monitoring and Flare Prediction Tool in SLE Patients

 , , , , ,
, , , , ,  ,
,
Abstract
1. Introduction
2. Patients and Methods
2.1. Patients Selection and Data Collection
2.2. Blood Samples
2.3. Anti-dsDNA Autoantibodies Detection and Complement Tests
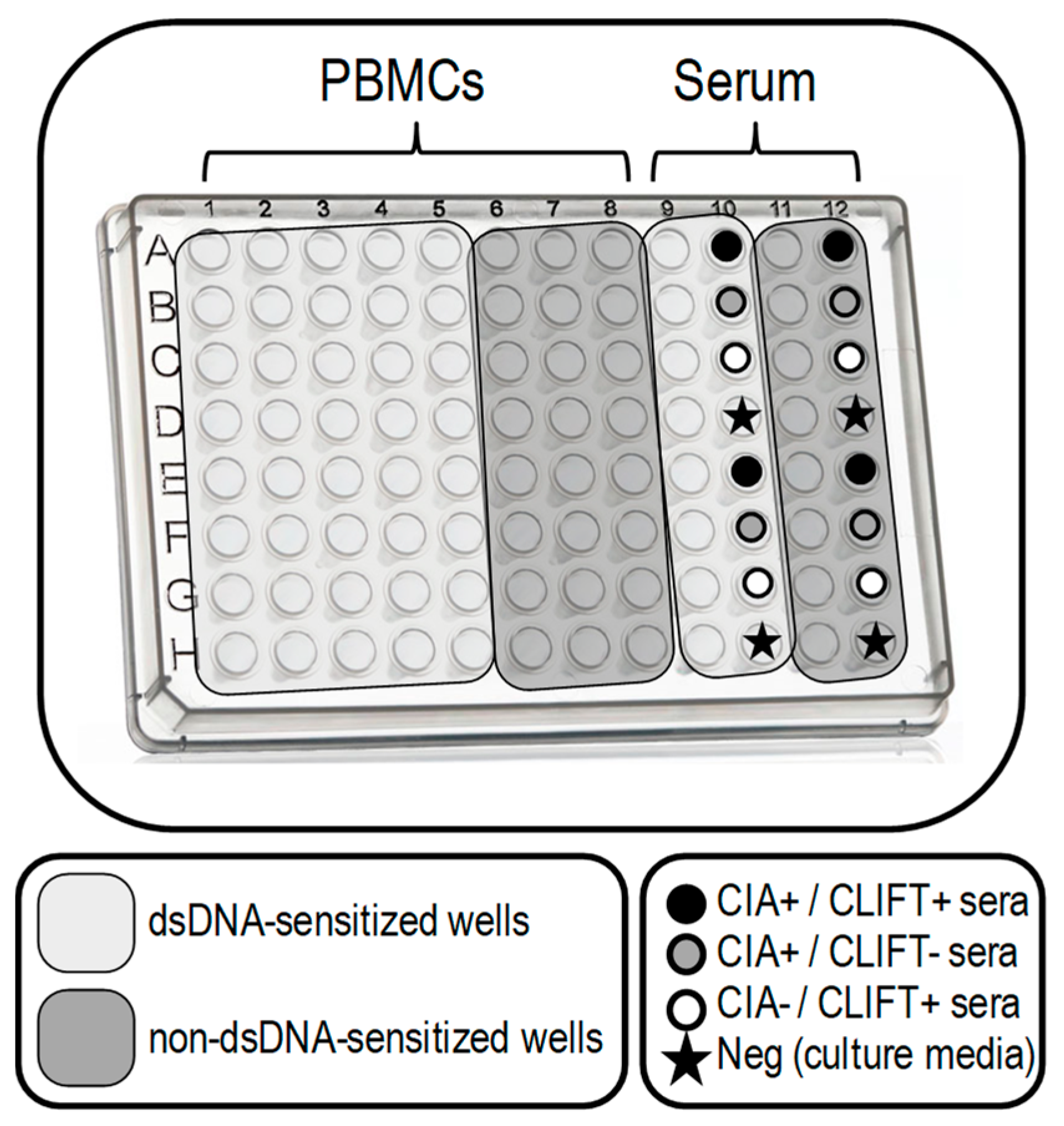
2.4. SLE-ELISpot Assay Conditions Standardization
2.5. SLE-ELISpot Assay
2.6. Statistics
3. Results
3.1. Study Cohort
3.2. SLE-ELISpot Improved Assay
- sens: average number of spots in dsDNA-sensitised wells
- non-sens: average number of spots in non-dsDNA-sensitised wells
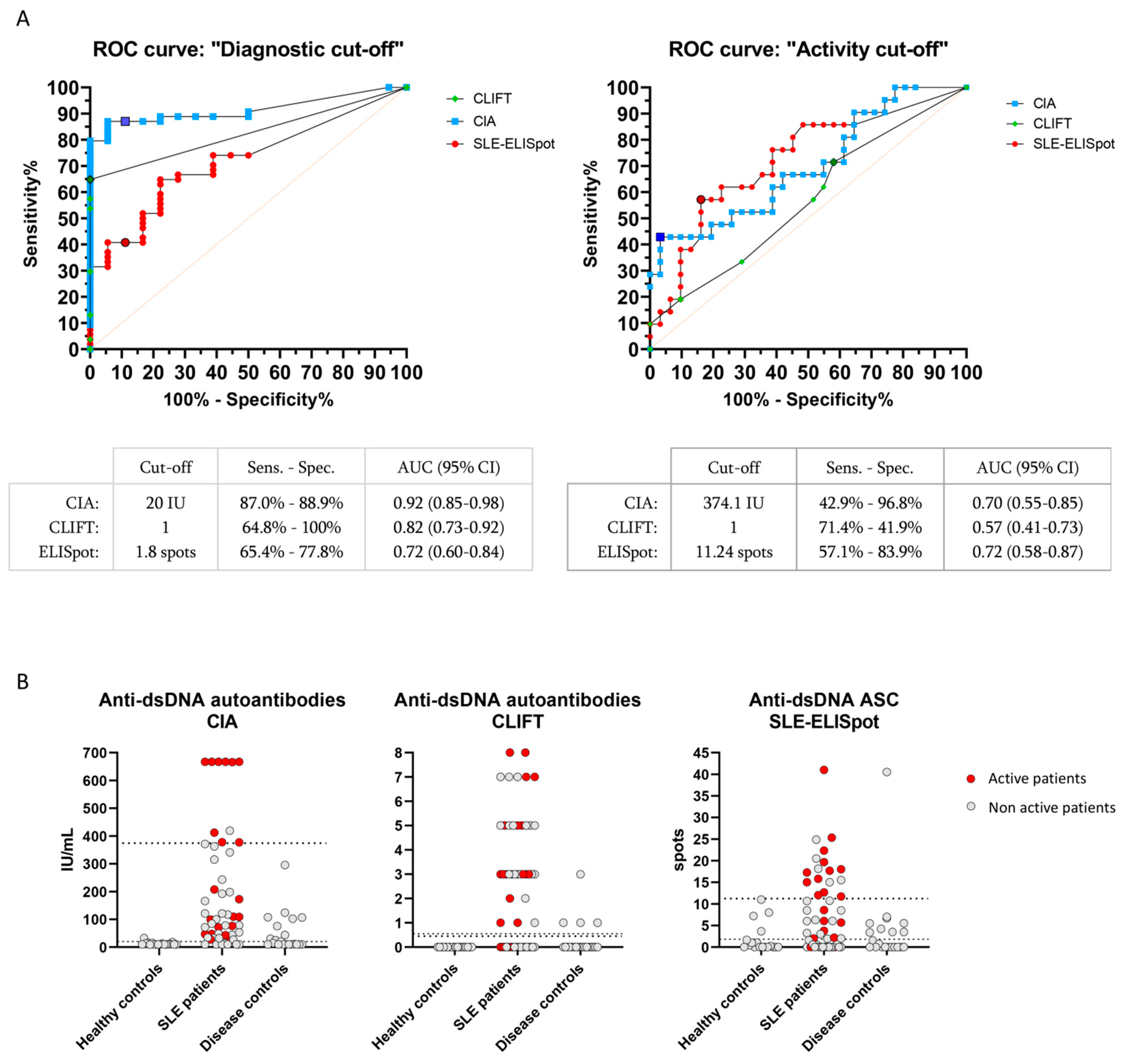
3.3. Clinical Performance of SLE-ELISpot, CIA and CLIFT
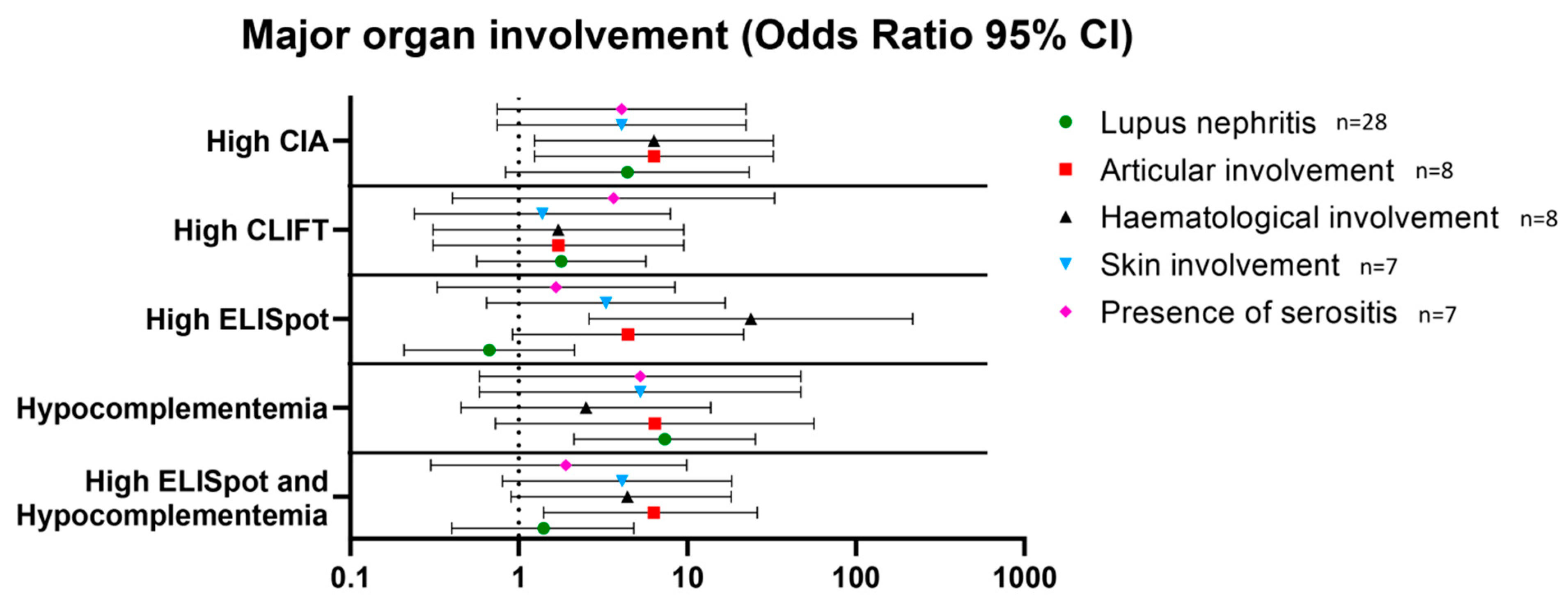
3.4. Association with Major Organ Involvement in SLE Patients
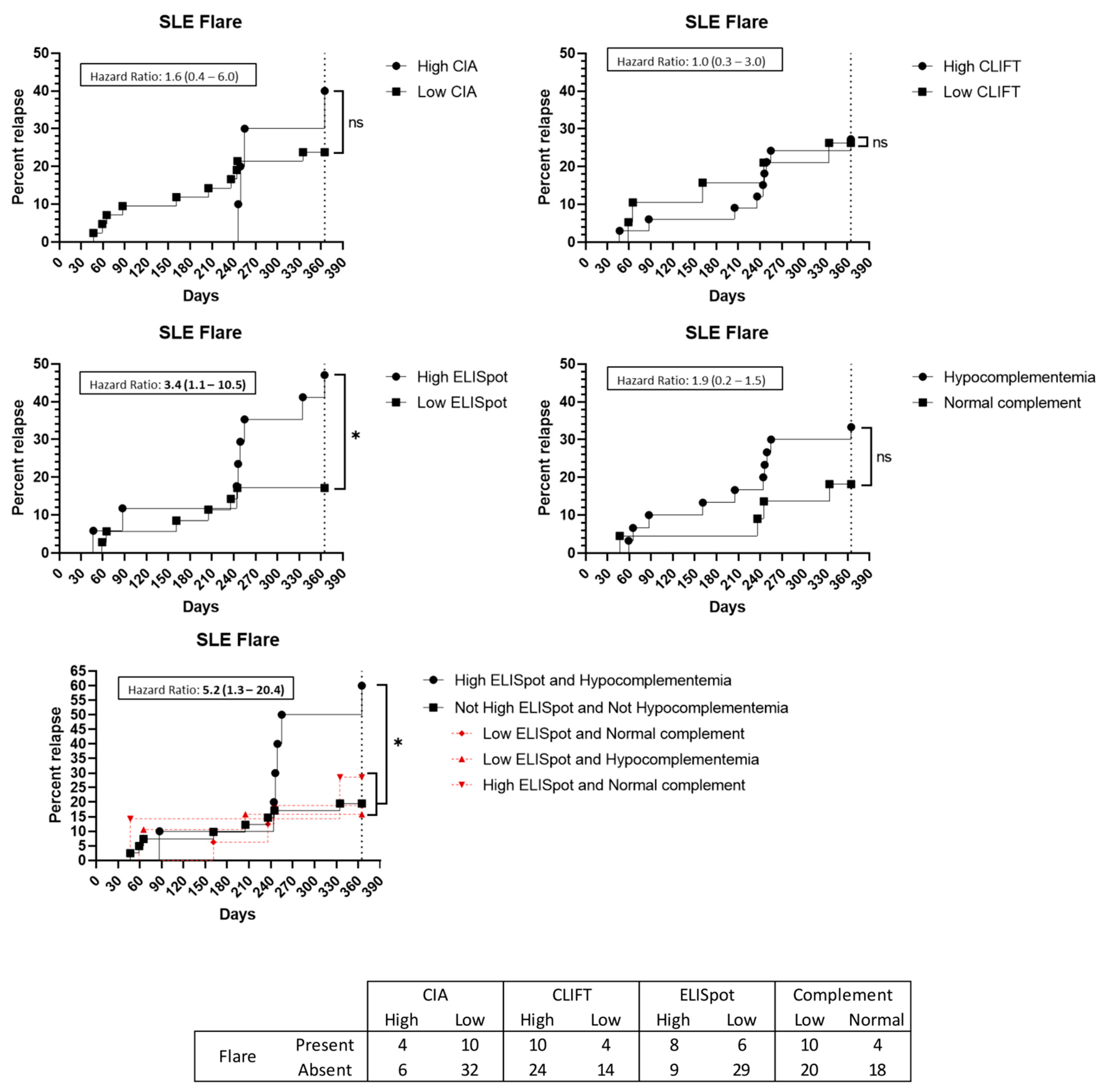
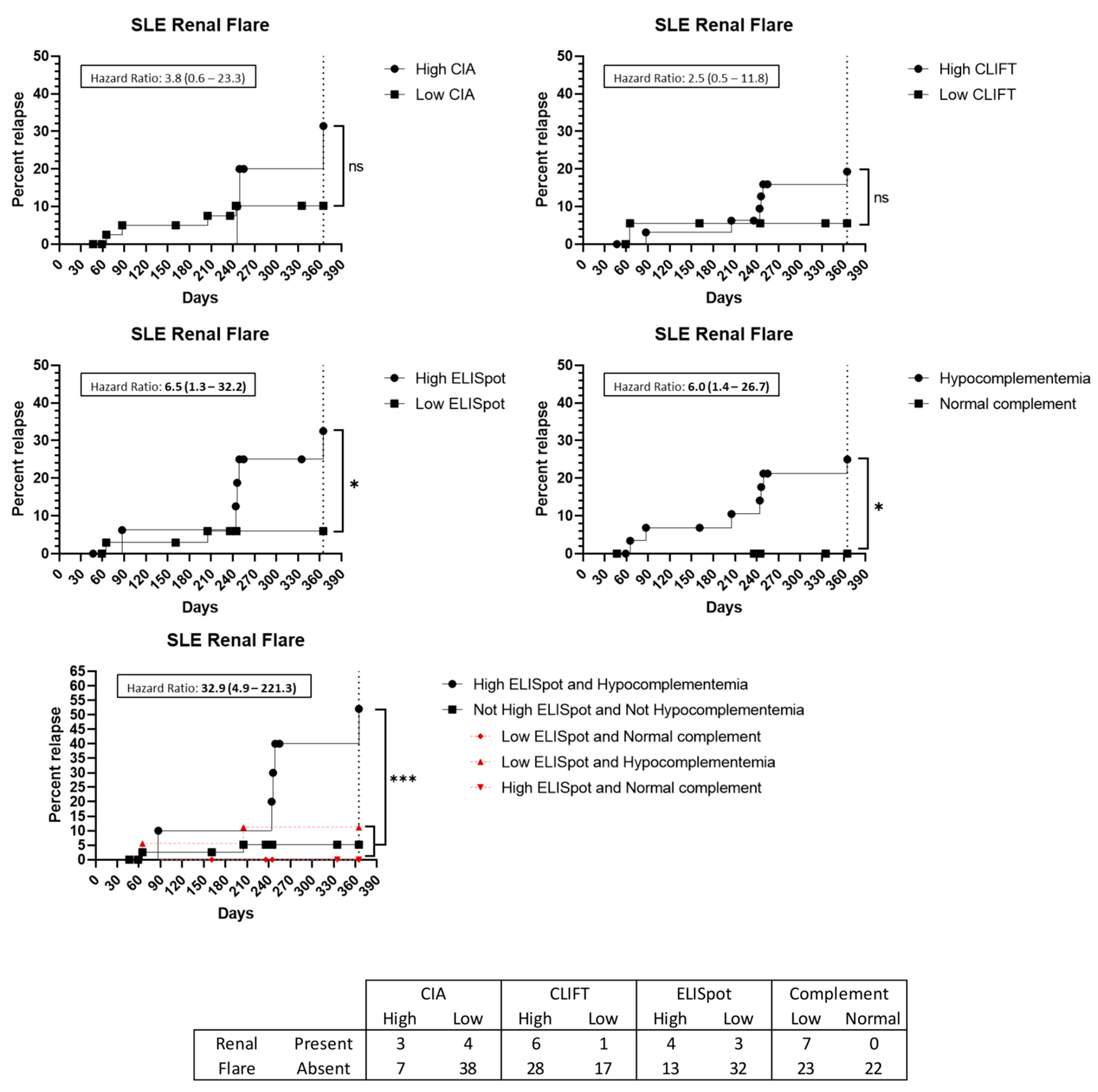
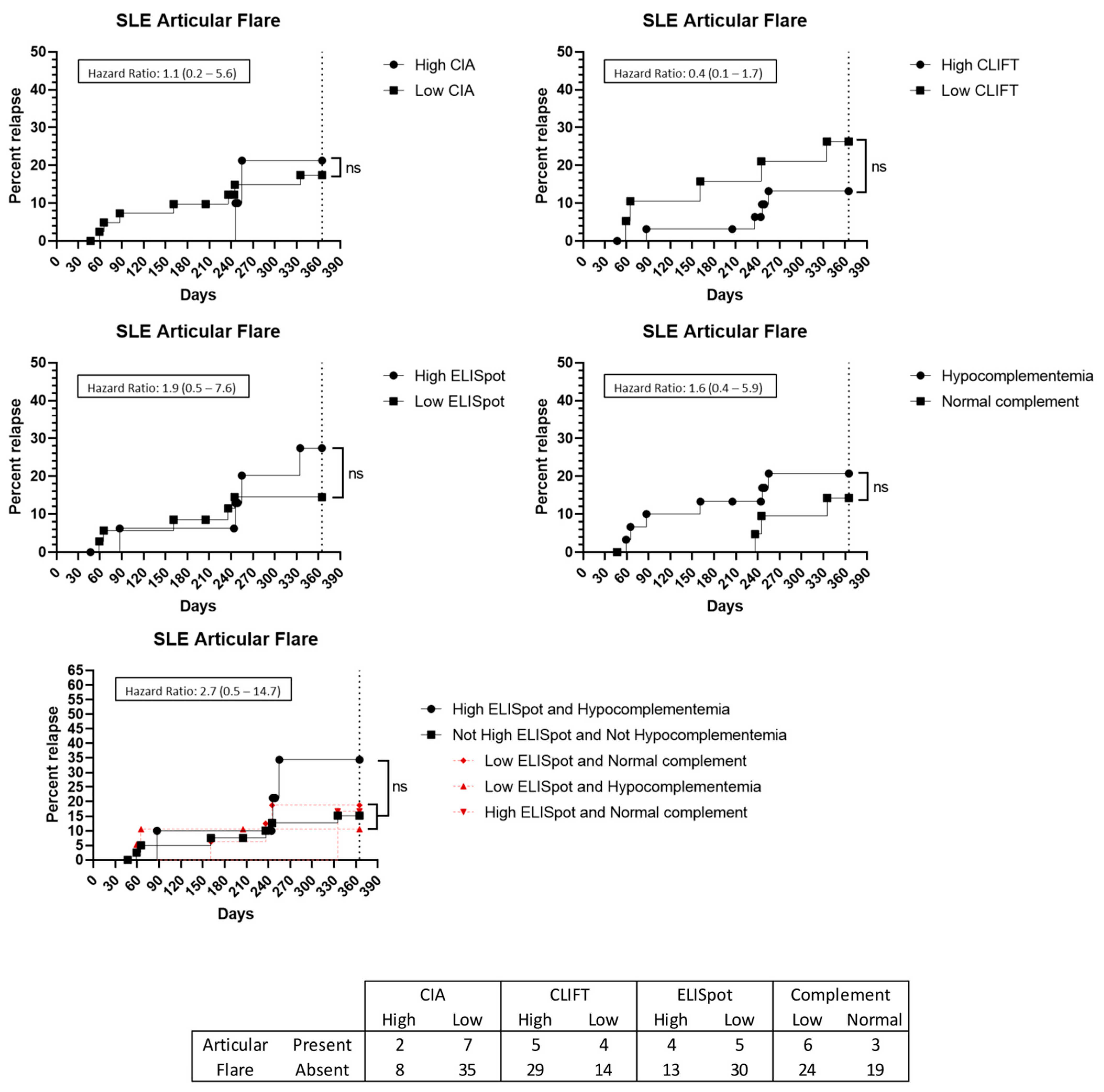
3.5. Flare Prediction Capacity
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Han, S.; Zhuang, H.; Shumyak, S.; Yang, L.; Reeves, W.H. Mechanisms of autoantibody production in systemic lupus erythematosus. Front. Immunol. 2015, 6, 228. [Google Scholar] [CrossRef] [PubMed]
- Aringer, M.; Costenbader, K.; Daikh, D.; Brinks, R.; Mosca, M.; Ramsey-Goldman, R.; Johnson, S.R. 2019 European League Against Rheumatism/American College of Rheumatology classification criteria for systemic lupus erythematosus. Ann. Rheum. Dis. 2019, 78, 1151–1159. [Google Scholar] [CrossRef] [PubMed]
- Correa-Rodríguez, M.; Pocovi-Gerardino, G.; Callejas-Rubio, J.L.; Ríos-Fernández, R.; Martín-Amada, M.; Cruz-Caparrós, M.G.; Ortego-Centeno, N. Clinical and serological associations of autoantibodies in patients with systemic lupus erythematosus. J. Investig. Med. 2021, 69, 1417–1425. [Google Scholar] [CrossRef] [PubMed]
- Tan, E.M.; Cohen, A.S.; Fries, J.F.; Masi, A.T.; Mcshane, D.J.; Rothfield, N.F.; Winchester, R.J. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1982, 25, 1271–1277. [Google Scholar] [CrossRef] [PubMed]
- Hochberg, M.C. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997, 40, 1725. [Google Scholar] [CrossRef]
- Petri, M.; Orbai, A.M.A.G. Derivation and Validation of Systemic Lupus International. Arthritis Rheum. 2012, 64, 2677–2686. [Google Scholar] [CrossRef]
- Gladman, D.D.; Ibañez, D.; Urowitz, M.B. Systemic Lupus Erythematosus Disease Activity Index 2000 Responder Index-50 Website. J. Rheumatol. 2002, 29, 288–291. [Google Scholar]
- Gensous, N.; Marti, A.; Barnetche, T.; Blanco, P.; Lazaro, E.; Seneschal, J.; Richez, C. Predictive biological markers of systemic lupus erythematosus flares: A systematic literature review. Arthritis Res. Ther. 2017, 19, 238. [Google Scholar] [CrossRef]
- Rekvig, O.P. The Anti-DNA Antibodies: Their Specificities for Unique DNA Structures and Their Unresolved Clinical Impact—A System Criticism and a Hypothesis. Front. Immunol. 2022, 12, 1–19. [Google Scholar] [CrossRef]
- Mumtaz, I.M.; Hoyer, B.F.; Panne, D.; Moser, K.; Winter, O.; Cheng, Q.Y.; Hiepe, F. Bone marrow of NZB/W mice is the major site for plasma cells resistant to dexamethasone and cyclophosphamide: Implications for the treatment of autoimmunity. J. Autoimmun. 2012, 39, 180–188. [Google Scholar] [CrossRef]
- Dörner, T.; Jacobi, A.M.; Lipsky, P.E. B cells in autoimmunity. Arthritis Res. Ther. 2009, 11, 247. [Google Scholar] [CrossRef]
- Winter, O.; Musiol, S.; Schablowsky, M.; Cheng, Q.; Khodadadi, L.; Hiepe, F. Analyzing pathogenic (double-stranded (ds) DNA-specific) plasma cells via immunofluorescence microscopy. Arthritis Res. Ther. 2015, 17, 1. [Google Scholar] [CrossRef]
- Hoyer, B.F.; Moser, K.; Hauser, A.E.; Peddinghaus, A.; Voigt, C.; Eilat, D.; Hiepe, F. Short-lived Plasmablasts and Long-lived Plasma Cells Contribute to Chronic Humoral Autoimmunity in NZB/W Mice. J. Exp. Med. 2004, 199, 1577–1584. [Google Scholar] [CrossRef]
- Yap, D.Y.H.; Chan, T.M. B cell abnormalities in systemic lupus erythematosus and lupus nephritis—Role in pathogenesis and effect of immunosuppressive treatments. Int. J. Mol. Sci. 2019, 20, 6231. [Google Scholar] [CrossRef] [PubMed]
- Malkiel, S.; Barlev, A.N.; Atisha-Fregoso, Y.; Suurmond, J.; Diamond, B. Plasma Cell Differentiation Pathways in Systemic Lupus Erythematosus. Front. Immunol. 2018, 9, 427. [Google Scholar] [CrossRef] [PubMed]
- Mathian, A.; Gallegos, M.; Pascual, V.; Banchereau, J.; Koutouzov, S. Interferon-α induces unabated production of short-lived plasma cells in pre-autoimmune lupus-prone (NZB × NZW)F1 mice but not in BALB/c mice. Eur. J. Immunol. 2011, 41, 863–872. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.D.; Tokoyoda, K.; Hoyer, B.; Alexander, T.; Khodadadi, L.; Mei, H.; Radbruch, A. Pathogenic memory plasma cells in autoimmunity. Curr. Opin. Immunol. 2019, 61, 86–91. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zou, Y.R.; Davidson, A. Plasma cells in systemic lupus erythematosus: The long and short of it all. Eur. J. Immunol. 2011, 41, 588–591. [Google Scholar] [CrossRef] [PubMed]
- Hanaoka, H.; Okazaki, Y.; Satoh, T.; Kaneko, Y.; Yasuoka, H.; Seta, N.; Kuwana, M. Circulating anti-double-stranded DNA antibody-secreting cells in patients with systemic lupus erythematosus: A novel biomarker for disease activity. Lupus 2012, 21, 1284–1293. [Google Scholar] [CrossRef] [PubMed]
- Czerkinsky, C.C.; Nilsson L, Å.; Nygren, H.; Ouchterlony, Ö.; Tarkowski, A. A solid-phase enzyme-linked immunospot (ELISPOT) assay for enumeration of specific antibody-secreting cells. J. Immunol. Methods. 1983, 65, 109–121. [Google Scholar] [CrossRef] [PubMed]
- Kuwana, M.; Okazaki, Y.; Kaburaki, J.; Kawakami, Y.; Ikeda, Y. Spleen Is a Primary Site for Activation of Platelet-Reactive T and B Cells in Patients with Immune Thrombocytopenic Purpura. J. Immunol. 2002, 168, 3675–3682. [Google Scholar] [CrossRef] [PubMed]
- Taddeo, A.; Khodadadi, L.; Voigt, C.; Mumtaz, I.M.; Cheng, Q.; Moser, K.; Hoyer, B.F. Long-lived plasma cells are early and constantly generated in New Zealand Black/New Zealand White F1 mice and their therapeutic depletion requires a combined targeting of autoreactive plasma cells and their precursors. Arthritis Res. Ther. 2015, 17, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Powell, W.E.; Hanna, S.J.; Hocter, C.N.; Robinson, E.; Lewis, M.; Dunseath, G.; Wong, F.S. Detecting autoreactive B cells in the peripheral blood of people with type 1 diabetes using ELISpot. J. Immunol. Methods. 2019, 471, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Tebruegge, M.; Ritz, N.; Koetz, K.; Noguera-Julian, A.; Seddon, J.A.; Welch, S.B.; Kampmann, B. Availability and use of molecular microbiological and immunological tests for the diagnosis of tuberculosis in Europe. PLoS ONE 2014, 9, e0099129. [Google Scholar] [CrossRef]
- Kaaijk, P.; Olivo Pimentel, V.; Emmelot, M.E.; Poelen, M.; Cevirgel, A.; Schepp, R.M.; de Wit, J. Children and Adults With Mild COVID-19: Dynamics of the Memory T Cell Response up to 10 Months. Front. Immunol. 2022, 13, 307. [Google Scholar]
- Ruperto, N.; Hanrahan, L.M.; Alarcón, G.S.; Belmont, H.M.; Brey, R.L.; Brunetta, P.; Merrill, J.T. International consensus for a definition of disease flare in lupus. Lupus 2011, 20, 453–462. [Google Scholar] [CrossRef]
- Weening, J.J.; D’agati, V.D.; Schwartz, M.M.; Seshan, S.V.; Alpers, C.E.; Appel, G.B.; Nagata, M. The Classification of Glomerulonephritis in Systemic Lupus Erythematosus Revisited. J. Am. Soc. Nephrol. 2004, 15, 241–250. [Google Scholar] [CrossRef]
- Weening, J.J.; D’agati, V.D.; Schwartz, M.M.; Seshan, S.V.; Alpers, C.E.; Appel, G.B.; Nagata, M. The classification of glomerulonephritis in systemic lupus erythematosus revisited. Kidney Int. 2004, 65, 521–530. [Google Scholar] [CrossRef]
- Austin, H.A., III; Muenz, L.R.; Joyce, K.M.; Antonovych, T.A.; Kullick, M.E.; Klippel, J.H.; Balow, J.E. Prognostic factors in lupus nephritis: Contribution of renal histologic data. Am. J. Med. 1983, 75, 382–391. [Google Scholar] [CrossRef]
- Yu, F.; Haas, M.; Glassock, R.; Zhao, M.H. Redefining lupus nephritis: Clinical implications of pathophysiologic subtypes. Nat. Rev. Nephrol. 2017, 13, 483–495. [Google Scholar] [CrossRef]
- Pan, N.; Amigues, I.; Lyman, S.; Duculan, R.; Aziz, F.; Crow, M.K.; Kirou, K.A. A surge in anti-dsDNA titer predicts a severe lupus flare within six months. Lupus 2014, 23, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Tselios, K.; Gladman, D.D.; Su, J.; Urowitz, M.B. Advanced chronic kidney disease in lupus nephritis: Is dialysis inevitable? J. Rheumatol. 2020, 47, 1366–1373. [Google Scholar] [CrossRef] [PubMed]
- Yurasov, S.; Tiller, T.; Tsuiji, M.; Velinzon, K.; Pascual, V.; Wardemann, H.; Nussenzweig, M.C. Persistent expression of autoantibodies in SLE patients in remission. J. Exp. Med. 2006, 203, 2255–2261. [Google Scholar] [CrossRef] [PubMed]
- Riveros Frutos, A.; Holgado, S.; Sanvisens Bergé, A.; Casas, I.; Olivé, A.; López-Longo, F.J.; Rúa-Figueroa, I. Late-onset versus early-onset systemic lupus: Characteristics and outcome in a national multicentre register (RELESSER). Rheumatology 2021, 60, 1793–1803. [Google Scholar] [CrossRef] [PubMed]
- Ohmura, K. Which is the best SLE activity index for clinical trials? Mod. Rheumatol. 2021, 31, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Tzovara, I.; Papadatou, I.; Tzanoudaki, M.; Spoulou, V. Development of a novel flow cytometry method for detecting pneumococcal-specific B cells. Cytom. Part A 2022, 101, 588–596. [Google Scholar] [CrossRef]
- Newell, K.L.; Waldran, M.J.; Thomas, S.J.; Endy, T.P.; Waickman, A.T. Simultaneous analysis of antigen-specific B and T cells after SARS-CoV-2 infection and vaccination. Cytom. Part A 2022, 101, 474–482. [Google Scholar] [CrossRef]
- Hernandez-Davies, J.E.; Dollinger, E.P.; Pone, E.J.; Felgner, J.; Liang, L.; Strohmeier, S.; Davies, D.H. Magnitude and breadth of antibody cross-reactivity induced by recombinant influenza hemagglutinin trimer vaccine is enhanced by combination adjuvants. Sci. Rep. 2022, 12, 9198. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control Group | SLE Group | Intra-SLE Group (p Value) | |||
|---|---|---|---|---|---|
| Healthy Controls (n = 18) | AID Controls (n = 21) | SLE (n = 31) | SLE Overlapping Other AID (n = 21) | ||
| Sex: female, n (%) | 16 (88.9%) | 17 (81.0%) | 29 (93.5%) | 19 (90.5%) | 0.999 |
| Age, mean ± SD | 32 ± 10 | 55 ± 13 | 41 ± 15 | 46 ± 16 | 0.183 |
| Anti-dsDNA autoantibodies (CIA), mean ± SD | 12.4 ± 5.6 | 49.6 ± 68.9 | 209.0 ± 223.4 | 163.8 ± 192.8 | 0.441 |
| Anti-dsDNA autoantibodies (CLIFT), n (%) | 0 (0.0%) | 4 (19.0%) | 21 (67.7%) | 13 (61.9%) | 0.769 |
| Hypocomplementemia, n (%) | - | - | 17 (54.8%) | 13 (61.9%) | 0.776 |
| clinical SLEDAI-2K, median [IQR] | - | - | 4 [2–13] | 4 [4–8] | 0.784 |
| Active disease (clinical SLEDAI-2K > 4), n (%) | - | - | 13 (41.9%) | 8 (38.1%) | 0.999 |
| Major organ involvement (at sampling) | |||||
| Renal involvement, n (%) | - | - | 19 (61.3%) | 9 (42.9%) | 0.259 |
| Active, n (%) | - | - | 10 (45.5%) | 6 (33.3%) | 0.526 |
| Chronic, n (%) | - | - | 16 (57.1%) | 8 (40.0%) | 0.380 |
| Articular involvement, n (%) | - | - | 4 (12.9%) | 4 (19.0%) | 0.700 |
| Haematological involvement, n (%) | - | - | 5 (16.1%) | 3 (14.3%) | 0.999 |
| Cutaneous involvement, n (%) | - | - | 4 (12.9%) | 3 (14.3%) | 0.999 |
| Presence of serositis, n (%) | - | - | 5 (16.1%) | 2 (9.5%) | 0.687 |
| Patients with a flare at follow-up, n (%) | - | - | 8 (25.8%) | 6 (28.6%) | 0.999 |
| Patients with renal flare, n (%) | - | - | 4 (12.9%) | 3 (14.3%) | 0.999 |
| Days between sample and flare, mean ± SD | - | - | 238 ± 99 | 150 ± 88 | 0.285 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pérez-Isidro, A.; Xipell, M.; Llobell, A.; De Moner, N.; Lledó, G.M.; Cervera, R.; Prieto-González, S.; Quintana, L.F.; Espinosa, G.; García-Ormaechea, M.; et al. Anti-dsDNA B-Cell ELISpot as a Monitoring and Flare Prediction Tool in SLE Patients. J. Clin. Med. 2023, 12, 1295. https://doi.org/10.3390/jcm12041295
Pérez-Isidro A, Xipell M, Llobell A, De Moner N, Lledó GM, Cervera R, Prieto-González S, Quintana LF, Espinosa G, García-Ormaechea M, et al. Anti-dsDNA B-Cell ELISpot as a Monitoring and Flare Prediction Tool in SLE Patients. Journal of Clinical Medicine. 2023; 12(4):1295. https://doi.org/10.3390/jcm12041295
Chicago/Turabian StylePérez-Isidro, Albert, Marc Xipell, Arturo Llobell, Noemí De Moner, Gema M. Lledó, Ricard Cervera, Sergio Prieto-González, Luis F. Quintana, Gerard Espinosa, Mila García-Ormaechea, and et al. 2023. "Anti-dsDNA B-Cell ELISpot as a Monitoring and Flare Prediction Tool in SLE Patients" Journal of Clinical Medicine 12, no. 4: 1295. https://doi.org/10.3390/jcm12041295
APA StylePérez-Isidro, A., Xipell, M., Llobell, A., De Moner, N., Lledó, G. M., Cervera, R., Prieto-González, S., Quintana, L. F., Espinosa, G., García-Ormaechea, M., Ruiz-Ortiz, E., & Viñas, O. (2023). Anti-dsDNA B-Cell ELISpot as a Monitoring and Flare Prediction Tool in SLE Patients. Journal of Clinical Medicine, 12(4), 1295. https://doi.org/10.3390/jcm12041295