
Characteristics of Congenital Adrenal Hyperplasia Diagnosed in Adulthood: A Literature Review and Case Series

Abstract
1. Introduction
2. Patients and Methods
2.1. Patients
2.2. Diagnosis
2.3. Results and Discussion
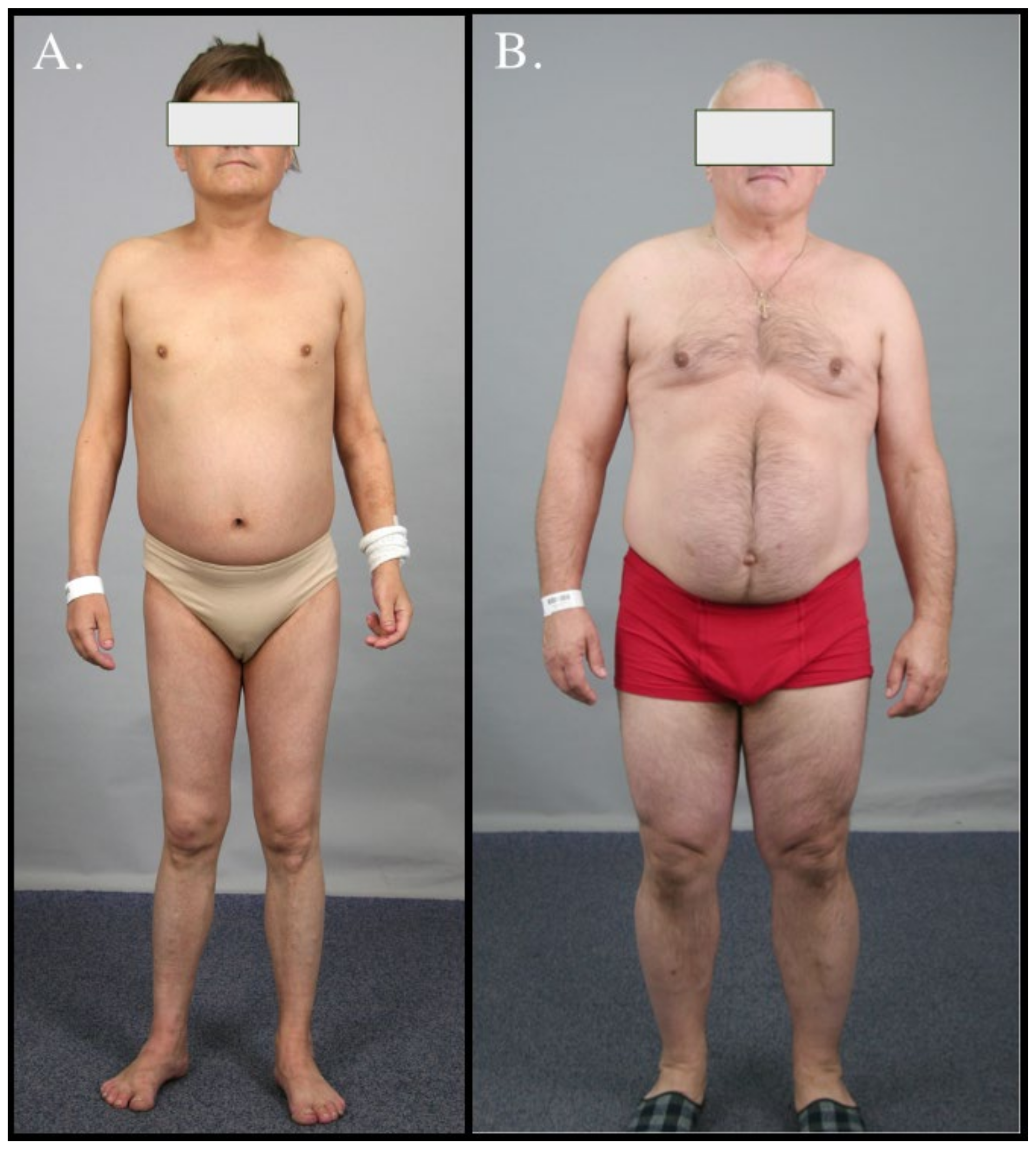
3. Case Series
3.1. Case 1
3.2. Case 2
3.3. Case 3
3.4. Case 4
3.5. Case 5
3.6. Case 6
3.7. Case 7
3.8. Case 8
4. Discussion
4.1. Nomenclature and Challenges in Congenital Adrenal Hyperplasia Classification
4.2. Genetic Testing
4.3. Adrenal Tumors
4.4. Impaired Growth and Short Stature
4.5. Fertility and Mental Health
4.6. Cardiovascular and Metabolic Health
4.7. Cognitive Function
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Momodu, L.B., II; Singh, G. Congenital Adrenal Hyperplasia. In StatPearls; StatPearls Publishing: Tampa, FL, USA, 2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK448098/ (accessed on 15 December 2022).
- Claahsen-van der Grinten, H.L.; Speiser, P.W.; Ahmed, S.F.; Arlt, W.; Auchus, R.J.; Falhammar, H. Congenital Adrenal Hyperplasia-Current Insights in Pathophysiology, Diagnostics, and Management. Endocr. Rev. 2022, 43, 91–159. [Google Scholar] [CrossRef] [PubMed]
- Merke, D.P.; Auchus, R.J. Congenital Adrenal Hyperplasia Due to 21-Hydroxylase Deficiency. N. Engl. J. Med. 2020, 383, 1248–1261. [Google Scholar] [CrossRef] [PubMed]
- Yau, M.; Khattab, A.; Yuen, T.; New, M. Congenital Adrenal Hyperplasia; Feingold, K.R., Anawalt, B., Boyce, A., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. Available online: https://www.ncbi.nlm.nih.gov/books/NBK278953/ (accessed on 10 December 2022).
- Auchus, R.J. Management considerations for the adult with congenital adrenal hyperplasia. Mol. Cell. Endocrinol. 2015, 408, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Hannah-Shmouni, F.; Morissette, R.; Sinaii, N.; Elman, M.; Prezant, T.R.; Chen, W.; Pulver, A.; Merke, D.P. Revisiting the prevalence of nonclassic congenital adrenal hyperplasia in US Ashkenazi Jews and Caucasians. Genet. Med. 2017, 19, 1276–1279. [Google Scholar] [CrossRef] [PubMed]
- Speiser, P.W. Nonclassic adrenal hyperplasia. Rev. Endocr. Metab. Disord. 2009, 10, 77–82. [Google Scholar] [CrossRef]
- McCann-Crosby, B.; Chen, M.J.; Lyons, S.K.; Lin, Y.; Axelrad, M.; Dietrich, J.E.; Sutton, V.R.; Macias, C.G.; Gunn, S.; Karaviti, L. Nonclassical congenital adrenal hyperplasia: Targets of treatment and transition. Pediatr. Endocrinol. Rev. 2014, 12, 224–238. [Google Scholar]
- Speiser, P.W. Medical treatment of classic and nonclassic congenital adrenal hyperplasia. Adv. Exp. Med. Biol. 2011, 707, 41–45. [Google Scholar] [CrossRef]
- Riedl, S.; Rohl, F.W.; Bonfig, W.; Bramswig, J.; Richter-Unruh, A.; Fricke-Otto, S.; Bettendorf, M.; Riepe, F.; Kriegshauser, G.; Schonau, E.; et al. Genotype/phenotype correlations in 538 congenital adrenal hyperplasia patients from Germany and Austria: Discordances in milder genotypes and in screened versus prescreening patients. Endocr. Connect. 2019, 8, 86–94. [Google Scholar] [CrossRef]
- Speiser, P.W.; Arlt, W.; Auchus, R.J.; Baskin, L.S.; Conway, G.S.; Merke, D.P.; Meyer-Bahlburg, H.F.L.; Miller, W.L.; Murad, M.H.; Oberfield, S.E.; et al. Congenital Adrenal Hyperplasia Due to Steroid 21-Hydroxylase Deficiency: An Endocrine Society Clinical Practice Guideline. J. Clin. Endocrinol. Metab. 2018, 103, 4043–4088. [Google Scholar] [CrossRef]
- Livadas, S.; Bothou, C. Management of the Female With Non-classical Congenital Adrenal Hyperplasia (NCCAH): A Patient-Oriented Approach. Front. Endocrinol. 2019, 10, 366. [Google Scholar] [CrossRef]
- Auchus, R.J.; Arlt, W. Approach to the patient: The adult with congenital adrenal hyperplasia. J. Clin. Endocrinol. Metab. 2013, 98, 2645–2655. [Google Scholar] [CrossRef]
- Falhammar, H.; Nordenstrom, A. Nonclassic congenital adrenal hyperplasia due to 21-hydroxylase deficiency: Clinical presentation, diagnosis, treatment, and outcome. Endocrine 2015, 50, 32–50. [Google Scholar] [CrossRef]
- Nimkarn, S.; Lin-Su, K.; Berglind, N.; Wilson, R.C.; New, M.I. Aldosterone-to-renin ratio as a marker for disease severity in 21-hydroxylase deficiency congenital adrenal hyperplasia. J. Clin. Endocrinol. Metab. 2007, 92, 137–142. [Google Scholar] [CrossRef]
- Liu, Q.; Wang, L.A.; Su, J.; Tong, D.; Lan, W.; Wang, L.; Liu, G.; Zhang, J.; Zhang, V.W.; Zhang, D.; et al. Giant bilateral adrenal myelolipomas in two Chinese families with congenital adrenal hyperplasia. Endocr. Connect. 2018, 7, 1136–1141. [Google Scholar] [CrossRef]
- El-Maouche, D.; Arlt, W.; Merke, D.P. Congenital adrenal hyperplasia. Lancet 2017, 390, 2194–2210. [Google Scholar] [CrossRef]
- Finkielstain, G.P.; Chen, W.; Mehta, S.P.; Fujimura, F.K.; Hanna, R.M.; Van Ryzin, C.; McDonnell, N.B.; Merke, D.P. Comprehensive genetic analysis of 182 unrelated families with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J. Clin. Endocrinol. Metab. 2011, 96, E161–E172. [Google Scholar] [CrossRef] [PubMed]
- White, P.C.; Vitek, A.; Dupont, B.; New, M.I. Characterization of frequent deletions causing steroid 21-hydroxylase deficiency. Proc. Natl. Acad. Sci. USA 1988, 85, 4436–4440. [Google Scholar] [CrossRef]
- New, M.I.; Abraham, M.; Gonzalez, B.; Dumic, M.; Razzaghy-Azar, M.; Chitayat, D.; Sun, L.; Zaidi, M.; Wilson, R.C.; Yuen, T. Genotype-phenotype correlation in 1,507 families with congenital adrenal hyperplasia owing to 21-hydroxylase deficiency. Proc. Natl. Acad. Sci. USA 2013, 110, 2611–2616. [Google Scholar] [CrossRef]
- Stikkelbroeck, N.M.; Hoefsloot, L.H.; de Wijs, I.J.; Otten, B.J.; Hermus, A.R.; Sistermans, E.A. CYP21 gene mutation analysis in 198 patients with 21-hydroxylase deficiency in The Netherlands: Six novel mutations and a specific cluster of four mutations. J. Clin. Endocrinol. Metab. 2003, 88, 3852–3859. [Google Scholar] [CrossRef]
- Bas, F.; Kayserili, H.; Darendeliler, F.; Uyguner, O.; Gunoz, H.; Yuksel Apak, M.; Atalar, F.; Bundak, R.; Wilson, R.C.; New, M.I.; et al. CYP21A2 gene mutations in congenital adrenal hyperplasia: Genotype-phenotype correlation in Turkish children. J. Clin. Res. Pediatr. Endocrinol. 2009, 1, 116–128. [Google Scholar] [CrossRef] [PubMed]
- Umana-Calderon, A.; Acuna-Navas, M.J.; Alvarado, D.; Jimenez, M.; Cavallo-Aita, F. CYP21A2 mutations in pediatric patients with congenital adrenal hyperplasia in Costa Rica. Mol. Genet. Metab. Rep. 2021, 27, 100728. [Google Scholar] [CrossRef] [PubMed]
- Wedell, A. Molecular genetics of congenital adrenal hyperplasia (21-hydroxylase deficiency): Implications for diagnosis, prognosis and treatment. Acta Paediatr. 1998, 87, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Krone, N.; Braun, A.; Roscher, A.A.; Knorr, D.; Schwarz, H.P. Predicting phenotype in steroid 21-hydroxylase deficiency? Comprehensive genotyping in 155 unrelated, well defined patients from southern Germany. J. Clin. Endocrinol. Metab. 2000, 85, 1059–1065. [Google Scholar] [CrossRef] [PubMed]
- El-Maouche, D.; Hannah-Shmouni, F.; Mallappa, A.; Hargreaves, C.J.; Avila, N.A.; Merke, D.P. Adrenal morphology and associated comorbidities in congenital adrenal hyperplasia. Clin. Endocrinol. 2019, 91, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Falhammar, H. Non-functioning adrenal incidentalomas caused by 21-hydroxylase deficiency or carrier status? Endocrine 2014, 47, 308–314. [Google Scholar] [CrossRef] [PubMed]
- Jaresch, S.; Kornely, E.; Kley, H.K.; Schlaghecke, R. Adrenal incidentaloma and patients with homozygous or heterozygous congenital adrenal hyperplasia. J. Clin. Endocrinol. Metab. 1992, 74, 685–689. [Google Scholar] [CrossRef]
- Nermoen, I.; Rorvik, J.; Holmedal, S.H.; Hykkerud, D.L.; Fougner, K.J.; Svartberg, J.; Husebye, E.S.; Lovas, K. High frequency of adrenal myelolipomas and testicular adrenal rest tumours in adult Norwegian patients with classical congenital adrenal hyperplasia because of 21-hydroxylase deficiency. Clin. Endocrinol. 2011, 75, 753–759. [Google Scholar] [CrossRef]
- Reisch, N.; Scherr, M.; Flade, L.; Bidlingmaier, M.; Schwarz, H.-P.; Müller-Lisse, U.; Reincke, M.; Quinkler, M.; Beuschlein, F. Total Adrenal Volume But Not Testicular Adrenal Rest Tumor Volume Is Associated with Hormonal Control in Patients with 21-Hydroxylase Deficiency. J. Clin. Endocrinol. Metab. 2010, 95, 2065–2072. [Google Scholar] [CrossRef]
- Nermoen, I.; Falhammar, H. Prevalence and Characteristics of Adrenal Tumors and Myelolipomas in Congenital Adrenal Hyperplasia: A Systematic Review and Meta-Analysis. Endocr. Pract. 2020, 26, 1351–1365. [Google Scholar] [CrossRef]
- Kenney, P.J.; Wagner, B.J.; Rao, P.; Heffess, C.S. Myelolipoma: CT and pathologic features. Radiology 1998, 208, 87–95. [Google Scholar] [CrossRef]
- Bonfig, W. Growth and development in children with classic congenital adrenal hyperplasia. Curr. Opin. Endocrinol. Diabetes Obes. 2017, 24, 39–42. [Google Scholar] [CrossRef]
- Bonfig, W.; Schwarz, H.P. Growth pattern of untreated boys with simple virilizing congenital adrenal hyperplasia indicates relative androgen insensitivity during the first six months of life. Horm. Res. Paediatr. 2011, 75, 264–268. [Google Scholar] [CrossRef]
- Claahsen-van der Grinten, H.L.; Noordam, K.; Borm, G.F.; Otten, B.J. Absence of increased height velocity in the first year of life in untreated children with simple virilizing congenital adrenal hyperplasia. J. Clin. Endocrinol. Metab. 2006, 91, 1205–1209. [Google Scholar] [CrossRef]
- Juan, L.; Huamei, M.; Zhe, S.; Yanhong, L.; Hongshan, C.; Qiuli, C.; Jun, Z.; Song, G.; Minlian, D. Near-final height in 82 Chinese patients with congenital adrenal hyperplasia due to classic 21-hydroxylase deficiency: A single-center study from China. J. Pediatr. Endocrinol. Metab. 2016, 29, 841–848. [Google Scholar] [CrossRef]
- Turner-Bowker, D.M.; Yaworsky, A.; Palladino, A.; Lamoureux, R.E.; Kelly, M.; Love, E.; Pleil, A.M.; Shields, A.; Loftus, J. Development and Psychometric Evaluation of the Life Interference Questionnaire for Growth Hormone Deficiency (LIQ-GHD) to Assess Growth Hormone Injection Burden in Children and Adults. Patient 2020, 13, 289–306. [Google Scholar] [CrossRef]
- Miranda, M.C.; Haddad, L.B.P.; Madureira, G.; Mendonca, B.B.; Bachega, T. Adverse Outcomes and Economic Burden of Congenital Adrenal Hyperplasia Late Diagnosis in the Newborn Screening Absence. J. Endocr. Soc. 2020, 4, bvz013. [Google Scholar] [CrossRef]
- Muthusamy, K.; Elamin, M.B.; Smushkin, G.; Murad, M.H.; Lampropulos, J.F.; Elamin, K.B.; Abu Elnour, N.O.; Gallegos-Orozco, J.F.; Fatourechi, M.M.; Agrwal, N.; et al. Clinical review: Adult height in patients with congenital adrenal hyperplasia: A systematic review and metaanalysis. J. Clin. Endocrinol. Metab. 2010, 95, 4161–4172. [Google Scholar] [CrossRef]
- Han, T.S.; Conway, G.S.; Willis, D.S.; Krone, N.; Rees, D.A.; Stimson, R.H.; Arlt, W.; Walker, B.R.; Ross, R.J. Relationship between final height and health outcomes in adults with congenital adrenal hyperplasia: United Kingdom congenital adrenal hyperplasia adult study executive (CaHASE). J. Clin. Endocrinol. Metab. 2014, 99, E1547–E1555. [Google Scholar] [CrossRef]
- Seth, A. Optimizing Stature in Congenital Adrenal Hyperplasia: Challenges and Solutions. Indian J. Pediatr. 2019, 86, 489–491. [Google Scholar] [CrossRef]
- King, T.F.; Lee, M.C.; Williamson, E.E.; Conway, G.S. Experience in optimizing fertility outcomes in men with congenital adrenal hyperplasia due to 21 hydroxylase deficiency. Clin. Endocrinol. 2016, 84, 830–836. [Google Scholar] [CrossRef]
- Finkielstain, G.P.; Kim, M.S.; Sinaii, N.; Nishitani, M.; Van Ryzin, C.; Hill, S.C.; Reynolds, J.C.; Hanna, R.M.; Merke, D.P. Clinical characteristics of a cohort of 244 patients with congenital adrenal hyperplasia. J. Clin. Endocrinol. Metab. 2012, 97, 4429–4438. [Google Scholar] [CrossRef] [PubMed]
- Aveiro-Lavrador, M.; De Sousa Lages, A.; Barros, L.; Paiva, I. Late diagnosis of classic congenital adrenal hyperplasia: Long-term consequences during adulthood. Endocrinol. Diabetes Metab. Case Rep. 2021, 2021. [Google Scholar] [CrossRef] [PubMed]
- Falhammar, H.; Nystrom, H.F.; Ekstrom, U.; Granberg, S.; Wedell, A.; Thoren, M. Fertility, sexuality and testicular adrenal rest tumors in adult males with congenital adrenal hyperplasia. Eur. J. Endocrinol. 2012, 166, 441–449. [Google Scholar] [CrossRef]
- Bidet, M.; Bellanne-Chantelot, C.; Galand-Portier, M.B.; Golmard, J.L.; Tardy, V.; Morel, Y.; Clauin, S.; Coussieu, C.; Boudou, P.; Mowzowicz, I.; et al. Fertility in women with nonclassical congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J. Clin. Endocrinol. Metab. 2010, 95, 1182–1190. [Google Scholar] [CrossRef] [PubMed]
- Jenkins-Jones, S.; Parviainen, L.; Porter, J.; Withe, M.; Whitaker, M.J.; Holden, S.E.; Morgan, C.L.; Currie, C.J.; Ross, R.J.M. Poor compliance and increased mortality, depression and healthcare costs in patients with congenital adrenal hyperplasia. Eur. J. Endocrinol. 2018, 178, 309–320. [Google Scholar] [CrossRef]
- Simeoli, C.; de Angelis, C.; Delli Veneri, A.; Menafra, D.; Di Paola, N.; Pivonello, C.; Di Somma, C.; Valerio, P.; Melis, D.; Alviggi, C.; et al. Severe impact of late diagnosis of congenital adrenal hyperplasia on gender identity, sexual orientation and function: Case report and review of the literature. Front. Genet. 2022, 13, 902844. [Google Scholar] [CrossRef]
- Dwiggins, M.; Brookner, B.; Fowler, K.; Veeraraghavan, P.; Gomez-Lobo, V.; Merke, D.P. Multidimensional Aspects of Female Sexual Function in Congenital Adrenal Hyperplasia: A Case-Control Study. J. Endocr. Soc. 2020, 4, bvaa131. [Google Scholar] [CrossRef]
- Meyer-Bahlburg, H.F.; Dolezal, C.; Baker, S.W.; Ehrhardt, A.A.; New, M.I. Gender development in women with congenital adrenal hyperplasia as a function of disorder severity. Arch. Sex. Behav. 2006, 35, 667–684. [Google Scholar] [CrossRef]
- Bailey, J.M.; Vasey, P.L.; Diamond, L.M.; Breedlove, S.M.; Vilain, E.; Epprecht, M. Sexual Orientation, Controversy, and Science. Psychol. Sci. Public Interest 2016, 17, 45–101. [Google Scholar] [CrossRef]
- Meyer-Bahlburg, H.F. What causes low rates of child-bearing in congenital adrenal hyperplasia? J. Clin. Endocrinol. Metab. 1999, 84, 1844–1847. [Google Scholar] [CrossRef]
- Gondim, R.; Teles, F.; Barroso, U., Jr. Sexual orientation of 46, XX patients with congenital adrenal hyperplasia: A descriptive review. J. Pediatr. Urol. 2018, 14, 486–493. [Google Scholar] [CrossRef]
- Kepczynska-Nyk, A.; Kurylowicz, A.; Nowak, A.; Bednarczuk, T.; Ambroziak, U. Sexual function in women with androgen excess disorders: Classic forms of congenital adrenal hyperplasia and polycystic ovary syndrome. J. Endocrinol. Investig. 2021, 44, 505–513. [Google Scholar] [CrossRef]
- Daae, E.; Feragen, K.B.; Nermoen, I.; Falhammar, H. Psychological adjustment, quality of life, and self-perceptions of reproductive health in males with congenital adrenal hyperplasia: A systematic review. Endocrine 2018, 62, 3–13. [Google Scholar] [CrossRef]
- Borges, J.H.; Santoro, R.I.; de Oliveira, D.M.; de Lemos-Marini, S.H.V.; Geloneze, B.; Guerra-Junior, G.; Goncalves, E.M. Cardiovascular dysfunction risk in young adults with congenital adrenal hyperplasia caused by 21-hydroxylase enzyme deficiency. Int. J. Clin. Pract. 2021, 75, e14233. [Google Scholar] [CrossRef]
- Falhammar, H.; Thoren, M. Clinical outcomes in the management of congenital adrenal hyperplasia. Endocrine 2012, 41, 355–373. [Google Scholar] [CrossRef]
- Zhang, H.J.; Yang, J.; Zhang, M.N.; Liu, C.Q.; Xu, M.; Li, X.J.; Yang, S.Y.; Li, X.Y. Metabolic disorders in newly diagnosed young adult female patients with simple virilizing 21-hydroxylase deficiency. Endocrine 2010, 38, 260–265. [Google Scholar] [CrossRef]
- Tamhane, S.; Rodriguez-Gutierrez, R.; Iqbal, A.M.; Prokop, L.J.; Bancos, I.; Speiser, P.W.; Murad, M.H. Cardiovascular and Metabolic Outcomes in Congenital Adrenal Hyperplasia: A Systematic Review and Meta-Analysis. J. Clin. Endocrinol. Metab. 2018, 103, 4097–4103. [Google Scholar] [CrossRef]
- Torky, A.; Sinaii, N.; Jha, S.; Desai, J.; El-Maouche, D.; Mallappa, A.; Merke, D.P. Cardiovascular Disease Risk Factors and Metabolic Morbidity in a Longitudinal Study of Congenital Adrenal Hyperplasia. J. Clin. Endocrinol. Metab. 2021, 106, e5247–e5257. [Google Scholar] [CrossRef]
- Barbot, M.; Mazzeo, P.; Lazzara, M.; Ceccato, F.; Scaroni, C. Metabolic syndrome and cardiovascular morbidity in patients with congenital adrenal hyperplasia. Front. Endocrinol. 2022, 13, 934675. [Google Scholar] [CrossRef]
- Arlt, W.; Willis, D.S.; Wild, S.H.; Krone, N.; Doherty, E.J.; Hahner, S.; Han, T.S.; Carroll, P.V.; Conway, G.S.; Rees, D.A.; et al. Health status of adults with congenital adrenal hyperplasia: A cohort study of 203 patients. J. Clin. Endocrinol. Metab. 2010, 95, 5110–5121. [Google Scholar] [CrossRef]
- Bachelot, A.; Plu-Bureau, G.; Thibaud, E.; Laborde, K.; Pinto, G.; Samara, D.; Nihoul-Fekete, C.; Kuttenn, F.; Polak, M.; Touraine, P. Long-term outcome of patients with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Horm. Res. 2007, 67, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Hashemi Dehkordi, E.; Khaheshi, S.; Mostofizadeh, N.; Hashemipour, M. Cardiovascular Risk Factors in Children and Adolescents with Congenital Adrenal Hyperplasia. Adv. Biomed. Res. 2021, 10, 19. [Google Scholar] [CrossRef]
- Zhao, Y.; Hou, L.; Gao, H.J.; Zhan, D.; Zhang, C.; Luo, X.P. Independent relationship between body mass index and LH peak value of GnRH stimulation test in ICPP girls: A cross-sectional study. J Huazhong Univ. Sci. Technol. Med. Sci. 2017, 37, 556–562. [Google Scholar] [CrossRef] [PubMed]
- Amr, N.H.; Baioumi, A.Y.; Serour, M.N.; Khalifa, A.; Shaker, N.M. Cognitive functions in children with congenital adrenal hyperplasia. Arch. Endocrinol. Metab. 2019, 63, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, L.; Gezelius, A.; Nordenstrom, A.; Hirvikoski, T.; Lajic, S. Cognitive impairment in adolescents and adults with congenital adrenal hyperplasia. Clin. Endocrinol. 2017, 87, 651–659. [Google Scholar] [CrossRef]
- Berenbaum, S.A. Cognitive function in congenital adrenal hyperplasia. Endocrinol. Metab. Clin. North Am. 2001, 30, 173–192. [Google Scholar] [CrossRef]
- Johannsen, T.H.; Ripa, C.P.; Reinisch, J.M.; Schwartz, M.; Mortensen, E.L.; Main, K.M. Impaired cognitive function in women with congenital adrenal hyperplasia. J. Clin. Endocrinol. Metab. 2006, 91, 1376–1381. [Google Scholar] [CrossRef]
- Puts, D.A.; McDaniel, M.A.; Jordan, C.L.; Breedlove, S.M. Spatial ability and prenatal androgens: Meta-analyses of congenital adrenal hyperplasia and digit ratio (2D:4D) studies. Arch. Sex. Behav. 2008, 37, 100–111. [Google Scholar] [CrossRef]
- Messina, V.; Karlsson, L.; Hirvikoski, T.; Nordenstrom, A.; Lajic, S. Cognitive Function of Children and Adolescents with Congenital Adrenal Hyperplasia: Importance of Early Diagnosis. J. Clin. Endocrinol. Metab. 2020, 105, e683–e691. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Case 1 | Case 2 | Case 3 | Case 4 | |
|---|---|---|---|---|
| Age at diagnosis [years] | 18 | 65 | 35 | 62 |
| BMI [kg/m2] | 33.6 | 24.8 | 20 | 23.1 |
| Height [cm ± SD] | 150 (±2.1) | 153 (±1.7) | 146 (±2.7) | 164 (±0.2) |
| HOMA-IR | 2.78 | 2.6 | 2.30 | 2.1 |
| Co-morbidities | None. | None. | Liver cirrhosis. | Parkinson’s disease and breast cancer. |
| Menstrual/fertility history | Primary amenorrhoea. | Primary amenorrhoea. | Primary amenorhhoea. | Oligomenorhoea and six miscarriages. |
| Ferriman–Gallwey score | 16 | 12 | 4 | 8 |
| Alopecia, acne | None. | None. | None. | None. |
| Genitalia | Clitoromegaly and vaginal stenosis. | Clitoromegaly and vaginal stenosis. | Common urogenital sinus. | Normal. |
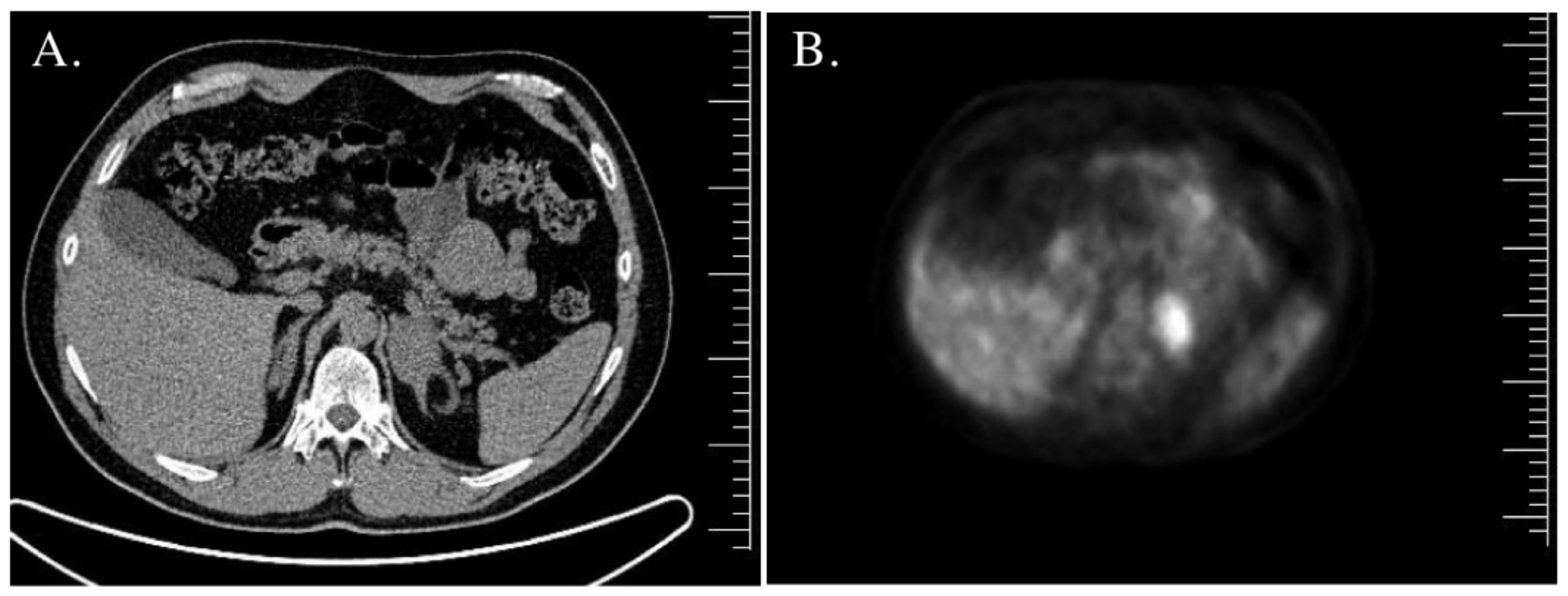
| Adrenal gland imaging | Normal. | CT—bilateral adrenal tumors, right—45 mm × 30 mm × 37 mm (20 HU), left—18 mm × 14 mm × 11 mm (30 HU). | MRI—right adrenal tumor, 85 mm × 57 mm × 70 mm; inhomogeneous, without signal loss in out-of-phase. Left adrenal hypertrophy. | MRI—left adrenal tumor, 44 mm × 38 mm; inhomogeneous, borderline signal loss in out-of-phase; 18FDG PET/CT—increased uptake. Right adrenal in normal size. |
| Testosterone [nmol/L] N: 0.29–1.67 | 9.9 | 5.15 | 27.90 | 3.34 |
| Androstenedione [ng/mL] N: 0.3–3.5 | 14.1 | 10.59 | NA | NA |
| DHEA-S [µg/dL] | >1000 (60.9–337) | 187 (9–246) | 670 (60.9–337) | 125.7 (18.9–205) |
| 17OHP [ng/mL] N: <1.7 | 23.9 | >20 | >20 | >20 |
| Urine 17-hydroksypreganolone [µg/24 h] N: 63–279 | 60800 | 2153.6 | 24007.9 | 1351.1 * |
| Urine pregnanetriol [µg/24 h] N: 179–992 | 51910 | 4306 | 19866.5 | 1241.5 * |
| Urine pregnantriolone [µg/24 h] N: 3.5–50 | 20040 | 1298.1 | 4799.6 | 193.9 * |
| Cortisol [µg/dL] after 250 µg tetracosactide acetate im | 0′ 11.6 30′ 12.87 60′ 14.13 | 0′ 14.96 30′ 18.59 | 0′ 11.27 30′ 14.92 60′ 16.08 | 0′ 19.01 30′ 20.19 60′ 21.64 |
| ACTH [pg/mL] N: 10–60 | 1518.8 | 34.5 | 200 | 10.3 |
| Genetic evaluation | I173N/deletion | c.293-13C>G/deletion | c.293-13C>G/ c.293-13C>G | NA |
| Sexual orientation | Heterosexual | Heterosexual | Homosexual | Heterosexual |
| Offspring [n] | 0 | 0 | 0 | 0 |
| Treatment | Prednisone 7.5 mg/day, vaginal calibration, GC stress-dosing treatment | Right adrenalectomy, GC stress-dosing treatment | Right adrenalectomy, GC stress-dosing treatment | Left adrenalectomy, GC stress-dosing treatment |
| Case 5 | Case 6 | Case 7 | Case 8 | |
|---|---|---|---|---|
| Age at diagnosis [years] | 32 | 52 | 44 | 81 |
| BMI [kg/m2] | 29.4 | 33.6 | 33.7 | 24.5 |
| Height [cm ± SD] | 178 (±0.1) | 164 (±2) | 168 (±1.6) | 160 (±2.7) |
| HOMA-IR | 2.23 | 2.15 | 2.87 | 2.76 |
| Co-morbidities | None. | Hypertension, DMt2, hyperlipidemia, and obesity. | Hyperlipidemia and obesity. | Hypertension and COPD. |
| Hypogonadic symptoms | Slight libido decrease. | Slight libido decrease. | Slight libido decrease. | None. |
| Testicular USG | Normal testicle size (right 27 mm × 21 mm × 47 mm, left 29 mm × 19 mm × 47 mm) and microcalcifications. | Decreased testicular volume (right testicle 21 mm × 15 mm × 32 mm, left testicle 17 mm × 13 mm × 32 mm) and TARTs. | Normal testicle size (right 35 mm × 20 mm × 47 mm, left 29 mm × 20 mm × 45 mm). | Normal testicle size (right 29 mm × 25 mm × 49 mm, left 34 mm × 25 mm × 45 mm). |
| Adrenal gland imaging | CT—right adrenal tumor 20 mm, 26 HU; MRI—signal loss in out-of-phase. Left adrenal normal in size. | CT—bilateral adrenal hypertrophy. | MRI—left adrenal tumor 44 mm—borderline signal loss in out-of-phase; right adrenal adenoma 16 mm × 10 mm × 7 mm; 18FDG PET/CT—increased uptake (SUVmax 7.6) in the left adrenal tumor. | CT—bilateral adrenal hypertrophy (left adrenal 42 mm × 30 mm, right adrenal 55 mm × 35 mm). |
| Testosterone [nmol/L] N: 8.64–29 | 8.12 | 17.22 | 5.42 | 17.4 |
| Androstenedione [ng/mL] | NA | >10 | 8.04 | NA |
| DHEA-S [µg/dL] N: 160–449 | 693.3 | 1363 | 276 | 56.3 |
| FSH [IU/L] N: 1.5–12.4 | 0.81 | 0.29 | 1.69 | 9.12 |
| LH [IU/L] N: 1.7–8.4 | 0.44 | <0.1 | 3.16 | 12 |
| ACTH [pg/mL] N: 10–65 | 158.1 | 241.8 | 70.5 | NA |
| 17OHP [ng/mL] N: <1.7 | 6.53 | >20 | 180.1 | 52.4 |
| Urine 17-hydroxypregnenolone [µg/24 h] N: 72–452 | 14444.1 | 52296.7 | 23138.8 | 2866.7 |
| Urine pregnanetriol [µg/24 h] N: 189–1737 | 19841.8 | 66298 | 37242.6 | 4268.2 |
| Urine pregnantriolone [µg/24 h] N: 6–66 | 5810.2 | 11569.5 | 20789 | 1598.7 |
| Urine free cortisol [µg/24 h] N: 13–120 | 431.7 | 108 | 648.3 | 65.3 |
| Cortisol [µg/dL] after 250 µg tetracosactide acetate im | 0′ 16.61 30′ 18.58 60′ 18.60 | 0′ 15.25 30′ 15.65 60′ 17.42 | 0′ 13.06 30′ 15.28 60′ 15.83 | 0′ 14.9 30′ 16.6 60′ 18.2 |
| Sexual orientation | Heterosexual | Heterosexual | Heterosexual | Heterosexual |
| Offspring [n] | 0 | 1 | 0 | 2 |
| Genetic evaluation | I173N/I173N | I173N/I173N | I173N/I173N | NA |
| Semen analysis | Oligozoospermia. | Azoospermia. | Oligozoospermia. | NA |
| Treatment | GC stress-dosing treatment. | GC stress-dosing treatment. | Left adrenalectomy and GC stress-dosing treatment. | GC stress-dosing treatment. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hubska, J.; Kępczyńska-Nyk, A.; Czady-Jurszewicz, K.; Ambroziak, U. Characteristics of Congenital Adrenal Hyperplasia Diagnosed in Adulthood: A Literature Review and Case Series. J. Clin. Med. 2023, 12, 653. https://doi.org/10.3390/jcm12020653
Hubska J, Kępczyńska-Nyk A, Czady-Jurszewicz K, Ambroziak U. Characteristics of Congenital Adrenal Hyperplasia Diagnosed in Adulthood: A Literature Review and Case Series. Journal of Clinical Medicine. 2023; 12(2):653. https://doi.org/10.3390/jcm12020653
Chicago/Turabian StyleHubska, Joanna, Anna Kępczyńska-Nyk, Katarzyna Czady-Jurszewicz, and Urszula Ambroziak. 2023. "Characteristics of Congenital Adrenal Hyperplasia Diagnosed in Adulthood: A Literature Review and Case Series" Journal of Clinical Medicine 12, no. 2: 653. https://doi.org/10.3390/jcm12020653
APA StyleHubska, J., Kępczyńska-Nyk, A., Czady-Jurszewicz, K., & Ambroziak, U. (2023). Characteristics of Congenital Adrenal Hyperplasia Diagnosed in Adulthood: A Literature Review and Case Series. Journal of Clinical Medicine, 12(2), 653. https://doi.org/10.3390/jcm12020653