
Revisiting Diagnosis and Treatment of Hypertrophic Cardiomyopathy: Current Practice and Novel Perspectives

 ,
,  , ,
, ,  ,
, 
 , and
, and 
Abstract
1. Introduction
1.1. Pathophysiology
1.2. Obstructive vs. Nonobstructive HCM
1.3. HCM Phenocopies
- Anderson–Fabry disease: this is a genetic disorder caused by mutations in the α-galactosidase A (GLA) gene that results in the accumulation of glycosphingolipids in various systems of the body. The disease is known to cause LVH, which can be diagnosed through echocardiography in almost 50% of male patients aged 30–40 [9]. LVH in Anderson–Fabry Disease happens due to an abnormal build-up of glycosphingolipids, leading to reduced α-galactosidase A activity. Most patients also exhibit abnormal ECG results, with voltage criteria for LVH, short PR interval and conduction disorders. Echocardiography shows concentric LVH, diastolic dysfunction, and systolic dysfunction in the later stages of the disease. Cardiovascular magnetic resonance (CMR) scans also show concentric LVH, with a late gadolinium enhancement (LGE) of the basal inferolateral wall being a characteristic feature. Moreover, LGE in these patients increases the risk of major cardiovascular events [10,11]. Most Fabry patients exhibit concentric LVH [12]; however, asymmetrical septal and apical hypertrophy can be observed. Cardiovascular involvement can be highly variable and mutation dependent, also affecting the eligibility of precision therapies such as oral chaperone therapy. Enzyme replacement therapy is dependent on serum alpha-1-galactosidase levels.
- Cardiac amyloidosis: this is a cardiomyopathy caused by the deposition of amyloid protein outside of the heart cells, affecting the myocardium. This condition can occur in amyloid light-chain (AL) amyloidosis and transthyretin amyloidosis. Cardiac involvement in amyloidosis is associated with poor prognosis and can lead to various symptoms, including chest pain, HF, arrhythmias, stroke, and signs and symptoms of autonomic dysfunction. The disease also affects other organs, with symptoms such as carpal tunnel syndrome, easy bruising, macroglossia, neuropathy, and hepatomegaly. The ECG in amyloid cardiomyopathy often shows low-voltage QRS complexes. Echocardiography typically reveals bi-ventricular hypertrophy, valve thickening, bi-atrial dilatation, and diastolic dysfunction. Strain and strain rate imaging using speckle tracking can help to differentiate amyloid cardiomyopathy from sarcomeric HCM. In most cases, the pattern of LVH in cardiac amyloid is concentric and non-obstructive, but some cases can present with asymmetric obstructive forms, mimicking sarcomeric forms of HCM. CMR has a central role in the non-invasive diagnosis of cardiac amyloidosis due to its ability to provide tissue characterisation in addition to high-resolution morphologic and functional assessment. While multiple LGE patterns have been described in cardiac amyloidosis, subendocardial and transmural distributions predominate. Both patterns are present to different extents in AL and ATTR cardiac amyloidosis, with subendocardial LGE being more prevalent in AL and transmural LGE more prevalent in ATTR cardiac amyloidosis. LGE initially appears predominant in the basal segments, but as the disease advances, biventricular transmural involvement occurs [13,14]. Additionally, it’s worth noting that wild-type ATTR features almost universal involvement of cardiac structures, while hereditary ATTR involvement varies. Both types of amyloidosis can mimic HCM. On the other hand, AL amyloidosis, the most prevalent form, affects the heart in approximately 50% of cases. It often presents as a non-ischemic dilated phenotype, resulting in global impairment of cardiac function.
- Mitochondrial cardiomyopathies: this is a heterogeneous group of conditions that arise due to genetic mutations in the mitochondrial DNA inherited maternally. This leads to impaired energy production and affects various systems in the body, including the central nervous system, heart, and skeletal system. The symptoms of the disease can manifest at any age, from infancy to adulthood. In around 25% of patients, non-obstructive cardiomyopathy with mild concentric hypertrophy is observed, significantly worsening the prognosis. Approximately 50% of these patients develop HF, and the mortality rate reaches 70% before the age of 30. In cases of HF, cardiac transplantation may be considered a treatment option.
- RAS-HCM: HCM phenotype can be associated with malformation syndromes, including Noonan syndrome and LEOPARD syndrome. These syndromes belong to a group of developmental disorders called RASopathies caused by mutations in genes involved in the RAS/mitogen-activated protein kinase (RAS/MAPK) pathway. Noonan syndrome is inherited in an autosomal dominant pattern and is characterised by various congenital heart defects, including hypertrophic cardiomyopathy, which can affect up to 25% of patients. Additionally, Noonan syndrome patients may have pulmonary and aortic valve stenosis and atrioventricular septal defects. LEOPARD syndrome is an allelic variant of Noonan syndrome and is characterised by a combination of clinical features, including lentigines, electrocardiographic abnormalities, ocular hypertelorism, pulmonic stenosis, abnormal male genitalia, retardation of growth, and deafness. A recent multi-omics study in myectomy tissue from HCM patients shows activation of the RAS-MAPK signalling, suggesting that RAS-HCM and sarcomeric HCM may, in fact, share some final common pathways. In a multicentre retrospective study aimed to understand the arrhythmic progression of RAS-HCM better and pinpoint shortcomings in its management, RAS-HCM was associated with heightened morbidity and a comparable risk of SCD to sarcomeric HCM [15]. Notably, there was a discernibly low frequency of ICD implantation among RAS-HCM patients, resulting in potentially preventable sudden deaths. Prospective studies are needed to identify risk factors for SCD and develop specific recommendations for ICD implantation in RAS-HCM [16].
- Hypertensive heart disease: LVH caused by hypertension can be challenging to differentiate from HCM (Table 2) caused by sarcomeric mutations, as there is a frequent overlap (up to 25%) in the patterns of hypertrophy seen in both conditions. Similarly, features of HCM, such as the systolic anterior motion (SAM) of the mitral valve anterior leaflets, chordal slack, friction or impact lesions (from chronic mitral–septal contact on septum) or dynamic LVOTO due to basal septal hypertrophy can also be observed in patients with LVH caused by hypertension, especially if untreated hypertension is severe and low preload. However, SAM in hypertensive LVH occurs at the end of systole, unlike in HCM, where it occurs earlier. Several echocardiographic techniques have been used to differentiate between the two conditions [17]. Tissue Doppler imaging can show more impairment of diastolic function in HCM and lower early diastolic velocities. Two-dimensional (2D) strain echocardiography can also aid in the diagnosis, as radial strain in the mid and apical short-axis segments is commonly reduced in HCM with sarcomeric mutations. Similarly, the systolic longitudinal strain has been found to be reduced in HCM, and it has value in distinguishing between HCM and hypertensive LVH. CMR imaging can identify typical patterns of fibrosis associated with HCM. Serum markers such as norepinephrine, atrial natriuretic peptide, and brain natriuretic peptide tend to be higher in HCM patients than in those with hypertensive LVH. A thorough clinical assessment of relatives may be crucial in making a diagnosis, as the identification of HCM in family members dramatically increases the likelihood that LVH has a genetic basis.
- Danon disease: this is a rare X-linked dominant genetic disorder caused by a deficiency in lysosome-associated membrane protein-2 (LAMP2), leading to lysosomal storage. The prevalence of the disease may be underestimated due to difficulties in diagnosis, but LAMP2 mutations have been identified in 1–8% of patients with suspected HCM who underwent genetic testing. The disease manifests in males with severe symptoms at an earlier age, while females may develop later onset and milder symptoms due to X-linked inheritance. Diagnosis is confirmed by molecular genetic screening that reveals a LAMP2 gene mutation. Clinical suspicion of the condition should prompt testing of serum creatine kinase and liver enzyme levels, which are usually raised in this condition. ECG may show ventricular pre-excitation with Wolff–Parkinson–White (WPW)-pattern in up to two-thirds of men and less than a third of women, along with very large voltage complexes in male teenagers, raising suspicion of the condition. Echocardiography typically shows severe concentric LVH, but asymmetric septal hypertrophy has also been observed. Skeletal muscle biopsy may show intra-sarcoplasmic periodic acid-Schiff-positive vacuoles. In late stages, the disease may progress to a dilated cardiomyopathy phenotype [18,19].
- Pompe disease: this is a genetic disorder that follows an autosomal recessive pattern of inheritance, resulting from the deficiency of acid maltase (acid alpha [α]-glucosidase) enzyme. The condition is characterised by the deposition of glycogen in multiple organs and can present in three different forms: infantile, juvenile, and adult. The infantile form is the most severe and can lead to death within two years due to extreme LVH, HF, hypotonia, macroglossia, and hepatomegaly. While dilated cardiomyopathy can occur in some patients, the infantile form of the disease often presents with asymmetric LVH and LVOTO. In contrast, later onset forms have milder cardiac involvement and present with proximal myopathy. Diagnosis can be confirmed by demonstrating enzyme deficiency in fibroblasts, lymphocytes, and/or urine, and skeletal muscle biopsy showing vacuolar glycogen deposition. ECG can show features of LVH as well as short PR interval with pre-excitation or conduction block [20].
- PRKAG2 cardiomyopathy: this is a genetic disorder with autosomal dominant inheritance caused by mutations in the PRKAG2 gene, which codes for the regulatory gamma-subunit of AMP-activated protein kinase (AMPK). This condition typically affects adolescents and young adults and is characterised by muscle weakness and imaging showing LVH with global hypokinesia. While the LVH in PRKAG2 cardiomyopathy is often associated with excess glycogen deposition and can be variable in severity, it can also be asymmetric, resembling the pattern seen in HCM caused by mutations in sarcomeric genes. However, PRKAG2 cardiomyopathy is different from sarcomeric HCM in that it progresses early to systolic dysfunction and dilated cardiomyopathy. This condition may also be associated with WPW syndrome and degeneration of the conduction system [21,22]. There are no known precision-based therapies for PRKAG2 cardiomyopathy.
- Cori–Forbes cardiomyopathy: this is a genetic disorder caused by mutations in the glycogen debranching enzyme (amylo-alpha-1, 6-glucosidase [AGL]) gene. It is inherited in an autosomal recessive pattern and can present in infants, adolescents, or young adults. Common clinical features of Forbes disease include muscle weakness, poor growth, and hypoglycemia. In addition, patients may develop concentric LVH, which can progress to dilated cardiomyopathy in later years [23].
- Athlete’s heart: in response to chronic high-intensity physical activity, the cardiovascular system activates a series of adaptative physiological mechanisms defined as the athlete’s heart, including a constellation of changes with increased biventricular mass, volume, and wall thickness. A stepwise approach to the cardiovascular assessment of athletes is essential to make sense of overlapping clinical phenotypes and eventually provide a correct differential diagnosis between HCM and adaptative cardiac response to exercise. Twelve-lead ECG enhances the sensitivity of the screening process by allowing early detection of cardiovascular conditions distinctively manifesting with ECG abnormalities. Echocardiography has a pivotal role in differentiating physiologic and pathologic responses to exercise, namely athlete’s heart, from HCM. Combining different methods, such as 2D and 3D measurements of cardiac size, volumes, wall thickness, mass index, tissue velocity, and myocardial strain imaging, cardiac ultrasound allows comprehensive morphologic and functional evaluation of the heart and distinction between physiologic and pathologic remodelling. In the presence of abnormal, uncertain, and/or controversial findings from the upstream diagnostic work up, CMR imaging can help distinguish between exercise-induced cardiac remodelling and cardiovascular pathology. CMR represents the current gold standard in the non-invasive assessment of cardiac morphology and quantification of volumes and flow and offers the opportunity for advanced myocardial tissue characterisation with excellent accuracy and precision [24,25].
2. Clinical Diagnosis and Imaging Tools
3. Sudden Cardiac Death Risk Assessment and Prevention
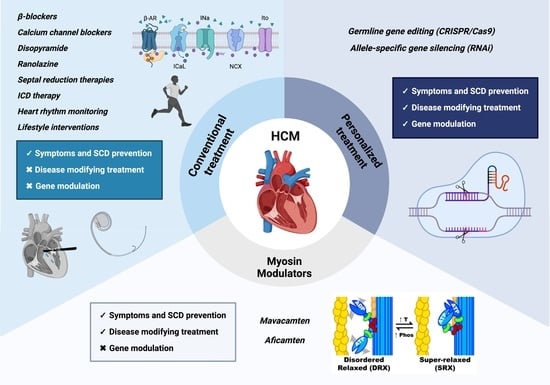
4. Therapeutic Approaches
4.1. Conventional Treatment
4.1.1. β-Blockers
4.1.2. Calcium Channel Blockers
4.1.3. Disopyramide
4.1.4. Cibenzoline
4.1.5. Late Sodium Channel Blockers
4.1.6. Angiotensin II Receptor Antagonists
4.1.7. Angiotensin Receptor Neprilysin Inhibitors
4.1.8. Septal Reduction Therapy
4.1.9. Lifestyle Interventions
4.2. Novel Therapeutic Approaches
4.2.1. Mavacamten
4.2.2. Mavacamten Trials
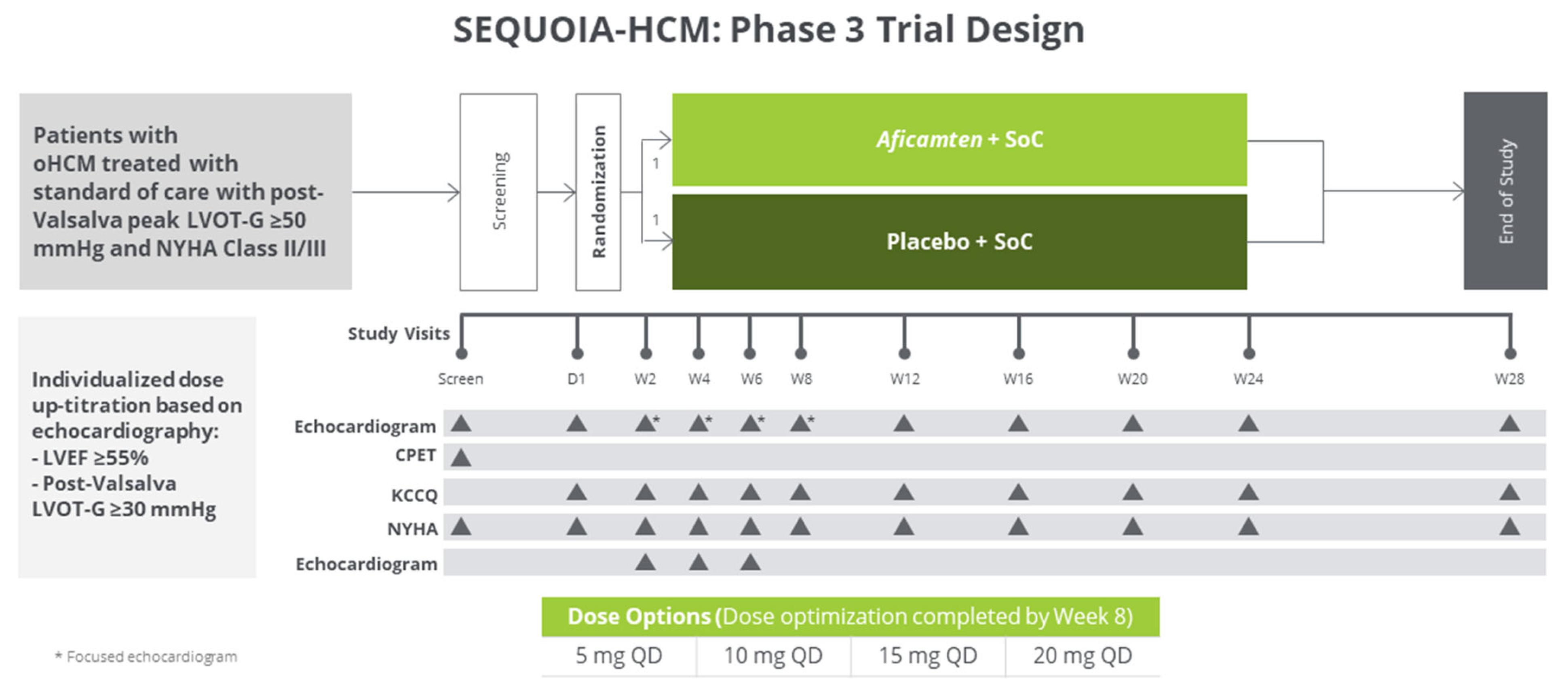
4.2.3. Aficamten
4.2.4. Aficamten Trials
5. Personalized Therapy
6. Artificial Intelligence: Hype or Hope?
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Palandri, C.; Santini, L.; Argirò, A.; Margara, F.; Doste, R.; Bueno-Orovio, A.; Olivotto, I.; Coppini, R. Pharmacological Management of Hypertrophic Cardiomyopathy: From Bench to Bedside. Drugs 2022, 82, 889–912. [Google Scholar] [CrossRef] [PubMed]
- Autore, C.; Francia, P.; Tini, G.; Musumeci, B. Old and New Therapeutic Solutions in the Treatment of Hypertrophic Cardiomyopathy. Eur. Heart J. Suppl. 2023, 25 (Suppl. B), B12–B15. [Google Scholar] [CrossRef] [PubMed]
- Ommen, S.R.; Mital, S.; Burke, M.A.; Day, S.M.; Deswal, A.; Elliott, P.; Evanovich, L.L.; Hung, J.; Joglar, J.A.; Kantor, P.; et al. 2020 AHA/ACC Guideline for the Diagnosis and Treatment of Patients with Hypertrophic Cardiomyopathy: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation 2020, 142, e558–e631. [Google Scholar] [CrossRef]
- Todde, G.; Canciello, G.; Borrelli, F.; Perillo, E.F.; Esposito, G.; Lombardi, R.; Losi, M.A. Diagnosis and Treatment of Obstructive Hypertrophic Cardiomyopathy. Cardiogenetics 2023, 13, 75–91. [Google Scholar] [CrossRef]
- Maron, B.J.; Desai, M.Y.; Nishimura, R.A.; Spirito, P.; Rakowski, H.; Towbin, J.A.; Rowin, E.J.; Maron, M.S.; Sherrid, M.V. Diagnosis and Evaluation of Hypertrophic Cardiomyopathy. J. Am. Coll. Cardiol. 2022, 79, 372–389. [Google Scholar] [CrossRef] [PubMed]
- Biddinger, K.J.; Jurgens, S.J.; Maamari, D.; Gaziano, L.; Choi, S.H.; Morrill, V.N.; Halford, J.L.; Khera, A.V.; Lubitz, S.A.; Ellinor, P.T.; et al. Rare and Common Genetic Variation Underlying the Risk of Hypertrophic Cardiomyopathy in a National Biobank. JAMA Cardiol. 2022, 7, 715–722. [Google Scholar] [CrossRef] [PubMed]
- Beltrami, M.; Fedele, E.; Fumagalli, C.; Mazzarotto, F.; Girolami, F.; Ferrantini, C.; Coppini, R.; Tofani, L.; Bertaccini, B.; Poggesi, C.; et al. Long-Term Prevalence of Systolic Dysfunction in MYBPC3 Versus MYH7-Related Hypertrophic Cardiomyopathy. Circ. Genomic. Precis. Med. 2023, 16, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Sankaranarayanan, R.; Fleming, J.E.; Garratt, J.C. Mimics of Hypertrophic Cardiomyopathy—Diagnostic Clues to Aid Early Identification of Phenocopies. Arrhythmia Electrophysiol. Rev. 2013, 2, 36–40. [Google Scholar] [CrossRef]
- Sachdev, B.; Takenaka, T.; Teraguchi, H.; Tei, C.; Lee, P.; McKenna, W.J.; Elliott, P.M. Prevalence of Anderson-Fabry Disease in Male Patients with Late Onset Hypertrophic Cardiomyopathy. Circulation 2002, 105, 1407–1411. [Google Scholar] [CrossRef]
- Ricci, F.; Bisaccia, G.; Mansour, D.; Molinari, L.V.; Di Mauro, M.; Renda, G.; Khanji, M.Y.; Gallina, S. Prognostic Significance of Late Gadolinium Enhancement in Fabry Disease—A Systematic Review and Meta-Analysis. Am. J. Cardiol. 2023, 202, 4–5. [Google Scholar] [CrossRef]
- Gange, C.A.; Link, M.S.; Maron, M.S. Utility of Cardiovascular Magnetic Resonance in the Diagnosis of Anderson-Fabry Disease. Circulation 2009, 120, e96–e97. [Google Scholar] [CrossRef] [PubMed]
- Linhart, A.; Elliott, P.M. The Heart in Anderson-Fabry Disease and Other Lysosomal Storage Disorders. Heart Br. Card. Soc. 2007, 93, 528–535. [Google Scholar] [CrossRef] [PubMed]
- Dorbala, S.; Ando, Y.; Bokhari, S.; Dispenzieri, A.; Falk, R.H.; Ferrari, V.A.; Fontana, M.; Gheysens, O.; Gillmore, J.D.; Glaudemans, A.W.J.M.; et al. ASNC/AHA/ASE/EANM/HFSA/ISA/SCMR/SNMMI Expert Consensus Recommendations for Multimodality Imaging in Cardiac Amyloidosis: Part 1 of 2-Evidence Base and Standardized Methods of Imaging. J. Nucl. Cardiol. 2019, 26, 2065–2123. [Google Scholar] [CrossRef] [PubMed]
- Dorbala, S.; Ando, Y.; Bokhari, S.; Dispenzieri, A.; Falk, R.H.; Ferrari, V.A.; Fontana, M.; Gheysens, O.; Gillmore, J.D.; Glaudemans, A.W.J.M.; et al. ASNC/AHA/ASE/EANM/HFSA/ISA/SCMR/SNMMI Expert Consensus Recommendations for Multimodality Imaging in Cardiac Amyloidosis: Part 2 of 2-Diagnostic Criteria and Appropriate Utilization. J. Nucl. Cardiol. 2020, 27, 659–673. [Google Scholar] [CrossRef] [PubMed]
- Lynch, A.; Tatangelo, M.; Ahuja, S.; Steve Fan, C.-P.; Min, S.; Lafreniere-Roula, M.; Papaz, T.; Zhou, V.; Armstrong, K.; Aziz, P.F.; et al. Risk of Sudden Death in Patients with RASopathy Hypertrophic Cardiomyopathy. J. Am. Coll. Cardiol. 2023, 81, 1035–1045. [Google Scholar] [CrossRef] [PubMed]
- Ackerman, M.J.; Garmany, R. RASopathy-Associated Cardiac Hypertrophy: A Shocking Gap in Care. J. Am. Coll. Cardiol. 2023, 81, 1046–1048. [Google Scholar] [CrossRef] [PubMed]
- Nagakura, T.; Takeuchi, M.; Yoshitani, H.; Nakai, H.; Nishikage, T.; Kokumai, M.; Otani, S.; Yoshiyama, M.; Yoshikawa, J. Hypertrophic Cardiomyopathy Is Associated with More Severe Left Ventricular Dyssynchrony than Is Hypertensive Left Ventricular Hypertrophy. Echocardiography 2007, 24, 677–684. [Google Scholar] [CrossRef]
- Boucek, D.; Jirikowic, J.; Taylor, M. Natural History of Danon Disease. Genet. Med. 2011, 13, 563–568. [Google Scholar] [CrossRef]
- Charron, P.; Villard, E.; Sébillon, P.; Laforêt, P.; Maisonobe, T.; Duboscq-Bidot, L.; Romero, N.; Drouin-Garraud, V.; Frébourg, T.; Richard, P.; et al. Danon’s Disease as a Cause of Hypertrophic Cardiomyopathy: A Systematic Survey. Heart Br. Card. Soc. 2004, 90, 842–846. [Google Scholar] [CrossRef]
- Fayssoil, A. Cardiomyopathy in Pompe’s Disease. Eur. J. Intern. Med. 2008, 19, 57–59. [Google Scholar] [CrossRef]
- Kelly, B.P.; Russell, M.W.; Hennessy, J.R.; Ensing, G.J. Severe Hypertrophic Cardiomyopathy in an Infant with a Novel PRKAG2 Gene Mutation: Potential Differences between Infantile and Adult Onset Presentation. Pediatr. Cardiol. 2009, 30, 1176–1179. [Google Scholar] [CrossRef] [PubMed]
- Wolf, C.M.; Arad, M.; Ahmad, F.; Sanbe, A.; Bernstein, S.A.; Toka, O.; Konno, T.; Morley, G.; Robbins, J.; Seidman, J.G.; et al. Reversibility of PRKAG2 Glycogen-Storage Cardiomyopathy and Electrophysiological Manifestations. Circulation 2008, 117, 144–154. [Google Scholar] [CrossRef] [PubMed]
- Galanti, K.; Di Marino, M.; Mantini, C.; Gallina, S.; Ricci, F. Burnt-out Cori-Forbes Cardiomyopathy. Eur. Heart J. Case Rep. 2023, 7, ytad326. [Google Scholar] [CrossRef] [PubMed]
- Maron, B.J. Distinguishing Hypertrophic Cardiomyopathy from Athlete’s Heart: A Clinical Problem of Increasing Magnitude and Significance. Heart Br. Card. Soc. 2005, 91, 1380–1382. [Google Scholar] [CrossRef] [PubMed]
- De Innocentiis, C.; Ricci, F.; Khanji, M.Y.; Aung, N.; Tana, C.; Verrengia, E.; Petersen, S.E.; Gallina, S. Athlete’s Heart: Diagnostic Challenges and Future Perspectives. Sports Med. 2018, 48, 2463–2477. [Google Scholar] [CrossRef] [PubMed]
- Ricci, F.; Banihashemi, B.; Pirouzifard, M.; Sundquist, J.; Sundquist, K.; Sutton, R.; Fedorowski, A.; Zöller, B. Familial Risk of Dilated and Hypertrophic Cardiomyopathy: A National Family Study in Sweden. ESC Heart Fail. 2023, 10, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, A.; Sharma, S. Hypertrophic Cardiomyopathy in Athletes. Eur. Cardiol. 2017, 12, 80–82. [Google Scholar] [CrossRef] [PubMed]
- Canepa, M.; Pozios, I.; Vianello, P.F.; Ameri, P.; Brunelli, C.; Ferrucci, L.; Abraham, T.P. Distinguishing Ventricular Septal Bulge versus Hypertrophic Cardiomyopathy in the Elderly. Heart Br. Card. Soc. 2016, 102, 1087–1094. [Google Scholar] [CrossRef]
- Sebastian, S.A.; Panthangi, V.; Singh, K.; Rayaroth, S.; Gupta, A.; Shantharam, D.; Rasool, B.Q.; Padda, I.; Co, E.L.; Johal, G. Hypertrophic Cardiomyopathy: Current Treatment and Future Options. Curr. Probl. Cardiol. 2023, 48, 101552. [Google Scholar] [CrossRef]
- Nagueh, S.F.; Phelan, D.; Abraham, T.; Armour, A.; Desai, M.Y.; Dragulescu, A.; Gilliland, Y.; Lester, S.J.; Maldonado, Y.; Mohiddin, S.; et al. Recommendations for Multimodality Cardiovascular Imaging of Patients with Hypertrophic Cardiomyopathy: An Update from the American Society of Echocardiography, in Collaboration with the American Society of Nuclear Cardiology, the Society for Cardiovascular Magnetic Resonance, and the Society of Cardiovascular Computed Tomography. J. Am. Soc. Echocardiogr. 2022, 35, 533–569. [Google Scholar] [CrossRef]
- Turvey, L.; Augustine, D.X.; Robinson, S.; Oxborough, D.; Stout, M.; Smith, N.; Harkness, A.; Williams, L.; Steeds, R.P.; Bradlow, W. Transthoracic Echocardiography of Hypertrophic Cardiomyopathy in Adults: A Practical Guideline from the British Society of Echocardiography. Echo Res. Pract. 2021, 8, G61–G86. [Google Scholar] [CrossRef] [PubMed]
- D’Andrea, A.; Caso, P.; Cuomo, S.; Salerno, G.; Scarafile, R.; Mita, C.; De Corato, G.; Sarubbi, B.; Scherillo, M.; Calabrò, R. Prognostic Value of Intra-Left Ventricular Electromechanical Asynchrony in Patients with Mild Hypertrophic Cardiomyopathy Compared with Power Athletes. Br. J. Sports Med. 2006, 40, 244–250; discussion 244–250. [Google Scholar] [CrossRef] [PubMed]
- Haland, T.F.; Almaas, V.M.; Hasselberg, N.E.; Saberniak, J.; Leren, I.S.; Hopp, E.; Edvardsen, T.; Haugaa, K.H. Strain Echocardiography Is Related to Fibrosis and Ventricular Arrhythmias in Hypertrophic Cardiomyopathy. Eur. Heart J. Cardiovasc. Imaging 2016, 17, 613–621. [Google Scholar] [CrossRef]
- Erden, M.; van Velzen, H.G.; Menting, M.E.; van den Bosch, A.E.; Ren, B.; Michels, M.; Vletter, W.B.; van Domburg, R.T.; Schinkel, A.F.L. Three-Dimensional Echocardiography for the Assessment of Left Ventricular Geometry and Papillary Muscle Morphology in Hypertrophic Cardiomyopathy. J. Ultrasound 2018, 21, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Arbelo, E.; Protonotarios, A.; Gimeno, J.R.; Arbustini, E.; Barriales-Villa, R.; Basso, C.; Bezzina, C.R.; Biagini, E.; Blom, N.A.; de Boer, R.A.; et al. 2023 ESC Guidelines for the Management of Cardiomyopathies: Developed by the Task Force on the Management of Cardiomyopathies of the European Society of Cardiology (ESC). Eur. Heart J. 2023, ehad194, online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Zeppenfeld, K.; Tfelt-Hansen, J.; De Riva, M.; Winkel, B.G.; Behr, E.R.; Blom, N.A.; Charron, P.; Corrado, D.; Dagres, N.; De Chillou, C. 2022 ESC Guidelines for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death. Eur. Heart J. 2022, 43, 3997–4126. [Google Scholar] [CrossRef] [PubMed]
- Penetrance of Hypertrophic Cardiomyopathy in Sarcomere Protein Mutation Carriers. Available online: https://pubmed.ncbi.nlm.nih.gov/32731933/ (accessed on 21 July 2023).
- Joy, G.; Kelly, C.I.; Webber, M.; Pierce, I.; Teh, I.; McGrath, L.; Velazquez, P.; Hughes, R.K.; Kotwal, H.; Das, A.; et al. Microstructural and Microvascular Phenotype of Sarcomere Mutation Carriers and Overt Hypertrophic Cardiomyopathy. Circulation 2023. ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Lillo, R.; Graziani, F.; Franceschi, F.; Iannaccone, G.; Massetti, M.; Olivotto, I.; Crea, F.; Liuzzo, G. Inflammation across the Spectrum of Hypertrophic Cardiac Phenotypes. Heart Fail. Rev. 2023, 28, 1065–1075. [Google Scholar] [CrossRef]
- Matthia, E.L.; Setteducato, M.L.; Elzeneini, M.; Vernace, N.; Salerno, M.; Kramer, C.M.; Keeley, E.C. Circulating Biomarkers in Hypertrophic Cardiomyopathy. J. Am. Heart Assoc. 2022, 11, e027618. [Google Scholar] [CrossRef]
- Lampert, R.; Ackerman, M.J.; Marino, B.S.; Burg, M.; Ainsworth, B.; Salberg, L.; Tome Esteban, M.T.; Ho, C.Y.; Abraham, R.; Balaji, S.; et al. Vigorous Exercise in Patients with Hypertrophic Cardiomyopathy. JAMA Cardiol. 2023, 8, 595–605. [Google Scholar] [CrossRef]
- Delise, P.; Mos, L.; Sciarra, L.; Basso, C.; Biffi, A.; Cecchi, F.; Colivicchi, F.; Corrado, D.; D’Andrea, A.; Di Cesare, E.; et al. Italian Cardiological Guidelines (COCIS) for Competitive Sport Eligibility in Athletes with Heart Disease: Update 2020. J. Cardiovasc. Med. 2021, 22, 874–891. [Google Scholar] [CrossRef] [PubMed]
- 2014 ESC Guidelines on Diagnosis and Management of Hypertrophic Cardiomyopathy: The Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur. Heart J. 2014, 35, 2733–2779. [CrossRef]
- Joy, G.; Moon, J.C.; Lopes, L.R. Detection of Subclinical Hypertrophic Cardiomyopathy. Nat. Rev. Cardiol. 2023, 20, 369–370. [Google Scholar] [CrossRef] [PubMed]
- Rapezzi, C. EXPLORER-HCM: Il mavacamten nel trattamento della cardiomiopatia ipertrofica ostruttiva sintomatica. G. Ital. Cardiol. 2021, 22, 30–32. [Google Scholar]
- Argirò, A.; Zampieri, M.; Marchi, A.; Franco, A.D.; Pàlinkàs, E.D.; Biagioni, G.; Chiti, C.; Mazzoni, C.; Fornaro, A.; Targetti, M.; et al. Approcci terapeutici nella cardiomiopatia ipertrofica: Dal controllo dei sintomi alla terapia di precisione. G. Ital. Cardiol. 2023, 24, 1–8. [Google Scholar]
- Rosenzveig, A.; Garg, N.; Rao, S.J.; Kanwal, A.K.; Kanwal, A.; Aronow, W.S.; Martinez, M.W. Current and Emerging Pharmacotherapy for the Management of Hypertrophic Cardiomyopathy. Expert Opin. Pharmacother. 2023, 24, 1349–1360. [Google Scholar] [CrossRef] [PubMed]
- Ammirati, E.; Contri, R.; Coppini, R.; Cecchi, F.; Frigerio, M.; Olivotto, I. Pharmacological Treatment of Hypertrophic Cardiomyopathy: Current Practice and Novel Perspectives: Pharmacological Treatment of HCM. Eur. J. Heart Fail. 2016, 18, 1106–1118. [Google Scholar] [CrossRef]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. 2021 ESC Guidelines for the Diagnosis and Treatment of Acute and Chronic Heart Failure. Eur. Heart J. 2021, 42, 3599–3726. [Google Scholar] [CrossRef]
- Frishman, W.H. Calcium Channel Blockers: Differences between Subclasses. Am. J. Cardiovasc. Drugs 2007, 7 (Suppl. S1), 17–23. [Google Scholar] [CrossRef]
- Coppini, R.; Santini, L.; Olivotto, I.; Ackerman, M.J.; Cerbai, E. Abnormalities in Sodium Current and Calcium Homoeostasis as Drivers of Arrhythmogenesis in Hypertrophic Cardiomyopathy. Cardiovasc. Res. 2020, 116, 1585–1599. [Google Scholar] [CrossRef]
- Gilligan, D.M.; Chan, W.L.; Joshi, J.; Clarke, P.; Fletcher, A.; Krikler, S.; Oakley, C.M. A Double-Blind, Placebo-Controlled Crossover Trial of Nadolol and Verapamil in Mild and Moderately Symptomatic Hypertrophic Cardiomyopathy. J. Am. Coll. Cardiol. 1993, 21, 1672–1679. [Google Scholar] [CrossRef] [PubMed]
- Sherrid, M.V.; Barac, I.; McKenna, W.J.; Elliott, P.M.; Dickie, S.; Chojnowska, L.; Casey, S.; Maron, B.J. Multicenter Study of the Efficacy and Safety of Disopyramide in Obstructive Hypertrophic Cardiomyopathy. J. Am. Coll. Cardiol. 2005, 45, 1251–1258. [Google Scholar] [CrossRef] [PubMed]
- Coppini, R.; Ferrantini, C.; Pioner, J.M.; Santini, L.; Wang, Z.J.; Palandri, C.; Scardigli, M.; Vitale, G.; Sacconi, L.; Stefàno, P.; et al. Electrophysiological and Contractile Effects of Disopyramide in Patients with Obstructive Hypertrophic Cardiomyopathy: A Translational Study. JACC Basic Transl. Sci. 2019, 4, 795–813. [Google Scholar] [CrossRef] [PubMed]
- Maurizi, N.; Chiriatti, C.; Fumagalli, C.; Targetti, M.; Passantino, S.; Antiochos, P.; Skalidis, I.; Chiti, C.; Biagioni, G.; Tomberli, A.; et al. Real-World Use and Predictors of Response to Disopyramide in Patients with Obstructive Hypertrophic Cardiomyopathy. J. Clin. Med. 2023, 12, 2725. [Google Scholar] [CrossRef] [PubMed]
- Morady, F.; Scheinman, M.M.; Desai, J. Disopyramide. Ann. Intern. Med. 1982, 96, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Hamada, M.; Shigematsu, Y.; Ikeda, S.; Hara, Y.; Okayama, H.; Kodama, K.; Ochi, T.; Hiwada, K. Class Ia Antiarrhythmic Drug Cibenzoline: A New Approach to the Medical Treatment of Hypertrophic Obstructive Cardiomyopathy. Circulation 1997, 96, 1520–1524. [Google Scholar] [CrossRef]
- Anan, R. Editorial: Cibenzoline for Left Ventricular Outflow Tract Obstruction in Tako-Tsubo Cardiomyopathy and Hypertrophic Cardiomyopathy. J. Cardiol. Cases 2015, 11, 158–159. [Google Scholar] [CrossRef][Green Version]
- Olivotto, I.; Camici, P.G.; Merlini, P.A.; Rapezzi, C.; Patten, M.; Climent, V.; Sinagra, G.; Tomberli, B.; Marin, F.; Ehlermann, P.; et al. Efficacy of Ranolazine in Patients with Symptomatic Hypertrophic Cardiomyopathy: The RESTYLE-HCM Randomized, Double-Blind, Placebo-Controlled Study. Circ. Heart Fail. 2018, 11, e004124. [Google Scholar] [CrossRef]
- Olivotto, I.; Hellawell, J.L.; Farzaneh-Far, R.; Blair, C.; Coppini, R.; Myers, J.; Belardinelli, L.; Maron, M.S. Novel Approach Targeting the Complex Pathophysiology of Hypertrophic Cardiomyopathy: The Impact of Late Sodium Current Inhibition on Exercise Capacity in Subjects with Symptomatic Hypertrophic Cardiomyopathy (LIBERTY-HCM) Trial. Circ. Heart Fail. 2016, 9, e002764. [Google Scholar] [CrossRef]
- Axelsson, A.; Iversen, K.; Vejlstrup, N.; Ho, C.; Norsk, J.; Langhoff, L.; Ahtarovski, K.; Corell, P.; Havndrup, O.; Jensen, M.; et al. Efficacy and Safety of the Angiotensin II Receptor Blocker Losartan for Hypertrophic Cardiomyopathy: The INHERIT Randomised, Double-Blind, Placebo-Controlled Trial. Lancet Diabetes Endocrinol. 2015, 3, 123–131. [Google Scholar] [CrossRef]
- Martin, T.G.; Juarros, M.A.; Leinwand, L.A. Regression of Cardiac Hypertrophy in Health and Disease: Mechanisms and Therapeutic Potential. Nat. Rev. Cardiol. 2023, 20, 347–363. [Google Scholar] [CrossRef] [PubMed]
- Olivotto, I.; Ashley, E.A. INHERIT (INHibition of the Renin Angiotensin System in Hypertrophic Cardiomyopathy and the Effect on Hypertrophy—A Randomised Intervention Trial with Losartan). Glob. Cardiol. Sci. Pract. 2015, 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.Y.; Day, S.M.; Axelsson, A.; Russell, M.W.; Zahka, K.; Lever, H.M.; Pereira, A.C.; Colan, S.D.; Margossian, R.; Murphy, A.M.; et al. Valsartan in Early-Stage Hypertrophic Cardiomyopathy: A Randomized Phase 2 Trial. Nat. Med. 2021, 27, 1818–1824. [Google Scholar] [CrossRef] [PubMed]
- Packer, M.; McMurray, J.J.; Desai, A.S.; Gong, J.; Lefkowitz, M.P.; Rizkala, A.R.; Rouleau, J.L.; Shi, V.C.; Solomon, S.D.; Swedberg, K.; et al. Angiotensin Receptor Neprilysin Inhibition Compared with Enalapril on the Risk of Clinical Progression in Surviving Patients with Heart Failure. Circulation 2015, 131, 54–61. [Google Scholar] [CrossRef]
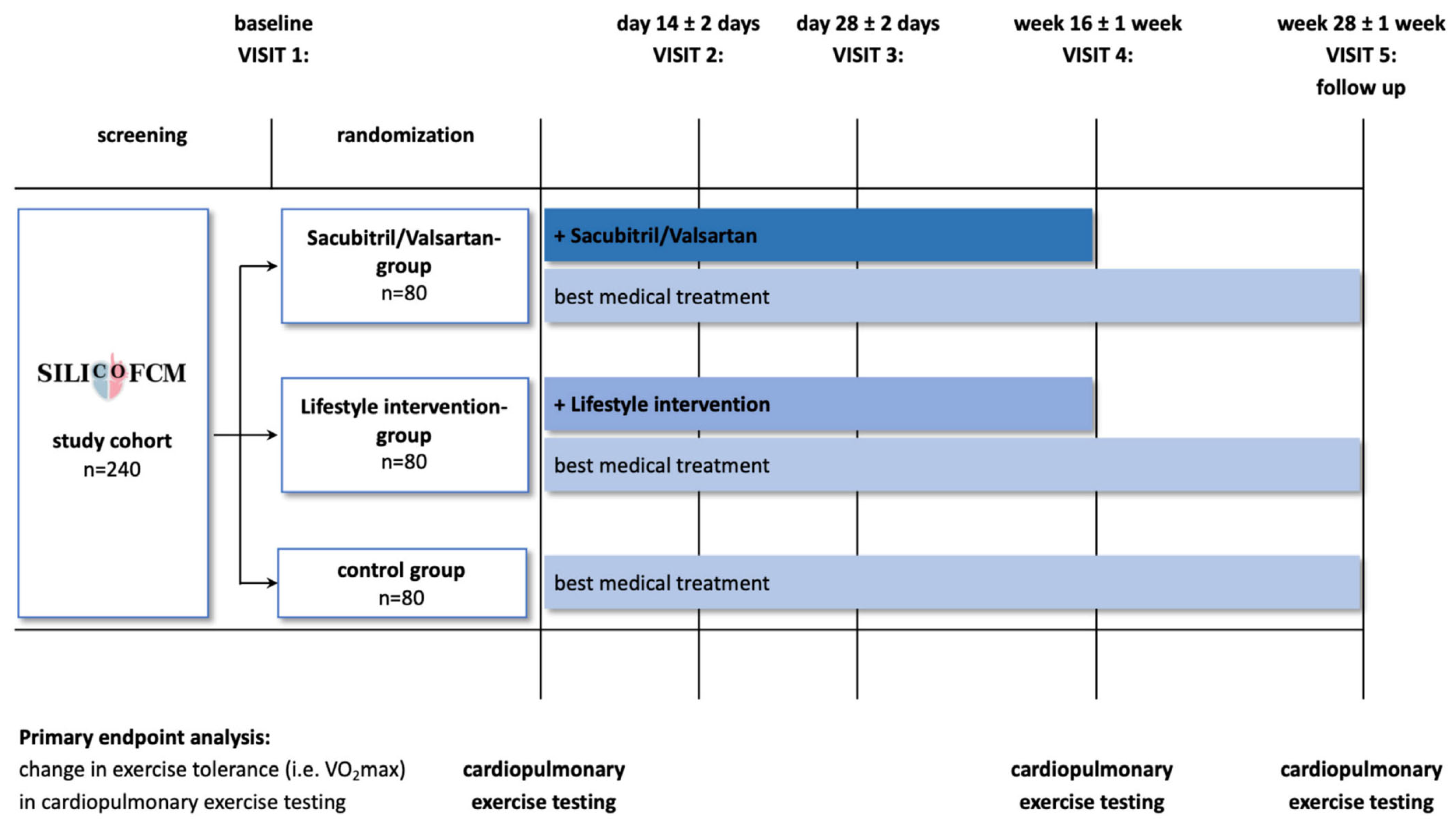
- Tafelmeier, M.; Baessler, A.; Wagner, S.; Unsoeld, B.; Preveden, A.; Barlocco, F.; Tomberli, A.; Popovic, D.; Brennan, P.; MacGowan, G.A.; et al. Design of the SILICOFCM Study: Effect of Sacubitril/Valsartan vs Lifestyle Intervention on Functional Capacity in Patients with Hypertrophic Cardiomyopathy. Clin. Cardiol. 2020, 43, 430–440. [Google Scholar] [CrossRef]
- Nishimura, R.A.; Ommen, S.R. Septal Reduction Therapy for Obstructive Hypertrophic Cardiomyopathy and Sudden Death. Circ. Cardiovasc. Interv. 2010, 3, 91–93. [Google Scholar] [CrossRef]
- Ommen, S.R.; Mital, S.; Burke, M.A.; Day, S.M.; Deswal, A.; Elliott, P.; Evanovich, L.L.; Hung, J.; Joglar, J.A.; Kantor, P.; et al. 2020 AHA/ACC Guideline for the Diagnosis and Treatment of Patients with Hypertrophic Cardiomyopathy: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. J. Am. Coll. Cardiol. 2020, 76, e159–e240. [Google Scholar] [CrossRef] [PubMed]
- Maron, B.J.; Desai, M.Y.; Nishimura, R.A.; Spirito, P.; Rakowski, H.; Towbin, J.A.; Dearani, J.A.; Rowin, E.J.; Maron, M.S.; Sherrid, M.V. Management of Hypertrophic Cardiomyopathy. J. Am. Coll. Cardiol. 2022, 79, 390–414. [Google Scholar] [CrossRef]
- Pelliccia, F.; Niccoli, G.; Gragnano, F.; Limongelli, G.; Moscarella, E.; Andò, G.; Esposito, A.; Stabile, E.; Ussia, G.P.; Tarantini, G.; et al. Alcohol Septal Ablation for Hypertrophic Obstructive Cardiomyopathy: A Contemporary Reappraisal. EuroIntervention 2019, 15, 411–417. [Google Scholar] [CrossRef]
- Gersh, B.J.; Maron, B.J.; Bonow, R.O.; Dearani, J.A.; Fifer, M.A.; Link, M.S.; Naidu, S.S.; Nishimura, R.A.; Ommen, S.R.; Rakowski, H.; et al. 2011 ACCF/AHA Guideline for the Diagnosis and Treatment of Hypertrophic Cardiomyopathy: A Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Developed in Collaboration with the American Association for Thoracic Surgery, American Society of Echocardiography, American Society of Nuclear Cardiology, Heart Failure Society of America, Heart Rhythm Society, Society for Cardiovascular Angiography and Interventions, and Society of Thoracic Surgeons. J. Am. Coll. Cardiol. 2011, 58, e212–e260. [Google Scholar] [CrossRef]
- Pelliccia, A. New Evidences Recommend an Active Lifestyle in Young HCM Patients. Int. J. Cardiol. 2022, 369, 82–83. [Google Scholar] [CrossRef] [PubMed]
- Stewart, S.; Mason, D.T.; Braunwald, E. Impaired Rate of Left Ventricular Filling in Idiopathic Hypertrophic Subaortic Stenosis and Valvular Aortic Stenosis. Circulation 1968, 37, 8–14. [Google Scholar] [CrossRef] [PubMed]
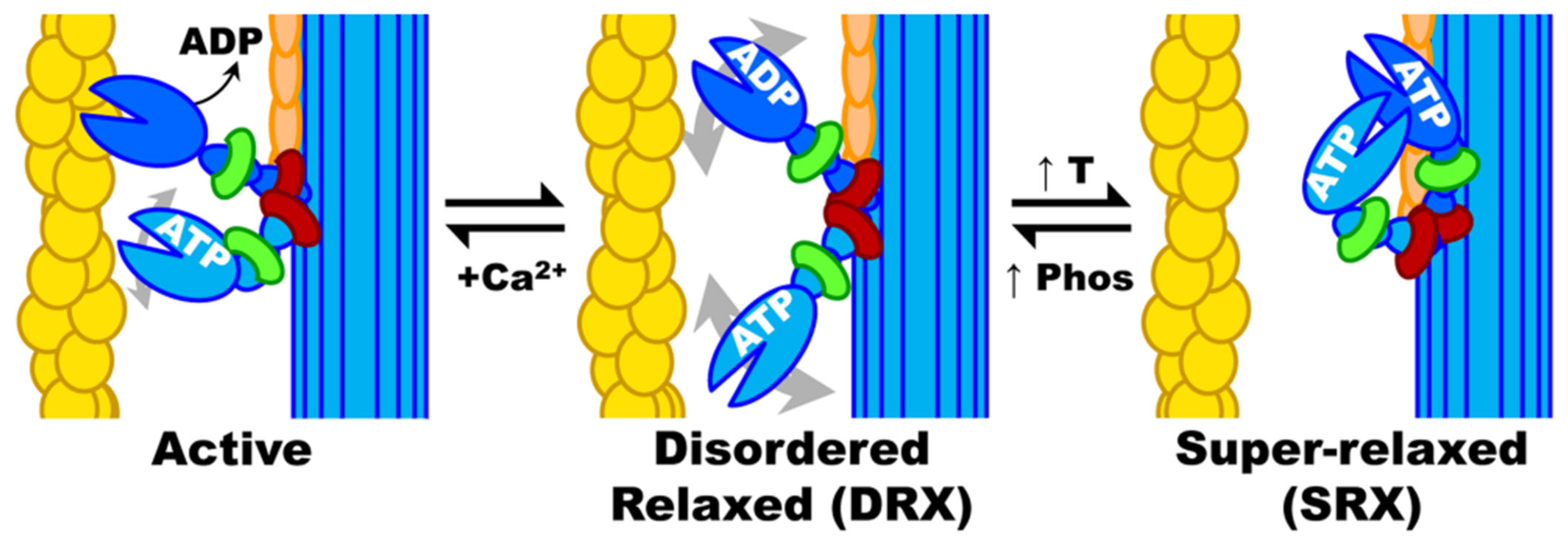
- Phung, L.; Karvinen, S.; Colson, B.; Thomas, D.; Lowe, D. Age Affects Myosin Relaxation States in Skeletal Muscle Fibers of Female but Not Male Mice. PLoS ONE 2018, 13, e0199062. [Google Scholar] [CrossRef] [PubMed]
- Toepfer, C.N.; Garfinkel, A.C.; Venturini, G.; Wakimoto, H.; Repetti, G.; Alamo, L.; Sharma, A.; Agarwal, R.; Ewoldt, J.F.; Cloonan, P.; et al. Myosin Sequestration Regulates Sarcomere Function, Cardiomyocyte Energetics, and Metabolism, Informing the Pathogenesis of Hypertrophic Cardiomyopathy. Circulation 2020, 141, 828–842. [Google Scholar] [CrossRef] [PubMed]
- Toepfer, C.N.; Wakimoto, H.; Garfinkel, A.C.; McDonough, B.; Liao, D.; Jiang, J.; Tai, A.C.; Gorham, J.M.; Lunde, I.G.; Lun, M.; et al. Hypertrophic Cardiomyopathy Mutations in MYBPC3 Dysregulate Myosin. Sci. Transl. Med. 2019, 11, eaat1199. [Google Scholar] [CrossRef] [PubMed]
- Volkmann, N.; Lui, H.; Hazelwood, L.; Trybus, K.M.; Lowey, S.; Hanein, D. The R403Q Myosin Mutation Implicated in Familial Hypertrophic Cardiomyopathy Causes Disorder at the Actomyosin Interface. PLoS ONE 2007, 2, e1123. [Google Scholar] [CrossRef] [PubMed]
- Green, E.M.; Wakimoto, H.; Anderson, R.L.; Evanchik, M.J.; Gorham, J.M.; Harrison, B.C.; Henze, M.; Kawas, R.; Oslob, J.D.; Rodriguez, H.M.; et al. A Small-Molecule Inhibitor of Sarcomere Contractility Suppresses Hypertrophic Cardiomyopathy in Mice. Science 2016, 351, 617–621. [Google Scholar] [CrossRef]
- Spudich, J.A. Hypertrophic and Dilated Cardiomyopathy: Four Decades of Basic Research on Muscle Lead to Potential Therapeutic Approaches to These Devastating Genetic Diseases. Biophys. J. 2014, 106, 1236–1249. [Google Scholar] [CrossRef]
- Ashrafian, H.; Redwood, C.; Blair, E.; Watkins, H. Hypertrophic Cardiomyopathy:A Paradigm for Myocardial Energy Depletion. Trends Genet. TIG 2003, 19, 263–268. [Google Scholar] [CrossRef]
- Schenk, A.; Fields, N. Mavacamten-A Targeted Therapy for Hypertrophic Cardiomyopathy. J. Cardiovasc. Pharmacol. 2023, 81, 317–326. [Google Scholar] [CrossRef]
- MyoKardia, Inc. A Phase 2 Open-Label Pilot Study to Evaluate Efficacy, Pharmacokinetics, Pharmacodynamics, Safety, and Tolerability of MYK-461 in Subjects with Symptomatic Hypertrophic Cardiomyopathy and Left Ventricular Outflow Tract Obstruction; Clinical Trial Registration NCT02842242. clinicaltrials.gov. 2021. Available online: https://clinicaltrials.gov/study/NCT02842242 (accessed on 12 July 2023).
- Heitner, S.B.; Jacoby, D.; Lester, S.J.; Owens, A.; Wang, A.; Zhang, D.; Lambing, J.; Lee, J.; Semigran, M.; Sehnert, A.J. Mavacamten Treatment for Obstructive Hypertrophic Cardiomyopathy: A Clinical Trial. Ann. Intern. Med. 2019, 170, 741–748. [Google Scholar] [CrossRef] [PubMed]
- Masri, A.; Lester, S.J.; Stendahl, J.; Hegde, S.M.; Sehnert, A.; Balaratnam, G.; Fox, S.; Wang, A. Long-Term Safety and Efficacy of Mavacamten in Patients (Pts) with Symptomatic Obstructive Hypertrophic Cardiomyopathy (Hcm): Updated Results from the Pioneer-Ole Study. J. Am. Coll. Cardiol. 2023, 81 (Suppl. S8), 346. [Google Scholar] [CrossRef]
- Heitner, S.B.; Lester, S.; Wang, A.; Hegde, S.M.; Fang, L.; Balaratnam, G.; Sehnert, A.J.; Jacoby, D. Abstract 13962: Precision Pharmacological Treatment for Obstructive Hypertrophic Cardiomyopathy with Mavacamten: One-Year Results From PIONEER-OLE. Circulation 2019, 140 (Suppl. S1), A13962. [Google Scholar] [CrossRef]
- Ho, C.Y.; Mealiffe, M.E.; Bach, R.G.; Bhattacharya, M.; Choudhury, L.; Edelberg, J.M.; Hegde, S.M.; Jacoby, D.; Lakdawala, N.K.; Lester, S.J.; et al. Evaluation of Mavacamten in Symptomatic Patients with Nonobstructive Hypertrophic Cardiomyopathy. J. Am. Coll. Cardiol. 2020, 75, 2649–2660. [Google Scholar] [CrossRef] [PubMed]
- Olivotto, I.; Oreziak, A.; Barriales-Villa, R.; Abraham, T.P.; Masri, A.; Garcia-Pavia, P.; Saberi, S.; Lakdawala, N.K.; Wheeler, M.T.; Owens, A.; et al. Mavacamten for Treatment of Symptomatic Obstructive Hypertrophic Cardiomyopathy (EXPLORER-HCM): A Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet 2020, 396, 759–769. [Google Scholar] [CrossRef] [PubMed]
- Desai, M.Y.; Owens, A.; Geske, J.B.; Wolski, K.; Naidu, S.S.; Smedira, N.G.; Cremer, P.C.; Schaff, H.; McErlean, E.; Sewell, C.; et al. Myosin Inhibition in Patients with Obstructive Hypertrophic Cardiomyopathy Referred for Septal Reduction Therapy. J. Am. Coll. Cardiol. 2022, 80, 95–108. [Google Scholar] [CrossRef]
- Rader, F.; Choudhury, L.; Saberi, S.; Fermin, D.; Wheeler, M.T.; Abraham, T.P.; Oreziak, A.; Garcia, P.P.; Zwas, D.; Sehnert, A.J.; et al. Long-Term Safety of Mavacamten in Patients with Obstructive Hypertrophic Cardiomyopathy: Interim Results of the Mava-Long Term Extension (Lte) Study. J. Am. Coll. Cardiol. 2021, 77 (Suppl. S1), 532. [Google Scholar] [CrossRef]
- Bristol-Myers Squibb. A Randomized, Double-Blind, Placebo-Controlled Clinical Study to Evaluate Mavacamten in Adults with Symptomatic Non-Obstructive Hypertrophic Cardiomyopathy; Clinical Trial Registration NCT05582395. Available online: https://clinicaltrials.gov/study/NCT05582395 (accessed on 16 July 2023).
- DeVries, J.H.; Irs, A.; Hillege, H.L. The European Medicines Agency Assessment of Mavacamten as Treatment of Symptomatic Obstructive Hypertrophic Cardiomyopathy in Adult Patients. Eur. Heart J. 2023, ehad429. [Google Scholar] [CrossRef]
- Chuang, C.; Collibee, S.; Ashcraft, L.; Wang, W.; Vander Wal, M.; Wang, X.; Hwee, D.T.; Wu, Y.; Wang, J.; Chin, E.R.; et al. Discovery of Aficamten (CK-274), a Next-Generation Cardiac Myosin Inhibitor for the Treatment of Hypertrophic Cardiomyopathy. J. Med. Chem. 2021, 64, 14142–14152. [Google Scholar] [CrossRef]
- Maron, M.S.; Masri, A.; Choudhury, L.; Olivotto, I.; Saberi, S.; Wang, A.; Garcia, P.P.; Lakdawala, N.K.; Nagueh, S.F.; Rader, F.; et al. Phase 2 Study of Aficamten in Patients with Obstructive Hypertrophic Cardiomyopathy. J. Am. Coll. Cardiol. 2023, 81, 34–45. [Google Scholar] [CrossRef]
- REDWOOD-HCM OLE: Aficamten Improves Patient-Measured Health Status. TCTMD.com. Available online: https://www.tctmd.com/news/redwood-hcm-ole-aficamten-improves-patient-measured-health-status (accessed on 18 July 2023).
- Owens, A.T.; Masri, A.; Abraham, T.P.; Choudhury, L.; Rader, F.; Symanski, J.D.; Turer, A.T.; Wong, T.C.; Tower-Rader, A.; Coats, C.J.; et al. Aficamten for Drug-Refractory Severe Obstructive Hypertrophic Cardiomyopathy in Patients Receiving Disopyramide: REDWOOD-HCM Cohort 3. J. Card. Fail. 2023. [Google Scholar] [CrossRef] [PubMed]
- Cytokinetics. A Phase 3, Multi-Center, Randomized, Double-Blind, Placebo-Controlled Trial to Evaluate the Efficacy and Safety of CK-3773274 in Adults with Symptomatic Hypertrophic Cardiomyopathy and Left Ventricular Outflow Tract Obstruction; Clinical Trial Registration NCT05186818. clinicaltrials.gov. 2023. Available online: https://clinicaltrials.gov/study/NCT05186818 (accessed on 16 July 2023).
- Cytokinetics, Incorporated. Cytokinetics Outlines Go-To-Market Strategy for Omecamtiv Mecarbil and Provides Updates on Cardiovascular Pipeline at Today’s Analyst & Investor Day. GlobeNewswire News Room. Available online: https://www.globenewswire.com/en/news-release/2021/10/07/2310261/35409/en/Cytokinetics-Outlines-Go-To-Market-Strategy-for-Omecamtiv-Mecarbil-and-Provides-Updates-on-Cardiovascular-Pipeline-at-Today-s-Analyst-Investor-Day.html (accessed on 18 July 2023).
- Corral-Acero, J.; Margara, F.; Marciniak, M.; Rodero, C.; Loncaric, F.; Feng, Y.; Gilbert, Y.; Fernandes, J.F.; Bukhari, H.A.; Wajdan, A.; et al. The ‘Digital Twin’ to enable the vision of precision cardiology. Eur. Heart J. 2020, 41, 4556–4564. [Google Scholar] [CrossRef] [PubMed]
- Passini, E.; Mincholé, A.; Coppini, R.; Cerbai, E.; Rodriguez, B.; Severi, S.; Bueno-Orovio, A. Mechanisms of Pro-Arrhythmic Abnormalities in Ventricular Repolarisation and Anti-Arrhythmic Therapies in Human Hypertrophic Cardiomyopathy. J. Mol. Cell. Cardiol. 2016, 96, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Sewanan, L.R.; Moore, J.R.; Lehman, W.; Campbell, S.G. Predicting Effects of Tropomyosin Mutations on Cardiac Muscle Contraction through Myofilament Modeling. Front. Physiol. 2016, 7, 473. [Google Scholar] [CrossRef] [PubMed]
- Margara, F.; Wang, Z.J.; Levrero-Florencio, F.; Santiago, A.; Vázquez, M.; Bueno-Orovio, A.; Rodriguez, B. In-Silico Human Electro-Mechanical Ventricular Modelling and Simulation for Drug-Induced pro-Arrhythmia and Inotropic Risk Assessment. Prog. Biophys. Mol. Biol. 2021, 159, 58–74. [Google Scholar] [CrossRef] [PubMed]
- Musunuru, K. Genome Editing: The Recent History and Perspective in Cardiovascular Diseases. J. Am. Coll. Cardiol. 2017, 70, 2808–2821. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Marti-Gutierrez, N.; Park, S.-W.; Wu, J.; Lee, Y.; Suzuki, K.; Koski, A.; Ji, D.; Hayama, T.; Ahmed, R.; et al. Correction of a Pathogenic Gene Mutation in Human Embryos. Nature 2017, 548, 413–419. [Google Scholar] [CrossRef]
- Bongianino, R.; Denegri, M.; Mazzanti, A.; Lodola, F.; Vollero, A.; Boncompagni, S.; Fasciano, S.; Rizzo, G.; Mangione, D.; Barbaro, S.; et al. Allele-Specific Silencing of Mutant MRNA Rescues Ultrastructural and Arrhythmic Phenotype in Mice Carriers of the R4496C Mutation in the Ryanodine Receptor Gene (RYR2). Circ. Res. 2017, 121, 525–536. [Google Scholar] [CrossRef]
- Reichart, D.; Newby, G.A.; Wakimoto, H.; Lun, M.; Gorham, J.M.; Curran, J.J.; Raguram, A.; DeLaughter, D.M.; Conner, D.A.; Marsiglia, J.D.C.; et al. Efficient in Vivo Genome Editing Prevents Hypertrophic Cardiomyopathy in Mice. Nat. Med. 2023, 29, 412–421. [Google Scholar] [CrossRef]
- Tison, G.H.; Siontis, K.C.; Abreau, S.; Attia, Z.; Agarwal, P.; Balasubramanyam, A.; Li, Y.; Sehnert, A.J.; Edelberg, J.M.; Friedman, P.A.; et al. Assessment of Disease Status and Treatment Response with Artificial Intelligence−Enhanced Electrocardiography in Obstructive Hypertrophic Cardiomyopathy. J. Am. Coll. Cardiol. 2022, 79, 1032–1034. [Google Scholar] [CrossRef]
- Tison, G.H.; Zhang, J.; Delling, F.N.; Deo, R.C. Automated and Interpretable Patient ECG Profiles for Disease Detection, Tracking, and Discovery. Circ. Cardiovasc. Qual. Outcomes 2019, 12, e005289. [Google Scholar] [CrossRef] [PubMed]
- Carrington, M.; Providência, R.; Chahal, C.A.A.; Ricci, F.; Epstein, A.E.; Gallina, S.; Fedorowski, A.; Sutton, R.; Khanji, M.Y. Monitoring and Diagnosis of Intermittent Arrhythmias: Evidence-Based Guidance and Role of Novel Monitoring Strategies. Eur. Heart J. Open 2022, 2, oeac072. [Google Scholar] [CrossRef] [PubMed]
- Carrington, M.; Providência, R.; Chahal, C.A.A.; Ricci, F.; Epstein, A.E.; Gallina, S.; Fedorowski, A.; Sutton, R.; Khanji, M.Y. Clinical Applications of Heart Rhythm Monitoring Tools in Symptomatic Patients and for Screening in High-Risk Groups. EP Eur. 2022, 24, 1721–1729. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inheritance Pattern | Ancillary Signs and Symptoms | ECG Abnormalities | Laboratory Findings | Echocardiography | CMR Imaging | |
|---|---|---|---|---|---|---|
| Sarcomeric HCM | AD | Uncommon | - High QRS voltages - LV strain pattern - Giant TWI - Q waves of pseudo-necrosis | - Moderate-to-severe LVH (asymmetrical and septal but potentially found at any location) - 10–20% can have RVH - Diastolic dysfunction - LVOT obstruction - Mitral valve abnormalities (mitral SAM, leaflets elongation, dysplasia, prolapse, chordal elongation, laxity, and hypermobility) - Papillary muscle abnormalities (hypertrophy, splaying and apical displaced insertion) - Atrial enlargement - Apical aneurysm | - LGE at RV insertion points and intramural patchy more frequently observe at sites of hypertrophied segments - Perfusion defects - Increased values of T1 mapping indices - Increased T2 mapping/T2w hyperintensity in hot-phase of disease | |
| Anderson–Fabry disease | X-linked | - Visual impairment - Sensorineural deafness - Paranesthesia/sensory abnormalities - Angiokeratoma - Higher stroke risk - Males > females, but females can be affected (lyonization) from mild conduction disease to overt oHCM | - Short PR interval - Pre-excitation - AV block | - In males, low or undetectable alpha-1-galactosidase - Proteinuria with/without reduced glomerular filtration rate | - Concentric LVH ((but can have LVOTO) - Increased atrioventricular valve thickness - Increased RV free wall thickness - Global hypokinesia (with/without LV dilatation) | - Basal-mid infero-lateral LGE - Low T1 mapping values (accumulation/hypertrophy phases) - T1 pseudonormalisation (advanced stage) - Progressive T1 mapping dispersion |
| Cardiac amyloidosis | AD (for h-TTR) | - Carpal tunnel syndrome (bilateral) - Visual impairment - Paranesthesia/sensory abnormalities -Autonomic dysfunction | - Low QRS voltage - AV block -Pseudoinfarct pattern - Bundle branch block | Proteinuria with/without reduced glomerular filtration rate | - Ground-glass appearance of myocardium - Thickened interatrial septum, atrioventricular valve thickness, RV free wall - Pericardial effusion - Global hypokinesia (with/without LV dilatation) - relative longitudinal apical sparing (cherry apex) | - Diffuse subendocardial LGE (zebra pattern) - Abnormal T1 nulling - Increased T1 mapping indices - Increased T2 mapping values (AL) |
| Mitochondrial cardiomyopathies | X-linked or matrilinear inheritance | - Sensorineural deafness - Learning difficulties/mental retardation - Visual impairment - Muscle weakness | - Short PR interval - Preexcitation - Abnormal CPET with low VO2 max | - ↑ CK - ↑ AST, ALT - Lactate | Global hypokinesia (with/without LV dilatation) | - Large amount non-ischemic LGE pattern, mostly confined to the basal LV inferolateral wall |
| Hypertensive heart disease | None | Uncommon | Isolated LVH | Microalbuminuria | - LVH symmetrical or eccentric (mild to moderate). - Normal systolic and diastolic function | Non-specific findings |
| Danon disease | X-linked | - Learning difficulties/cognitive impairment - Visual impairment | - Short PR - Pre-excitation - AV block - Extreme LVH (Sokolow > 100) | - ↑ CK - ↑ AST, ALT | - Extreme concentric LVH - Global hypokinesia (with/without LV dilatation) | - Large amount subendocardial or transmural scarring, with typical septal sparing |
| Athlete’s heart | None | Uncommon | Isolated LVH TWI in anterior precordial leads with J point elevation (consider ethnicity) | - LVH (mild to moderate) - Normal systolic function - Normal diastolic function | - Absence of LGE or junctional pattern - Low/normal T1 mapping indices |
| Imaging Features (Echo, CMR, CT) | HCM | Hypertensive Heart Disease |
|---|---|---|
| LVH | Severe, asymmetric IVS/ILW ratio > 1.3 | Mild (<15 mm, except blacks and chronic renal failure), concentric or midly asymmetric |
| LVOTO | Frequent | Rare |
| Sigmoid septum | Rare | Frequent |
| Mitral valve and papillary muscle abnormalities | Frequent | Rare |
| Crypts | Common | Rare |
| Basal-apical muscle bundles | Frequent | Rare |
| Severe longitudinal systolic dysfunction | Frequent | Rare |
| Severe strain abnormalities | Frequent | Less frequent |
| LGE | Frequent, RV insertion points, and intramural with patchy enhancement being the most common pattern | Less frequent, non-subendocardial, no specific pattern |
| Class of Drug and Mechanism of Action | Drug and Daily Dose | Side Effect | Patients | Notes |
|---|---|---|---|---|
| BBs Blockade of β-receptors | Metoprolol 25–200 mg Atenolol 25–100 mg Bisoprolol 1.25–10 mg Nadolol 20–80 mg | Hypotension Bradycardia Bronchoconstriction Fatigue Limb ischemia | oHCM, noHCM | To titrate focusing on symptoms to the maximally tolerated dose |
| CCBs Blockade of L-type calcium channels | Verapamil 120–240 mg Diltiazem 120–240 mg | Headache, dizziness Constipation Flushing HF Conduction disturbances | oHCM, noHCM | Combination of CCBs and BBs is generally not recommended |
| Disopyramide (Class IA antiarrhythmic) Blockade of INaL. Other minor effects on peak INa, L-type calcium channels, ryanodine receptors and IKr | 300–600 mg | Antagonism of muscarinic receptors (dry mouth, constipation, urinary hesitancy, etc.) | Refractory symptomatic oHCM despite BBs or CCBs | It can increase ventricular rate response in patients with atrial fibrillation (use concomitantly with AV blocking agent) |
| Cibenzoline (Class IA antiarrhythmic) Blockade of INaL and peak INa | 100–400 mg | Lower inhibitory activity on muscarinic receptors than disopyramide | oHCM | Used in Japan and Korea. Not listed in ESC or AHA/ACC Guidelines |
| Late sodium channel blockers Blockade of INaL | Ranolazine Eleclazine | Dizziness Headache Nausea | Not approved | Not listed in ESC or AHA/ACC Guidelines |
| Potassium channel blockers Dofetilide Sotalol | Caution renal failure, reduce dose Monitor QTc interval as risk of life-threatening QT prolongation and TdP | Particularly useful for AF Sotalol in low doses has beta blocking effects and higher doses class III Singh–Vaughan Williams effects Exhibits reverse use-dependent effects (i.e., more potent when bradycardic) | ||
| ARBs Blockade of AT1 receptor | Valsartan 80–320 mg | Hypotension Cough Hyperkalaemia Worsen kidney function | noHCM with HFrEF | Further evidence is desirable in early HCM (VANISH) |
| ARNIs Blockade of AT1 receptor and neprilysin | Sacubitril/Valsartan 24/26 to 97/103 mg | Hypotension Cough Hyperkalaemia Worsen kidney function | noHCM with HFrEF | Further evidence is desirable in symptomatic noHCM with HFpEF (SILICOFCM study) |
| Mavacamten (myosin inhibitor) Allosteric inhibition of cardiac myosin ATPase | 2.5–15 mg | Decrease in ejection fraction | NYHA II-III oHCM (in America) | Inducer of CYP3A4, CYP2C9, and CYP2C19. Long half-life (7–10 days) |
| Aficamten (myosin inhibitor) Allosteric inhibition of cardiac Myosin ATPase | Not established | Decrease in ejection fraction | Not yet approved | Absence of interaction with CYP3A4, CYP2C9, and CYP2C19. Short half-life (2 days) |
| Trial | Study Design | Study Population | Dose and Timeline | Sample Size | Results |
|---|---|---|---|---|---|
| EXPLORER-HCM [76] | Randomized double blind placebo control | Obstructive HCM NYHA II–III | Mavacamten 2.5–15 mg 30 weeks | 251 | Primary endpoint achieved in 37% of mavacamten patients vs. 17% placebo patients |
| MAVERICK-HCM [75] | Randomized double blind placebo control | Non-obstructive HCM NYHA class II–III | Mavacamten 16 weeks | 59 | Median NT-proBNP reduced by 53% in mavacamten group vs. 1% in placebo group Median TnI reduced by 34% in mavacamten group vs. 4% in placebo group |
| REDWOOD-HCM [81] Cohorts 1 and 2 | Randomized double blind placebo control | Obstructive HCM NYHA II–III | Aficamten 5–15 mg (Cohort 1) 10–30 mg (Cohort 2) 10 weeks | 41 | Resting LVOTO gradient reduction: median difference of −40 ± 27 mmHg −43 ± 37 mmHg Post-Valsalva LVOTO gradient reduction: −36 ± 27 mmHg −53 ± 44 mmHg |
| REDWOOD-HCM Cohort 3 [84] | Open label | Obstructive HCM NYHA I–III On disopyramide | Aficamten 5–15 mg 10 weeks | 13 | Resting LVOTO gradient reduction: −28 ± 3.2 mmHg Post-Valsalva LVOTO gradient reduction: −27 ± 5.9 mmHg |
| REDWOOD-HCM Cohort 4 [83] | Open label | Non-obstructive HCM NYHA II–III | Aficamten 5–15 mg 10 weeks | 41 | NT-proBNP reduction (mean reduction by 66%, p < 0.0001), TnI reduction (−21%, p < 0.01) |
| VALOR-HCM [77] | Randomized double blind placebo control | Obstructive HCM, referred or under active consideration for SRT | Mavacamten 2.5–15 mg 32 weeks | 112 | After 16 weeks, 17.9% of mavacamten patients eligible to SRT vs. 76.8% in placebo group (p < 0.001) |
| PIONEER-HCM [71] Cohort A | Open label | Diagnosed with HCM, resting LVOT gradient ≥ 30 mmHg and post-exercise peak LVOTO gradient ≥ 50 mmHg | Mavacamten 10–15 mg 12 weeks | 11 | pVO2 improvement: +3.5 mL/kg/min, (95%CI:1.2;5.9); Peak exercise LVOTO gradient reduction: −90 mmHg (95%CI: −138; −41) |
| PIONEER-HCM [71] Cohort B | Open label | Obstructive HCM NYHA II–III | Mavacamten 2–5 mg/die 12 weeks | 10 | pVO2 improvement +1.7 mL/kg/min, (95%CI: 0.0–3.3) Peak exercise LVOTO gradient reduction: −25 mmHg (95%CI: −47; −3.0) |
| FOREST-HCM [82] | Open label | Obstructive HCM | Aficamten 5–20 mg 48 weeks | 38 | Resting LVOTO gradient reduction: −32 ± 28 Resting LVOTO gradient reduction: −47 ± 28 |
| MAVA-LTE [78] | Open label | Obstructive HCM | Mavacamten 2.5–15 mg 36 weeks | 137 | Resting LVOTO gradient reduction: −27.4 ± 33 Resting LVOTO gradient reduction: −45.7 ± 39.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ottaviani, A.; Mansour, D.; Molinari, L.V.; Galanti, K.; Mantini, C.; Khanji, M.Y.; Chahal, A.A.; Zimarino, M.; Renda, G.; Sciarra, L.; et al. Revisiting Diagnosis and Treatment of Hypertrophic Cardiomyopathy: Current Practice and Novel Perspectives. J. Clin. Med. 2023, 12, 5710. https://doi.org/10.3390/jcm12175710
Ottaviani A, Mansour D, Molinari LV, Galanti K, Mantini C, Khanji MY, Chahal AA, Zimarino M, Renda G, Sciarra L, et al. Revisiting Diagnosis and Treatment of Hypertrophic Cardiomyopathy: Current Practice and Novel Perspectives. Journal of Clinical Medicine. 2023; 12(17):5710. https://doi.org/10.3390/jcm12175710
Chicago/Turabian StyleOttaviani, Andrea, Davide Mansour, Lorenzo V. Molinari, Kristian Galanti, Cesare Mantini, Mohammed Y. Khanji, Anwar A. Chahal, Marco Zimarino, Giulia Renda, Luigi Sciarra, and et al. 2023. "Revisiting Diagnosis and Treatment of Hypertrophic Cardiomyopathy: Current Practice and Novel Perspectives" Journal of Clinical Medicine 12, no. 17: 5710. https://doi.org/10.3390/jcm12175710
APA StyleOttaviani, A., Mansour, D., Molinari, L. V., Galanti, K., Mantini, C., Khanji, M. Y., Chahal, A. A., Zimarino, M., Renda, G., Sciarra, L., Pelliccia, F., Gallina, S., & Ricci, F. (2023). Revisiting Diagnosis and Treatment of Hypertrophic Cardiomyopathy: Current Practice and Novel Perspectives. Journal of Clinical Medicine, 12(17), 5710. https://doi.org/10.3390/jcm12175710