
Development of CAR T Cell Therapy in Children—A Comprehensive Overview

 ,
, 
Abstract
:1. Introduction
2. Methods
3. Molecular Architecture of CAR Receptors
4. Exponential Evolution in CAR T Cell Development
5. The Evolution of CAR Receptors
6. Link of CAR Architecture and Function
7. CD19—A Curse and Blessing
7.1. CD19 Antigen
7.2. CD19 in Comparison to Other Leukemia-Associated Antigens
8. A Paradigm without a Shift—Affinity and CAR Performance
8.1. Comparison of the T Cell Receptor and a Chimeric Antigen Receptor
8.2. Requirement for CAR Optimization per Antigen
9. Immunogenicity of CAR Products
10. Comparison of FDA/EMA-Approved CAR-T Cell Products
11. State-of-the-Art CAR T Cell Therapy in Children
11.1. Clinical Indication for CD19-CAR-T Cell Product Tisagenlecleucel (CTL019, Kymriah®)
11.2. Tisagenlecleucel Therapy
11.3. Follow-Up Patient Care Post Tisagenlecleucel Infusion
11.4. Allogeneic HSCT versus CD19-CAR-T Cell Therapy
11.5. Cytokine Release Syndrome
11.6. Neurotoxicity
11.7. Macrophage-Associated Hyperinflammation
11.8. B-Cell Aplasia
11.9. Relapse Patterns in Pediatric CD19-CAR-T Cell Therapy
11.10. CAR T Cell Trials on Alternative Targets in B-Lineage Malignancies
11.11. CAR T Cell Therapy for T Cell Malignancies
11.12. CAR T Cell Therapy for AML
11.13. CAR T Cell Therapy in Solid Tumors
12. Novel CAR T Technologies—The Antigen Question
12.1. Indirect CAR Technologies
12.2. Technologies to Improve the Safety of CAR T Cells
12.3. Armored Modules to Increase CAR T Cell Performance in TME
13. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Singh, A.K.; McGuirk, J.P. CAR T cells: Continuation in a revolution of immunotherapy. Lancet Oncol. 2020, 21, e168–e178. [Google Scholar] [CrossRef]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric Antigen Receptor T Cells for Sustained Remissions in Leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef] [PubMed]
- Spadoni, C. Pediatric Drug Development: Challenges and Opportunities. Curr. Res. Clin. Exp. 2018, 90, 119–122. [Google Scholar] [CrossRef] [PubMed]
- Joseph, P.D.; Craig, J.C.; Caldwell, P.H.Y. Clinical trials in children. Br. J. Clin. Pharmacol. 2015, 79, 357–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kakaje, A.; Alhalabi, M.; Ghareeb, A.; Karam, B.; Mansour, B.; Zahra, B.; Hamdan, O. Rates and trends of childhood acute lymphoblastic leukaemia: An epidemiology study. Sci. Rep. 2020, 10, 6756. [Google Scholar] [CrossRef] [Green Version]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Terwilliger, T.; Abdul-Hay, M. Acute lymphoblastic leukemia: A comprehensive review and 2017 update. Blood Cancer J. 2017, 7, e577. [Google Scholar] [CrossRef] [Green Version]
- Inaba, H.; Mullighan, C.G. Pediatric acute lymphoblastic leukemia. Haematologica 2020, 105, 2524–2539. [Google Scholar] [CrossRef]
- Reismüller, B.; Peters, C.; Dworzak, M.N.; Pötschger, U.; Urban, C.; Meister, B.; Schmitt, K.; Dieckmann, K.; Gadner, H.; Attarbaschi, A.; et al. Outcome of children and adolescents with a second or third relapse of acute lymphoblastic leukemia (ALL): A population-based analysis of the Austrian ALL-BFM (Berlin-Frankfurt-Münster) study group. J. Pediatr. Hematol. Oncol. 2013, 35, e200–e204. [Google Scholar] [CrossRef]
- Lemal, R.; Tournilhac, O. State-of-the-art for CAR T-cell therapy for chronic lymphocytic leukemia in 2019. J. ImmunoTher. Cancer 2019, 7, 202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jäger, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2018, 380, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Pui, C.-H.; Evans, W.E. A 50-year journey to cure childhood acute lymphoblastic leukemia. Semin. Hematol. 2013, 50, 185–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brissot, E.; Rialland, F.; Cahu, X.; Strullu, M.; Corradini, N.; Thomas, C.; Blin, N.; Thebaud, E.; Chevallier, P.; Moreau, P.; et al. Improvement of overall survival after allogeneic hematopoietic stem cell transplantation for children and adolescents: A three-decade experience of a single institution. Bone Marrow Transplant. 2016, 51, 267–272. [Google Scholar] [CrossRef] [Green Version]
- Handgretinger, R.; Zugmaier, G.; Henze, G.; Kreyenberg, H.; Lang, P.; Von Stackelberg, A. Complete remission after blinatumomab-induced donor T-cell activation in three pediatric patients with post-transplant relapsed acute lymphoblastic leukemia. Leukemia 2011, 25, 181–184. [Google Scholar] [CrossRef] [Green Version]
- Patrick, S.; Peter, L.; Gerhard, Z.; Martin, E.; Hermann, K.; Kai-Erik, W.; Judith, F.; Matthias, P.; Heiko-Manuel, T.; Christina, K.; et al. Pediatric posttransplant relapsed/refractory B-precursor acute lymphoblastic leukemia shows durable remission by therapy with the T-cell engaging bispecific antibody blinatumomab. Haematologica 2014, 99, 1212–1219. [Google Scholar]
- Lee, D.W.; Kochenderfer, J.N.; Stetler-Stevenson, M.; Cui, Y.K.; Delbrook, C.; Feldman, S.A.; Fry, T.J.; Orentas, R.; Sabatino, M.; Shah, N.N.; et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: A phase 1 dose-escalation trial. Lancet 2015, 385, 517–528. [Google Scholar] [CrossRef]
- Kebriaei, P.; Singh, H.; Huls, M.H.; Figliola, M.J.; Bassett, R.; Olivares, S.; Jena, B.; Dawson, M.J.; Kumaresan, P.R.; Su, S.; et al. Phase I trials using Sleeping Beauty to generate CD19-specific CAR T cells. J. Clin. Investig. 2016, 126, 3363–3376. [Google Scholar] [CrossRef]
- Turtle, C.J.; Hanafi, L.-A.; Berger, C.; Gooley, T.A.; Cherian, S.; Hudecek, M.; Sommermeyer, D.; Melville, K.; Pender, B.; Budiarto, T.M.; et al. CD19 CAR–T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J. Clin. Investig. 2016, 126, 2123–2138. [Google Scholar] [CrossRef] [Green Version]
- Turtle, C.J.; Hay, K.; Hanafi, L.-A.; Li, D.; Cherian, S.; Chen, X.; Wood, B.; Lozanski, A.; Byrd, J.C.; Heimfeld, S.; et al. Durable Molecular Remissions in Chronic Lymphocytic Leukemia Treated With CD19-Specific Chimeric Antigen Receptor–Modified T Cells After Failure of Ibrutinib. J. Clin. Oncol. 2017, 35, 3010–3020. [Google Scholar] [CrossRef]
- KYMRIAH® (Tisagenlecleucel) [Package Insert]; Novartis: Basel, Switzerland, 2017.
- YESCARTA® (Axicabtagene Ciloleucel) [Package Insert]; Kite: Los Angeles, CA, USA, 2017.
- BREYANZI® (Lisocabtagene Maraleucel) [Package Insert]; Bristol Myers Squibb: New York, NY, USA, 2021.
- TECARTUS® (Brexucabtagene Autoleucel) [Package Insert]; Kite: Los Angeles, CA, USA, 2021.
- ABECMA® (Idecabtagene Vicleucel) [Package Insert]; Myers Squibb: New York, NY, USA, 2021.
- Quintás-Cardama, A. What CAR Will Win the CD19 Race? Mol. Cancer Ther. 2019, 18, 498–506. [Google Scholar] [CrossRef] [Green Version]
- Schuster, S.J.; Maziarz, R.T.; Rusch, E.S.; Li, J.; Signorovitch, J.E.; Romanov, V.V.; Locke, F.L.; Maloney, D.G. Grading and management of cytokine release syndrome in patients treated with tisagenlecleucel in the JULIET trial. Blood Adv. 2020, 4, 1432–1439. [Google Scholar] [CrossRef]
- Sheth, V.S.; Gauthier, J. Taming the beast: CRS and ICANS after CAR T-cell therapy for ALL. Bone Marrow Transplant. 2021, 56, 552–566. [Google Scholar] [CrossRef]
- Wudhikarn, K.; Palomba, M.L.; Pennisi, M.; Garcia-Recio, M.; Flynn, J.R.; Devlin, S.M.; Afuye, A.; Silverberg, M.L.; Maloy, M.A.; Shah, G.L.; et al. Infection during the first year in patients treated with CD19 CAR T cells for diffuse large B cell lymphoma. Blood Cancer J. 2020, 10, 79. [Google Scholar] [CrossRef]
- Majzner, R.G.; Mackall, C.L. Tumor Antigen Escape from CAR T-cell Therapy. Cancer Discov. 2018, 8, 1219–1226. [Google Scholar] [CrossRef] [Green Version]
- Wagner, J.; Wickman, E.; DeRenzo, C.; Gottschalk, S. CAR T Cell Therapy for Solid Tumors: Bright Future or Dark Reality? Mol. Ther. 2020, 28, 2320–2339. [Google Scholar] [CrossRef]
- Chen, G.M.; Chen, C.; Das, R.K.; Gao, P.; Chen, C.-H.; Bandyopadhyay, S.; Ding, Y.-Y.; Uzun, Y.; Yu, W.; Zhu, Q.; et al. Integrative Bulk and Single-Cell Profiling of Premanufacture T-cell Populations Reveals Factors Mediating Long-Term Persistence of CAR T-cell Therapy. Cancer Discov. 2021, 11, 2186–2199. [Google Scholar] [CrossRef]
- Kasakovski, D.; Xu, L.; Li, Y. T cell senescence and CAR-T cell exhaustion in hematological malignancies. J. Hematol. Oncol. 2018, 11, 91. [Google Scholar] [CrossRef]
- Martinez, M.; Moon, E.K. CAR T Cells for Solid Tumors: New Strategies for Finding, Infiltrating, and Surviving in the Tumor Microenvironment. Front. Immunol. 2019, 10, 128. [Google Scholar] [CrossRef] [Green Version]
- Gross, G.; Waks, T.; Eshhar, Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc. Natl. Acad. Sci. USA 1989, 86, 10024–10028. [Google Scholar] [CrossRef] [Green Version]
- Milone, M.C.; Fish, J.D.; Carpenito, C.; Carroll, R.G.; Binder, G.K.; Teachey, D.; Samanta, M.; Lakhal, M.; Gloss, B.; Danet-Desnoyers, G.; et al. Chimeric Receptors Containing CD137 Signal Transduction Domains Mediate Enhanced Survival of T Cells and Increased Antileukemic Efficacy In Vivo. Mol. Ther. 2009, 17, 1453–1464. [Google Scholar] [CrossRef]
- Gong, M.C.; Latouche, J.-B.; Krause, A.; Heston, W.D.; Bander, N.H.; Sadelain, M. Cancer Patient T Cells Genetically Targeted to Prostate-Specific Membrane Antigen Specifically Lyse Prostate Cancer Cells and Release Cytokines in Response to Prostate-Specific Membrane Antigen. Neoplasia 1999, 1, 123–127. [Google Scholar] [CrossRef] [Green Version]
- Eshhar, Z.; Waks, T.; Gross, G.; Schindler, D.G. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc. Natl. Acad. Sci. USA 1993, 90, 720–724. [Google Scholar] [CrossRef] [Green Version]
- Goverman, J.; Gomez, S.M.; Segesman, K.D.; Hunkapiller, T.; Laug, W.E.; Hood, L. Chimeric immunoglobulin-T cell receptor proteins form functional receptors: Implications for T cell receptor complex formation and activation. Cell 1990, 60, 929–939. [Google Scholar] [CrossRef]
- Ochi, T.; Maruta, M.; Tanimoto, K.; Kondo, F.; Yamamoto, T.; Kurata, M.; Fujiwara, H.; Masumoto, J.; Takenaka, K.; Yasukawa, M. A single-chain antibody generation system yielding CAR-T cells with superior antitumor function. Commun. Biol. 2021, 4, 273. [Google Scholar] [CrossRef]
- Kang, T.H.; Seong, B.L. Solubility, Stability, and Avidity of Recombinant Antibody Fragments Expressed in Microorganisms. Front. Microbiol. 2020, 11, 1927. [Google Scholar] [CrossRef]
- Asensio, M.A.; Lim, Y.W.; Wayham, N.; Stadtmiller, K.; Edgar, R.C.; Leong, J.; Leong, R.; Mizrahi, R.A.; Adams, M.S.; Simons, J.F.; et al. Antibody repertoire analysis of mouse immunization protocols using microfluidics and molecular genomics. mAbs 2019, 11, 870–883. [Google Scholar] [CrossRef]
- Lonberg, N. Fully human antibodies from transgenic mouse and phage display platforms. Curr. Opin. Immunol. 2008, 20, 450–459. [Google Scholar] [CrossRef]
- Zajc, C.U.; Salzer, B.; Taft, J.M.; Reddy, S.T.; Lehner, M.; Traxlmayr, M.W. Driving CARs with alternative navigation tools—The potential of engineered binding scaffolds. FEBS J. 2021, 288, 2103–2118. [Google Scholar] [CrossRef]
- Asaadi, Y.; Jouneghani, F.F.; Janani, S.; Rahbarizadeh, F. A comprehensive comparison between camelid nanobodies and single chain variable fragments. Biomark. Res. 2021, 9, 87. [Google Scholar] [CrossRef]
- Schmidts, A.; Ormhøj, M.; Choi, B.D.; Taylor, A.O.; Bouffard, A.A.; Scarfò, I.; Larson, R.C.; Frigault, M.J.; Gallagher, K.; Castano, A.P.; et al. Rational design of a trimeric APRIL-based CAR-binding domain enables efficient targeting of multiple myeloma. Blood Adv. 2019, 3, 3248–3260. [Google Scholar] [CrossRef]
- Wang, D.; Starr, R.; Chang, W.-C.; Aguilar, B.; Alizadeh, D.; Wright, S.L.; Yang, X.; Brito, A.; Sarkissian, A.; Ostberg, J.R.; et al. Chlorotoxin-directed CAR T cells for specific and effective targeting of glioblastoma. Sci. Transl. Med. 2020, 12, eaaw2672. [Google Scholar] [CrossRef]
- Jayaraman, J.; Mellody, M.P.; Hou, A.J.; Desai, R.P.; Fung, A.W.; Pham, A.H.T.; Chen, Y.Y.; Zhao, W. CAR-T design: Elements and their synergistic function. EBioMedicine 2020, 58, 102931. [Google Scholar] [CrossRef]
- Zhang, H.; Zhao, P.; Huang, H. Engineering better chimeric antigen receptor T cells. Exp. Hematol. Oncol. 2020, 9, 34. [Google Scholar] [CrossRef]
- Ng, Y.-Y.; Tay, J.C.; Li, Z.; Wang, J.; Zhu, J.; Wang, S. T Cells Expressing NKG2D CAR with a DAP12 Signaling Domain Stimulate Lower Cytokine Production While Effective in Tumor Eradication. Mol. Ther. 2020, 29, 75–85. [Google Scholar] [CrossRef]
- Guedan, S.; Calderon, H.; Posey, A.D., Jr.; Maus, M.V. Engineering and Design of Chimeric Antigen Receptors. Mol. Ther. Methods Clin. Dev. 2019, 12, 145–156. [Google Scholar] [CrossRef] [Green Version]
- Tyagarajan, S.; Spencer, T.; Smith, J. Optimizing CAR-T Cell Manufacturing Processes during Pivotal Clinical Trials. Mol. Ther. Methods Clin. Dev. 2020, 16, 136–144. [Google Scholar] [CrossRef] [Green Version]
- Jackson, Z.; Roe, A.; Sharma, A.A.; Lopes, F.B.T.P.; Talla, A.; Kleinsorge-Block, S.; Zamborsky, K.; Schiavone, J.; Manjappa, S.; Schauner, R.; et al. Automated Manufacture of Autologous CD19 CAR-T Cells for Treatment of Non-hodgkin Lymphoma. Front. Immunol. 2020, 11, 1941. [Google Scholar] [CrossRef]
- Jürgens, B.; Clarke, N.S. Evolution of CAR T-cell immunotherapy in terms of patenting activity. Nat. Biotechnol. 2019, 37, 370–375. [Google Scholar] [CrossRef]
- Yang, J.; Kim, B.; Kim, G.Y.; Jung, G.Y.; Seo, S.W. Synthetic biology for evolutionary engineering: From perturbation of genotype to acquisition of desired phenotype. Biotechnol. Biofuels 2019, 12, 113. [Google Scholar] [CrossRef] [Green Version]
- Kunjapur, A.M.; Pfingstag, P.; Thompson, N.C. Gene synthesis allows biologists to source genes from farther away in the tree of life. Nat. Commun. 2018, 9, 4425. [Google Scholar] [CrossRef] [Green Version]
- Petrenko, V.A. Landscape Phage: Evolution from Phage Display to Nanobiotechnology. Viruses 2018, 10, 311. [Google Scholar] [CrossRef] [Green Version]
- Winter, G.; Harris, W.J. Humanized antibodies. Trends Pharmacol. Sci. 1993, 14, 139–143. [Google Scholar] [CrossRef]
- Zinsli, L.V.; Stierlin, N.; Loessner, M.J.; Schmelcher, M. Deimmunization of protein therapeutics—Recent advances in experimental and computational epitope prediction and deletion. Comput. Struct. Biotechnol. J. 2021, 19, 315–329. [Google Scholar] [CrossRef]
- Laffleur, B.; Pascal, V.; Sirac, C.; Cogné, M. Production of Human or Humanized Antibodies in Mice. Mol. Radio-Oncol. 2012, 901, 149–159. [Google Scholar]
- Zhou, X.; Tu, S.; Wang, C.; Huang, R.; Deng, L.; Song, C.; Yue, C.; He, Y.; Yang, J.; Liang, Z.; et al. Phase I Trial of Fourth-Generation Anti-CD19 Chimeric Antigen Receptor T Cells Against Relapsed or Refractory B Cell Non-Hodgkin Lymphomas. Front. Immunol. 2020, 11, 564099. [Google Scholar] [CrossRef]
- Chmielewski, M.; Kopecky, C.; Hombach, A.A.; Abken, H. IL-12 Release by Engineered T Cells Expressing Chimeric Antigen Receptors Can Effectively Muster an Antigen-Independent Macrophage Response on Tumor Cells That Have Shut Down Tumor Antigen Expression. Cancer Res. 2011, 71, 5697–5706. [Google Scholar] [CrossRef] [Green Version]
- Eyquem, J.; Mansilla-Soto, J.; Giavridis, T.; van der Stegen, S.J.C.; Hamieh, M.; Cunanan, K.M.; Odak, A.; Gönen, M.; Sadelain, M. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 2017, 543, 113–117. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.W.; Cho, J.-Y. Recent Advances in Allogeneic CAR-T Cells. Biomolecules 2020, 10, 263. [Google Scholar] [CrossRef] [Green Version]
- Savoldo, B.; Ramos, C.A.; Liu, E.; Mims, M.P.; Keating, M.J.; Carrum, G.; Kamble, R.T.; Bollard, C.M.; Gee, A.P.; Mei, Z.; et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor–modified T cells in lymphoma patients. J. Clin. Investig. 2011, 121, 1822–1826. [Google Scholar] [CrossRef] [Green Version]
- Maher, J.; Brentjens, R.J.; Gunset, G.; Rivière, I.; Sadelain, M. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRζ/CD28 receptor. Nat. Biotechnol. 2002, 20, 70–75. [Google Scholar] [CrossRef]
- Xie, G.; Dong, H.; Liang, Y.; Ham, J.D.; Rizwan, R.; Chen, J. CAR-NK cells: A promising cellular immunotherapy for cancer. EBioMedicine 2020, 59, 102975. [Google Scholar] [CrossRef]
- Davila, M.L.; Brentjens, R.J. CD19-Targeted CAR T cells as novel cancer immunotherapy for relapsed or refractory B-cell acute lymphoblastic leukemia. Clin. Adv. Hematol. Oncol. HO 2016, 14, 802–808. [Google Scholar]
- Sommermeyer, D.; Hudecek, M.; Kosasih, P.L.; Gogishvili, T.; Maloney, D.G.; Turtle, C.J.; Riddell, S.R. Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia 2016, 30, 492–500. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Huang, S.; Xiao, X.; Sun, Q.; Liang, X.; Chen, S.; Zhao, Z.; Huo, Z.; Tu, S.; Li, Y. Challenges and Clinical Strategies of CAR T-Cell Therapy for Acute Lymphoblastic Leukemia: Overview and Developments. Front. Immunol. 2021, 11, 569117. [Google Scholar] [CrossRef]
- Hartmann, J.; Schüßler-Lenz, M.; Bondanza, A.; Buchholz, C.J. Clinical development of CAR T cells—challenges and opportunities in translating innovative treatment concepts. EMBO Mol. Med. 2017, 9, 1183–1197. [Google Scholar] [CrossRef]
- Mariuzza, R.A.; Agnihotri, P.; and Orban, J. The structural basis of T-cell receptor (TCR) activation: An enduring enigma. J. Biol. Chem. 2020, 295, 914–925. [Google Scholar] [CrossRef]
- Li, R.; Ma, C.; Cai, H.; Chen, W. The CAR T-Cell Mechanoimmunology at a Glance. Adv. Sci. 2020, 7, 2002628. [Google Scholar] [CrossRef]
- Davenport, A.J.; Cross, R.S.; Watson, K.A.; Liao, Y.; Shi, W.; Prince, H.M.; Beavis, P.A.; Trapani, J.A.; Kershaw, M.H.; Ritchie, D.S.; et al. Chimeric antigen receptor T cells form nonclassical and potent immune synapses driving rapid cytotoxicity. Proc. Natl. Acad. Sci. USA 2018, 115, E2068–E2076. [Google Scholar] [CrossRef] [Green Version]
- Papadopoulou, M.; Tieppo, P.; McGovern, N.; Gosselin, F.; Chan, J.K.Y.; Goetgeluk, G.; Dauby, N.; Cogan, A.; Donner, C.; Ginhoux, F.; et al. TCR Sequencing Reveals the Distinct Development of Fetal and Adult Human Vγ9Vδ2 T Cells. J. Immunol. 2019, 203, 1468–1479. [Google Scholar] [CrossRef] [Green Version]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef]
- Scheuermann, R.; Racila, E. CD19 Antigen in Leukemia and Lymphoma Diagnosis and Immunotherapy. Leuk. Lymphoma 1995, 18, 385–397. [Google Scholar] [CrossRef]
- Maude, S.L.; Teachey, D.; Porter, D.L.; Grupp, S.A. CD19-targeted chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Blood 2015, 125, 4017–4023. [Google Scholar] [CrossRef] [Green Version]
- Hammer, O. CD19 as an attractive target for antibody-based therapy. mAbs 2012, 4, 571–577. [Google Scholar] [CrossRef] [Green Version]
- Spiegel, J.Y.; Patel, S.; Muffly, L.; Hossain, N.M.; Oak, J.; Baird, J.H.; Frank, M.J.; Shiraz, P.; Sahaf, B.; Craig, J.; et al. CAR T cells with dual targeting of CD19 and CD22 in adult patients with recurrent or refractory B cell malignancies: A phase 1 trial. Nat. Med. 2021, 27, 1419–1431. [Google Scholar] [CrossRef]
- Ginaldi, L.; De Martinis, M.; Matutes, E.; Farahat, N.; Morilla, R.; Catovsky, D. Levels of expression of CD19 and CD20 in chronic B cell leukaemias. J. Clin. Pathol. 1998, 51, 364–369. [Google Scholar] [CrossRef] [Green Version]
- Seidel, U.J.E.; Schlegel, P.; Grosse-Hovest, L.; Hofmann, M.; Aulwurm, S.; Pyz, E.; Schuster, F.R.; Meisel, R.; Ebinger, M.; Feuchtinger, T.; et al. Reduction of Minimal Residual Disease in Pediatric B-lineage Acute Lymphoblastic Leukemia by an Fc-optimized CD19 Antibody. Mol. Ther. 2016, 24, 1634–1643. [Google Scholar] [CrossRef] [Green Version]
- Qin, H.; Ramakrishna, S.; Nguyen, S.; Fountaine, T.J.; Ponduri, A.; Stetler-Stevenson, M.; Yuan, C.M.; Haso, W.; Shern, J.F.; Shah, N.N.; et al. Preclinical Development of Bivalent Chimeric Antigen Receptors Targeting Both CD19 and CD22. Mol. Ther. Oncolytics 2018, 11, 127–137. [Google Scholar] [CrossRef] [Green Version]
- Fry, T.J.; Shah, N.N.; Orentas, R.J.; Stetler-Stevenson, M.; Yuan, C.M.; Ramakrishna, S.; Wolters, P.; Martin, S.; Delbrook, C.; Yates, B.; et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat. Med. 2018, 24, 20–28. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, K.; Terakura, S.; Martens, A.C.; van Meerten, T.; Uchiyama, S.; Imai, M.; Sakemura, R.; Goto, T.; Hanajiri, R.; Imahashi, N.; et al. Target Antigen Density Governs the Efficacy of Anti–CD20-CD28-CD3 ζ Chimeric Antigen Receptor–Modified Effector CD8+ T Cells. J. Immunol. 2015, 194, 911–920. [Google Scholar] [CrossRef] [Green Version]
- Majzner, R.G.; Rietberg, S.P.; Sotillo, E.; Dong, R.; Vachharajani, V.T.; Labanieh, L.; Myklebust, J.H.; Kadapakkam, M.; Weber, E.W.; Tousley, A.M.; et al. Tuning the Antigen Density Requirement for CAR T-cell Activity. Cancer Discov. 2020, 10, 702–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ormhøj, M.; Scarfò, I.; Cabral, M.L.; Bailey, S.; Lorrey, S.J.; Bouffard, A.A.; Castano, A.P.; Larson, R.C.; Riley, L.S.; Schmidts, A.; et al. Chimeric Antigen Receptor T Cells Targeting CD79b Show Efficacy in Lymphoma with or without Cotargeting CD19. Clin. Cancer Res. 2019, 25, 7046–7057. [Google Scholar] [CrossRef] [Green Version]
- Ding, S.; Mao, X.; Cao, Y.; Wang, N.; Xu, H.; Zhou, J. Targeting CD79b for Chimeric Antigen Receptor T-Cell Therapy of B-Cell Lymphomas. Target. Oncol. 2020, 15, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Teachey, D.T.; Hunger, S.P. Anti-CD7 CAR T cells for T-ALL: Impressive early-stage efficacy. Nat. Rev. Clin. Oncol. 2021, 18, 677–678. [Google Scholar] [CrossRef] [PubMed]
- Maciocia, P.; A Wawrzyniecka, P.; Philip, B.; Ricciardelli, I.; Akarca, A.U.; Onuoha, S.C.; Legut, M.; Cole, D.; Sewell, A.K.; Gritti, G.; et al. Targeting the T cell receptor β-chain constant region for immunotherapy of T cell malignancies. Nat. Med. 2017, 23, 1416–1423. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Tan, Y.; Wang, G.; Deng, B.; Ling, Z.; Song, W.; Seery, S.; Zhang, Y.; Peng, S.; Xu, J.; et al. Donor-Derived CD7 Chimeric Antigen Receptor T Cells for T-Cell Acute Lymphoblastic Leukemia: First-in-Human, Phase I Trial. J. Clin. Oncol. 2021, 39, 3340–3351. [Google Scholar] [CrossRef] [PubMed]
- Cummins, K.; Gill, S. Will CAR T cell therapy have a role in AML? Promises and pitfalls. Semin. Hematol. 2019, 56, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Gill, S.; Tasian, S.; Ruella, M.; Shestova, O.; Li, Y.; Porter, D.L.; Carroll, M.; Danet-Desnoyers, G.; Scholler, J.; Grupp, S.A.; et al. Preclinical targeting of human acute myeloid leukemia and myeloablation using chimeric antigen receptor–modified T cells. Blood 2014, 123, 2343–2354. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.Y.; Yu, K.-R.; Kenderian, S.S.; Ruella, M.; Chen, S.; Shin, T.-H.; Aljanahi, A.A.; Schreeder, D.; Klichinsky, M.; Shestova, O.; et al. Genetic Inactivation of CD33 in Hematopoietic Stem Cells to Enable CAR T Cell Immunotherapy for Acute Myeloid Leukemia. Cell 2018, 173, 1439–1453.e19. [Google Scholar] [CrossRef] [Green Version]
- Hou, A.J.; Chen, L.C.; Chen, Y.Y. Navigating CAR-T cells through the solid-tumour microenvironment. Nat. Rev. Drug Discov. 2021, 20, 531–550. [Google Scholar] [CrossRef]
- Teplyakov, A.; Obmolova, G.; Luo, J.; Gilliland, G.L. Crystal structure of B-cell co-receptor CD19 in complex with antibody B43 reveals an unexpected fold. Proteins Struct. Funct. Bioinform. 2018, 86, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Ghorashian, S.; Kramer, A.M.; Onuoha, S.; Wright, G.; Bartram, J.; Richardson, R.; Albon, S.J.; Casanovas-Company, J.; Castro, F.; Popova, B.; et al. Enhanced CAR T cell expansion and prolonged persistence in pediatric patients with ALL treated with a low-affinity CD19 CAR. Nat. Med. 2019, 25, 1408–1414. [Google Scholar] [CrossRef] [PubMed]
- Long, A.H.; Haso, W.M.; Shern, J.F.; Wanhainen, K.M.; Murgai, M.; Ingaramo, M.; Smith, J.P.; Walker, A.J.; Kohler, M.E.; Venkateshwara, V.R.; et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat. Med. 2015, 21, 581–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, K.C.; Degano, M.; Stanfield, R.L.; Brunmark, A.; Jackson, M.R.; Peterson, P.A.; Teyton, L.; Wilson, I.A. An αβ T Cell Receptor Structure at 2.5 υ and Its Orientation in the TCR-MHC Complex. Science 1996, 274, 209–219. [Google Scholar] [CrossRef]
- Garboczi, D.N.; Ghosh, P.; Utz, U.; Fan, Q.R.; Biddison, W.E.; Wiley, D.C. Structure of the complex between human T-cell receptor, viral peptide and HLA-A2. Nature 1996, 384, 134–141. [Google Scholar] [CrossRef]
- Watanabe, N.; Bajgain, P.; Sukumaran, S.; Ansari, S.; Heslop, H.E.; Rooney, C.M.; Brenner, M.K.; Leen, A.M.; Vera, J.F. Fine-tuning the CAR spacer improves T-cell potency. OncoImmunology 2016, 5, e1253656. [Google Scholar] [CrossRef] [Green Version]
- Long, A.H.; Haso, W.M.; Orentas, R.J. Lessons learned from a highly-active CD22-specific chimeric antigen receptor. OncoImmunology 2013, 2, e23621. [Google Scholar] [CrossRef] [Green Version]
- James, S.E.; Greenberg, P.D.; Jensen, M.C.; Lin, Y.; Wang, J.; Till, B.G.; Raubitschek, A.A.; Forman, S.J.; Press, O.W. Antigen Sensitivity of CD22-Specific Chimeric TCR Is Modulated by Target Epitope Distance from the Cell Membrane. J. Immunol. 2008, 180, 7028–7038. [Google Scholar] [CrossRef] [Green Version]
- Haso, W.; Lee, D.W.; Shah, N.N.; Stetler-Stevenson, M.; Yuan, C.M.; Pastan, I.H.; Dimitrov, D.S.; Morgan, R.A.; Fitzgerald, D.J.; Barrett, D.M.; et al. Anti-CD22–chimeric antigen receptors targeting B-cell precursor acute lymphoblastic leukemia. Blood 2013, 121, 1165–1174. [Google Scholar] [CrossRef] [Green Version]
- Möricke, A.; Ratei, R.; Ludwig, W.-D.; Harbott, J.; Borkhardt, A.; Viehmann, S.; Zimmermann, M.; Gadner, H.; Riehm, H.; Schrappe, M. Prognostic Factors in CD10 Negative Precursor B-Cell Acute Lymphoblastic Leukemia in Children: Data from Three Consecutive Trials ALL-BFM 86, 90, and 95. Blood 2004, 104, 1957. [Google Scholar] [CrossRef]
- Sędek, Ł.; Bulsa, J.; Sonsala, A.; Twardoch, M.; Wieczorek, M.; Malinowska, I.; Derwich, K.; Niedźwiecki, M.; Sobol-Milejska, G.; Kowalczyk, J.R.; et al. The immunophenotypes of blast cells in B-cell precursor acute lymphoblastic leukemia: How different are they from their normal counterparts? Cytom. Part B Clin. Cytom. 2014, 86, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Baird, J.H.; Frank, M.J.; Craig, J.; Patel, S.; Spiegel, J.Y.; Sahaf, B.; Oak, J.S.; Younes, S.F.; Ozawa, M.G.; Yang, E.; et al. CD22-directed CAR T-cell therapy induces complete remissions in CD19-directed CAR–refractory large B-cell lymphoma. Blood 2021, 137, 2321–2325. [Google Scholar] [CrossRef] [PubMed]
- Schneider, D.; Xiong, Y.; Wu, D.; Hu, P.; Alabanza, L.; Steimle, B.; Dropulić, B. Trispecific CD19-CD20-CD22-targeting duoCAR-T cells eliminate antigen-heterogeneous B cell tumors in preclinical models. Sci. Transl. Med. 2021, 13, eabc6401. [Google Scholar] [CrossRef] [PubMed]
- Fousek, K.; Watanabe, J.; Joseph, S.K.; George, A.; An, X.; Byrd, T.T.; Morris, J.; Luong, A.; Martínez-Paniagua, M.A.; Sanber, K.; et al. CAR T-cells that target acute B-lineage leukemia irrespective of CD19 expression. Leukemia 2021, 35, 75–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramakrishna, S.; Highfill, S.L.; Walsh, Z.; Nguyen, S.M.; Lei, H.; Shern, J.F.; Qin, H.; Kraft, I.L.; Stetler-Stevenson, M.; Yuan, C.M.; et al. Modulation of Target Antigen Density Improves CAR T-cell Functionality and Persistence. Clin. Cancer Res. 2019, 25, 5329–5341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, C.; Zhang, Y.; Liu, Y.; Ji, X.; Zhang, W.; Guo, Y.; Han, X.; Ti, D.; Dai, H.; Wang, C. Optimized tandem CD19/CD20 CAR-engineered T cells in refractory/relapsed B-cell lymphoma. Blood 2020, 136, 1632–1644. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Feng, K.; Tong, C.; Jia, H.; Liu, Y.; Wang, Y.; Ti, D.; Yang, Q.; Wu, Z.; Han, W. Efficiency and side effects of anti-CD38 CAR T cells in an adult patient with relapsed B-ALL after failure of bi-specific CD19/CD22 CAR T cell treatment. Cell. Mol. Immunol. 2020, 17, 430–432. [Google Scholar] [CrossRef]
- Jyoti, N.; Maria, T.; de Regina, J.-K.; Ruud, W.J.R.; Pino, J.P.; Huipin, Y.; de Bruijn, J.D.; Ossenkoppele, G.J.; Zweegman, S.; Smit, L.; et al. CD38 as a therapeutic target for adult acute myeloid leukemia and T-cell acute lymphoblastic leukemia. Haematologica 2019, 104, e100–e103. [Google Scholar]
- Haubner, S.; Perna, F.; Köhnke, T.; Schmidt, C.; Berman, S.; Augsberger, C.; Schnorfeil, F.M.; Krupka, C.; Lichtenegger, F.S.; Liu, X.; et al. Coexpression profile of leukemic stem cell markers for combinatorial targeted therapy in AML. Leukemia 2019, 33, 64–74. [Google Scholar] [CrossRef]
- Tambaro, F.P.; Singh, H.; Jones, E.; Rytting, M.; Mahadeo, K.M.; Thompson, P.; Daver, N.; DiNardo, C.; Kadia, T.; Garcia-Manero, G.; et al. Autologous CD33-CAR-T cells for treatment of relapsed/refractory acute myelogenous leukemia. Leukemia 2021, 35, 3282–3286. [Google Scholar] [CrossRef]
- Mardiana, S.; Gill, S. CAR T Cells for Acute Myeloid Leukemia: State of the Art and Future Directions. Front. Oncol. 2020, 10, 697. [Google Scholar] [CrossRef] [PubMed]
- Baroni, M.L.; Martinez, D.S.; Aguera, F.G.; Ho, H.R.; Castella, M.; Zanetti, S.; Hernandez, T.V.; De La Guardia, R.D.; Castaño, J.; Anguita, E.; et al. 41BB-based and CD28-based CD123-redirected T-cells ablate human normal hematopoiesis in vivo. J. Immunother. Cancer 2020, 8, e000845. [Google Scholar] [CrossRef] [PubMed]
- Styczyński, J.; Ebmt, F.T.I.D.W.P.; Tridello, G.; Koster, L.; Iacobelli, S.; Van Biezen, A.; Van Der Werf, S.; Mikulska, M.; Gil, L.; Cordonnier, C.; et al. Death after hematopoietic stem cell transplantation: Changes over calendar year time, infections and associated factors. Bone Marrow Transplant. 2020, 55, 126–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Jiang, S.; Fang, C.; Yang, S.; Olalere, D.; Pequignot, E.C.; Cogdill, A.; Li, N.; Ramones, M.; Granda, B.; et al. Affinity-Tuned ErbB2 or EGFR Chimeric Antigen Receptor T Cells Exhibit an Increased Therapeutic Index against Tumors in Mice. Cancer Res. 2015, 75, 3596–3607. [Google Scholar] [CrossRef] [Green Version]
- Du, H.; Hirabayashi, K.; Ahn, S.; Kren, N.P.; Montgomery, S.A.; Wang, X.; Tiruthani, K.; Mirlekar, B.; Michaud, D.; Greene, K.; et al. Antitumor Responses in the Absence of Toxicity in Solid Tumors by Targeting B7-H3 via Chimeric Antigen Receptor T Cells. Cancer Cell 2019, 35, 221–237.e8. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Sun, Q.; Liang, X.; Chen, Z.; Zhang, X.; Zhou, X.; Li, M.; Tu, H.; Liu, Y.; Tu, S.; et al. Mechanisms of Relapse After CD19 CAR T-Cell Therapy for Acute Lymphoblastic Leukemia and Its Prevention and Treatment Strategies. Front. Immunol. 2019, 10, 2664. [Google Scholar] [CrossRef] [Green Version]
- Kagoya, Y.; Tanaka, S.; Guo, T.; Anczurowski, M.; Wang, C.-H.; Saso, K.; Butler, M.O.; Minden, M.D.; Hirano, N. A novel chimeric antigen receptor containing a JAK–STAT signaling domain mediates superior antitumor effects. Nat. Med. 2018, 24, 352–359. [Google Scholar] [CrossRef]
- Salter, A.I.; Rajan, A.; Kennedy, J.J.; Ivey, R.G.; Shelby, S.A.; Leung, I.; Templeton, M.L.; Muhunthan, V.; Voillet, V.; Sommermeyer, D.; et al. Comparative analysis of TCR and CAR signaling informs CAR designs with superior antigen sensitivity and in vivo function. Sci. Signal. 2021, 14, eabe2606. [Google Scholar] [CrossRef]
- Watanabe, K.; Kuramitsu, S.; Posey, A.D., Jr.; June, C.H. Expanding the Therapeutic Window for CAR T Cell Therapy in Solid Tumors: The Knowns and Unknowns of CAR T Cell Biology. Front. Immunol. 2018, 9, 2486. [Google Scholar] [CrossRef] [Green Version]
- Dong, R.; A Libby, K.; Blaeschke, F.; Fuchs, W.; Marson, A.; Vale, R.D.; Su, X. Rewired signaling network in T cells expressing the chimeric antigen receptor ( CAR ). EMBO J. 2020, 39, e104730. [Google Scholar] [CrossRef]
- Stopfer, L.E.; Gajadhar, A.S.; Patel, B.; Gallien, S.; Frederick, D.T.; Boland, G.M.; Sullivan, R.J.; White, F.M. Absolute quantification of tumor antigens using embedded MHC-I isotopologue calibrants. Proc. Natl. Acad. Sci. USA 2021, 118, 2111173118. [Google Scholar] [CrossRef] [PubMed]
- Croft, N.; Smith, S.A.; Wong, Y.C.; Tan, C.T.; Dudek, N.L.; Flesch, I.E.A.; Lin, L.C.W.; Tscharke, D.C.; Purcell, A.W. Kinetics of Antigen Expression and Epitope Presentation during Virus Infection. PLOS Pathog. 2013, 9, e1003129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, A.J.; Majzner, R.G.; Zhang, L.; Wanhainen, K.; Long, A.H.; Nguyen, S.M.; Lopomo, P.; Vigny, M.; Fry, T.J.; Orentas, R.J.; et al. Tumor Antigen and Receptor Densities Regulate Efficacy of a Chimeric Antigen Receptor Targeting Anaplastic Lymphoma Kinase. Mol. Ther. 2017, 25, 2189–2201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heitzeneder, S.; Bosse, K.R.; Zhu, Z.; Zhelev, D.; Majzner, R.G.; Radosevich, M.T.; Dhingra, S.; Sotillo, E.; Buongervino, S.; Pascual-Pasto, G.; et al. GPC2-CAR T cells tuned for low antigen density mediate potent activity against neuroblastoma without toxicity. Cancer Cell 2022, 40, 53–69.e9. [Google Scholar] [CrossRef] [PubMed]
- A Morgan, R.; Yang, J.C.; Kitano, M.; E Dudley, M.; Laurencot, C.M.; A Rosenberg, S. Case Report of a Serious Adverse Event Following the Administration of T Cells Transduced With a Chimeric Antigen Receptor Recognizing ERBB2. Mol. Ther. 2010, 18, 843–851. [Google Scholar] [CrossRef]
- Hill, J.A.; Giralt, S.; Torgerson, T.R.; Lazarus, H.M. CAR-T—And a side order of IgG, to go?—Immunoglobulin replacement in patients receiving CAR-T cell therapy. Blood Rev. 2019, 38, 100596. [Google Scholar] [CrossRef]
- Seitz, C.M.; Mittelstaet, J.; Atar, D.; Hau, J.; Reiter, S.; Illi, C.; Kieble, V.; Engert, F.; Drees, B.; Bender, G.; et al. Novel adapter CAR-T cell technology for precisely controllable multiplex cancer targeting. OncoImmunology 2021, 10, 2003532. [Google Scholar] [CrossRef]
- Sommermeyer, D.; Hill, T.; Shamah, S.M.; Salter, A.; Chen, Y.; Mohler, K.M.; Riddell, S.R. Fully human CD19-specific chimeric antigen receptors for T-cell therapy. Leukemia 2017, 31, 2191–2199. [Google Scholar] [CrossRef] [Green Version]
- Muzard, J.; Bouabdelli, M.; Zahid, M.; Ollivier, V.; Lacapère, J.J.; Jandrot-Perrus, M.; Billiald, P. Design and humanization of a murine scFv that blocks human platelet glycoprotein VI in vitro. FEBS J. 2009, 276, 4207–4222. [Google Scholar] [CrossRef]
- Lam, N.; Trinklein, N.D.; Buelow, B.; Patterson, G.H.; Ojha, N.; Kochenderfer, J.N. Anti-BCMA chimeric antigen receptors with fully human heavy-chain-only antigen recognition domains. Nat. Commun. 2020, 11, 283. [Google Scholar] [CrossRef] [Green Version]
- Holliger, P.; Hudson, P.J. Engineered antibody fragments and the rise of single domains. Nat. Biotechnol. 2005, 23, 1126–1136. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.C.; Popplewell, L.; Cooper, L.J.; DiGiusto, D.; Kalos, M.; Ostberg, J.R.; Forman, S.J. Antitransgene Rejection Responses Contribute to Attenuated Persistence of Adoptively Transferred CD20/CD19-Specific Chimeric Antigen Receptor Redirected T Cells in Humans. Biol. Blood Marrow Transplant. 2010, 16, 1245–1256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Li, C.; Xia, J.; Li, P.; Cao, J.; Pan, B.; Tan, X.; Li, H.; Qi, K.; Wang, X.; et al. Humoral immune reconstitution after anti-BCMA CAR T-cell therapy in relapsed/refractory multiple myeloma. Blood Adv. 2021, 5, 5290–5299. [Google Scholar] [CrossRef]
- Doan, A.; Pulsipher, M.A. Hypogammaglobulinemia due to CAR T-cell therapy. Pediatr. Blood Cancer 2018, 65, e26914. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Jensen, P.E. The role of B lymphocytes as antigen-presenting cells. Arch. Immunol. Ther. Exp. 2008, 56, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Harris, D.T.; Kranz, D.M. Adoptive T Cell Therapies: A Comparison of T Cell Receptors and Chimeric Antigen Receptors. Trends Pharmacol. Sci. 2016, 37, 220–230. [Google Scholar] [CrossRef] [Green Version]
- De Groot, A.S.; Terry, F.; Cousens, L.; Martin, W. Beyond humanization and de-immunization: Tolerization as a method for reducing the immunogenicity of biologics. Expert Rev. Clin. Pharmacol. 2013, 6, 651–662. [Google Scholar] [CrossRef] [Green Version]
- Augustyniak, D.; Majkowska-Skrobek, G.; Roszkowiak, J.; Dorotkiewicz-Jach, A. Defensive and Offensive Cross-Reactive Antibodies Elicited by Pathogens: The Good, the Bad and the Ugly. Curr. Med. Chem. 2017, 24, 4002–4037. [Google Scholar] [CrossRef]
- Polymeros, D.; Bogdanos, D.P.; Day, R.; Arioli, D.; Vergani, D.; Forbes, A. Does Cross-Reactivity between Mycobacterium avium paratuberculosis and Human Intestinal Antigens Characterize Crohn’s Disease? Gastroenterology 2006, 131, 85–96. [Google Scholar] [CrossRef]
- Larmonier, C.B.; Shehab, K.W.; Ghishan, F.K.; Kiela, P. T Lymphocyte Dynamics in Inflammatory Bowel Diseases: Role of the Microbiome. BioMed Res. Int. 2015, 2015, 504638. [Google Scholar] [CrossRef] [Green Version]
- Bartelds, G.M.; Krieckaert, C.L.M.; Nurmohamed, M.T.; van Schouwenburg, P.A.; Lems, W.F.; Twisk, J.W.R.; Dijkmans, B.A.C.; Aarden, L.; jan Wolbink, G. Development of Antidrug Antibodies Against Adalimumab and Association With Disease Activity and Treatment Failure During Long-term Follow-up. JAMA 2011, 305, 1460–1468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abramson, J.S.; Palomba, M.L.; Gordon, L.I.; Lunning, M.A.; Wang, M.; Arnason, J.; Mehta, A.; Purev, E.; Maloney, D.G.; Andreadis, C.; et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): A multicentre seamless design study. Lancet 2020, 396, 839–852. [Google Scholar] [CrossRef]
- Wang, M.; Munoz, J.; Goy, A.; Locke, F.L.; Jacobson, C.A.; Hill, B.T.; Timmerman, J.M.; Holmes, H.; Jaglowski, S.; Flinn, I.W.; et al. KTE-X19 CAR T-Cell Therapy in Relapsed or Refractory Mantle-Cell Lymphoma. N. Engl. J. Med. 2020, 382, 1331–1342. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Munshi, N.C.; Anderson, L.D., Jr.; Shah, N.; Madduri, D.; Berdeja, J.; Lonial, S.; Raje, N.; Lin, Y.; Siegel, D.; Oriol, A.; et al. Idecabtagene Vicleucel in Relapsed and Refractory Multiple Myeloma. N. Engl. J. Med. 2021, 384, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Geyer, M.B.; Brentjens, R.J. CD19-targeted CAR T-cell therapeutics for hematologic malignancies: Interpreting clinical outcomes to date. Blood 2016, 127, 3312–3320. [Google Scholar] [CrossRef] [PubMed]
- Kenderian, S.; Ruella, M.; Shestova, O.; Klichinsky, M.; Aikawa, V.; Morrissette, J.J.D.; Scholler, J.; Song, D.; Porter, D.L.; Carroll, M.C.; et al. CD33-specific chimeric antigen receptor T cells exhibit potent preclinical activity against human acute myeloid leukemia. Leukemia 2015, 29, 1637–1647. [Google Scholar] [CrossRef]
- Zhong, Q.; Zhu, Y.-M.; Zheng, L.-L.; Shen, H.-J.; Ou, R.-M.; Liu, Z.; She, Y.-L.; Chen, R.; Li, C.; Huang, J.; et al. Chimeric Antigen Receptor-T Cells with 4-1BB Co-Stimulatory Domain Present a Superior Treatment Outcome than Those with CD28 Domain Based on Bioinformatics. Acta Haematol. 2018, 140, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Shao, Z.; Schwarz, H. CD137 ligand, a member of the tumor necrosis factor family, regulates immune responses via reverse signal transduction. J. Leukoc. Biol. 2011, 89, 21–29. [Google Scholar] [CrossRef]
- Esensten, J.H.; Helou, Y.A.; Chopra, G.; Weiss, A.; Bluestone, J.A. CD28 Costimulation: From Mechanism to Therapy. Immunity 2016, 44, 973–988. [Google Scholar] [CrossRef] [Green Version]
- Colombetti, S.; Basso, V.; Mueller, D.; Mondino, A. Prolonged TCR/CD28 Engagement Drives IL-2-Independent T Cell Clonal Expansion through Signaling Mediated by the Mammalian Target of Rapamycin. J. Immunol. 2006, 176, 2730–2738. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Condomines, M.; Van Der Stegen, S.J.C.; Perna, F.; Kloss, C.C.; Gunset, G.; Plotkin, J.; Sadelain, M. Structural Design of Engineered Costimulation Determines Tumor Rejection Kinetics and Persistence of CAR T Cells. Cancer Cell 2015, 28, 415–428. [Google Scholar] [CrossRef] [Green Version]
- Kawalekar, O.U.; O’Connor, R.S.; Fraietta, J.A.; Guo, L.; Mcgettigan, S.E.; Posey, A.D.; Patel, P.R.; Guedan, S.; Scholler, J.; Keith, B.; et al. Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity 2016, 44, 380–390. [Google Scholar] [CrossRef] [Green Version]
- Ying, Z.; He, T.; Wang, X.; Zheng, W.; Lin, N.; Tu, M.; Xie, Y.; Ping, L.; Zhang, C.; Liu, W.; et al. Parallel Comparison of 4-1BB or CD28 Co-stimulated CD19-Targeted CAR-T Cells for B Cell Non-Hodgkin’s Lymphoma. Mol. Ther. Oncolytics 2019, 15, 60–68. [Google Scholar] [CrossRef] [Green Version]
- Drent, E.; Poels, R.; Ruiter, R.; Van De Donk, N.W.C.J.; Zweegman, S.; Yuan, H.; de Bruijn, J.; Sadelain, M.; Lokhorst, H.M.; Groen, R.W.J.; et al. Combined CD28 and 4-1BB Costimulation Potentiates Affinity-tuned Chimeric Antigen Receptor–engineered T Cells. Clin. Cancer Res. 2019, 25, 4014–4025. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Boucher, J.C.; Kotani, H.; Park, K.; Zhang, Y.; Shrestha, B.; Wang, X.; Guan, L.; Beatty, N.; Abate-Daga, D.; et al. 4-1BB enhancement of CAR T function requires NF-κB and TRAFs. JCI Insight 2018, 3, e121322. [Google Scholar] [CrossRef] [Green Version]
- Porter, D.L.; Hwang, W.-T.; Frey, N.V.; Lacey, S.F.; Shaw, P.A.; Loren, A.W.; Bagg, A.; Marcucci, K.T.; Shen, A.; Gonzalez, V.; et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci. Transl. Med. 2015, 7, 303ra139. [Google Scholar] [CrossRef] [Green Version]
- Park, J.H.; Rivière, I.; Gonen, M.; Wang, X.; Sénéchal, B.; Curran, K.J.; Sauter, C.; Wang, Y.; Santomasso, B.; Mead, E.; et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 449–459. [Google Scholar] [CrossRef]
- Gökbuget, N.; Stanze, D.; Beck, J.; Diedrich, H.; Horst, H.-A.; Hüttmann, A.; Kobbe, G.; Kreuzer, K.-A.; Leimer, L.; Reichle, A.; et al. Outcome of relapsed adult lymphoblastic leukemia depends on response to salvage chemotherapy, prognostic factors, and performance of stem cell transplantation. Blood 2012, 120, 2032–2041. [Google Scholar] [CrossRef]
- Martin, A.; Morgan, E.; Hijiya, N. Relapsed or refractory pediatric acute lymphoblastic leukemia: Current and emerging treatments. Paediatr. Drugs 2012, 14, 377–387. [Google Scholar] [CrossRef]
- Sun, W.; Malvar, J.; Sposto, R.; Verma, A.; Wilkes, J.J.; Dennis, R.; Heym, K.; Laetsch, T.W.; Widener, M.; Rheingold, S.R.; et al. Outcome of children with multiply relapsed B-cell acute lymphoblastic leukemia: A therapeutic advances in childhood leukemia & lymphoma study. Leukemia 2018, 32, 2316–2325. [Google Scholar] [PubMed]
- Locatelli, F.; Whitlock, J.A.; Peters, C.; Chen-Santel, C.; Chia, V.; Dennis, R.M.; Heym, K.M.; Katz, A.J.; Kelsh, M.A.; Sposto, R.; et al. Blinatumomab versus historical standard therapy in pediatric patients with relapsed/refractory Ph-negative B-cell precursor acute lymphoblastic leukemia. Leukemia 2020, 34, 2473–2478. [Google Scholar] [CrossRef] [PubMed]
- Parker, K.R.; Migliorini, D.; Perkey, E.; Yost, K.E.; Bhaduri, A.; Bagga, P.; Haris, M.; Wilson, N.E.; Liu, F.; Gabunia, K.; et al. Single-Cell Analyses Identify Brain Mural Cells Expressing CD19 as Potential Off-Tumor Targets for CAR-T Immunotherapies. Cell 2020, 183, 126–142.e17. [Google Scholar] [CrossRef] [PubMed]
- Mejstrikova, E.; Klinger, M.; Markovic, A.; Zugmaier, G.; Locatelli, F. CD19 expression in pediatric patients with relapsed/refractory B-cell precursor acute lymphoblastic leukemia pre- and post-treatment with blinatumomab. Pediatr. Blood Cancer 2021, 68, e29323. [Google Scholar] [CrossRef] [PubMed]
- Neelapu, S.S. CAR-T efficacy: Is conditioning the key? Blood 2019, 133, 1799–1800. [Google Scholar] [CrossRef]
- Harrison, R.P.; Rafiq, Q.A.; Medcalf, N. Centralised versus decentralised manufacturing and the delivery of healthcare products: A United Kingdom exemplar. Cytotherapy 2018, 20, 873–890. [Google Scholar] [CrossRef]
- Maude, S.L. Future directions in chimeric antigen receptor T cell therapy. Curr. Opin. Pediatr. 2017, 29, 27–33. [Google Scholar] [CrossRef]
- Gardner, R.A.; Finney, O.; Annesley, C.; Brakke, H.; Summers, C.; Leger, K.; Bleakley, M.; Brown, C.; Mgebroff, S.; Kelly-Spratt, K.S.; et al. Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood 2017, 129, 3322–3331. [Google Scholar] [CrossRef]
- Pasquini, M.C.; Hu, Z.-H.; Curran, K.; Laetsch, T.; Locke, F.; Rouce, R.; Pulsipher, M.A.; Phillips, C.L.; Keating, A.; Frigault, M.J.; et al. Real-world evidence of tisagenlecleucel for pediatric acute lymphoblastic leukemia and non-Hodgkin lymphoma. Blood Adv. 2020, 4, 5414–5424. [Google Scholar] [CrossRef]
- EHA2021 Virtual Congress Abstract Book. HemaSphere 2021, 5, e566. [CrossRef]
- Diorio, C.; Maude, S.L. CAR T cells vs allogeneic HSCT for poor-risk ALL. Hematology 2020, 2020, 501–507. [Google Scholar] [CrossRef] [PubMed]
- Gaynon, P.S.; Harris, R.E.; Altman, A.J.; Bostrom, B.C.; Breneman, J.C.; Hawks, R.; Steele, D.; Zipf, T.; Stram, D.O.; Villaluna, D.; et al. Bone Marrow Transplantation Versus Prolonged Intensive Chemotherapy for Children With Acute Lymphoblastic Leukemia and an Initial Bone Marrow Relapse Within 12 Months of the Completion of Primary Therapy: Children’s Oncology Group Study CCG-1941. J. Clin. Oncol. 2006, 24, 3150–3156. [Google Scholar] [CrossRef] [PubMed]
- Locatelli, F.; Zugmaier, G.; Mergen, N.; Bader, P.; Jeha, S.; Schlegel, P.-G.; Bourquin, J.-P.; Handgretinger, R.; Brethon, B.; Rössig, C.; et al. Blinatumomab In pediatric relapsed/refractory B-cell acute lymphoblastic leukemia: RIALTO expanded access study final analysis. Blood Adv. 2022, 6, 1004–1014. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Hu, Y.; Mei, H. Consolidative allogeneic hematopoietic stem cell transplantation after chimeric antigen receptor T-cell therapy for relapsed/refractory B-cell acute lymphoblastic leukemia: Who? When? Why? Biomark. Res. 2020, 8, 66. [Google Scholar] [CrossRef]
- Hierlmeier, S.; Eyrich, M.; Wölfl, M.; Schlegel, P.-G.; Wiegering, V. Early and late complications following hematopoietic stem cell transplantation in pediatric patients—A retrospective analysis over 11 years. PLoS ONE 2018, 13, e0204914. [Google Scholar] [CrossRef] [Green Version]
- Shah, N.N.; Highfill, S.L.; Shalabi, H.; Yates, B.; Jin, J.; Wolters, P.L.; Ombrello, A.; Steinberg, S.M.; Martin, S.; Delbrook, C.; et al. CD4/CD8 T-Cell Selection Affects Chimeric Antigen Receptor (CAR) T-Cell Potency and Toxicity: Updated Results From a Phase I Anti-CD22 CAR T-Cell Trial. J. Clin. Oncol. 2020, 38, 1938–1950. [Google Scholar] [CrossRef]
- Lee, D.W.; A Gardner, R.; Porter, D.L.; Louis, C.U.; Ahmed, N.; Jensen, M.C.; Grupp, S.A.; Mackall, C.L. Current concepts in the diagnosis and management of cytokine release syndrome. Blood 2014, 124, 188–195. [Google Scholar] [CrossRef] [Green Version]
- Hay, K.A.; Hanafi, L.-A.; Li, D.; Gust, J.; Liles, W.C.; Wurfel, M.M.; López, J.A.; Chen, J.; Chung, D.; Harju-Baker, S.; et al. Kinetics and biomarkers of severe cytokine release syndrome after CD19 chimeric antigen receptor–modified T-cell therapy. Blood 2017, 130, 2295–2306. [Google Scholar] [CrossRef] [Green Version]
- Fitzgerald, J.C.; Weiss, S.; Maude, S.L.; Barrett, D.M.; Lacey, S.F.; Melenhorst, J.J.; Shaw, P.; Berg, R.A.; June, C.H.; Porter, D.L.; et al. Cytokine Release Syndrome After Chimeric Antigen Receptor T Cell Therapy for Acute Lymphoblastic Leukemia. Crit. Care Med. 2017, 45, e124–e131. [Google Scholar] [CrossRef]
- Neelapu, S.S.; Tummala, S.; Kebriaei, P.; Wierda, W.; Gutierrez, C.; Locke, F.L.; Komanduri, K.V.; Lin, Y.; Jain, N.; Daver, N.; et al. Chimeric antigen receptor T-cell therapy—Assessment and management of toxicities. Nat. Rev. Clin. Oncol. 2018, 15, 47–62. [Google Scholar] [CrossRef]
- Wei, J.; Liu, Y.; Wang, C.; Zhang, Y.; Tong, C.; Dai, G.; Wang, W.; Rasko, J.; Melenhorst, J.J.; Qian, W.; et al. The model of cytokine release syndrome in CAR T-cell treatment for B-cell non-Hodgkin lymphoma. Signal Transduct. Target. Ther. 2020, 5, 134. [Google Scholar] [CrossRef] [PubMed]
- Gust, J.; Hay, K.A.; Hanafi, L.-A.; Li, D.; Myerson, D.; Gonzalez-Cuyar, L.F.; Yeung, C.; Liles, W.C.; Wurfel, M.; Lopez, J.A.; et al. Endothelial Activation and Blood–Brain Barrier Disruption in Neurotoxicity after Adoptive Immunotherapy with CD19 CAR-T Cells. Cancer Discov. 2017, 7, 1404–1419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegler, E.L.; Kenderian, S.S. Neurotoxicity and Cytokine Release Syndrome after Chimeric Antigen Receptor T Cell Therapy: Insights Into Mechanisms and Novel Therapies. Front. Immunol. 2020, 11, 1973. [Google Scholar] [CrossRef] [PubMed]
- Gust, J.; Ponce, R.; Liles, W.C.; Garden, G.A.; Turtle, C.J. Cytokines in CAR T Cell–Associated Neurotoxicity. Front. Immunol. 2020, 11, 577027. [Google Scholar] [CrossRef] [PubMed]
- Nellan, A.; McCully, C.M.L.; Garcia, R.C.; Jayaprakash, N.; Widemann, B.C.; Lee, D.W.; Warren, K.E. Improved CNS exposure to tocilizumab after cerebrospinal fluid compared to intravenous administration in rhesus macaques. Blood 2018, 132, 662–666. [Google Scholar] [CrossRef]
- Riegler, L.L.; Jones, G.P.; Lee, D.W. Current approaches in the grading and management of cytokine release syndrome after chimeric antigen receptor T-cell therapy. Ther. Clin. Risk Manag. 2019, 15, 323–335. [Google Scholar] [CrossRef] [Green Version]
- Wudhikarn, K.; Bansal, R.; Khurana, A.; Hathcock, M.; Ruff, M.; Carabenciov, I.D.; Braksick, S.A.; Bennani, N.N.; Paludo, J.; Villasboas, J.C.; et al. Characteristics, outcomes, and risk factors of ICANS after axicabtagene ciloleucel: Does age matter? J. Clin. Oncol. 2021, 39, e19556. [Google Scholar] [CrossRef]
- Sievers, S.; Watson, G.; Johncy, S.; Adkins, S. Recognizing and Grading CAR T-Cell Toxicities: An Advanced Practitioner Perspective. Front. Oncol. 2020, 10, 885. [Google Scholar] [CrossRef]
- Zhao, X.; Yang, J.; Zhang, X.; Lu, X.-A.; Xiong, M.; Zhang, J.; Zhou, X.; Qi, F.; He, T.; Ding, Y.; et al. Efficacy and Safety of CD28- or 4-1BB-Based CD19 CAR-T Cells in B Cell Acute Lymphoblastic Leukemia. Mol. Ther. Oncolytics 2020, 18, 272–281. [Google Scholar] [CrossRef]
- JCAR015 in ALL: A Root-Cause Investigation. Cancer Discov. 2018, 8, 4–5.
- Henderson, L.A.; Cron, R.Q. Macrophage Activation Syndrome and Secondary Hemophagocytic Lymphohistiocytosis in Childhood Inflammatory Disorders: Diagnosis and Management. Pediatr. Drugs 2020, 22, 29–44. [Google Scholar] [CrossRef] [PubMed]
- Voskoboinik, I.; Whisstock, J.C.; Trapani, J.A. Perforin and granzymes: Function, dysfunction and human pathology. Nat. Rev. Immunol. 2015, 15, 388–400. [Google Scholar] [CrossRef] [PubMed]
- Gadoury-Levesque, V.; Dong, L.; Su, R.; Chen, J.; Zhang, K.; Risma, K.A.; Marsh, R.A.; Sun, M. Frequency and spectrum of disease-causing variants in 1892 patients with suspected genetic HLH disorders. Blood Adv. 2020, 4, 2578–2594. [Google Scholar] [CrossRef] [PubMed]
- Grom, A.A.; Horne, A.; De Benedetti, F. Macrophage activation syndrome in the era of biologic therapy. Nat. Rev. Rheumatol. 2016, 12, 259–268. [Google Scholar] [CrossRef]
- Canna, S.W.; de Jesus, A.A.; Gouni, S.; Brooks, S.R.; Marrero, B.; Liu, Y.; DiMattia, M.A.; Zaal, K.J.M.; Sanchez, G.A.M.; Kim, H.; et al. An activating NLRC4 inflammasome mutation causes autoinflammation with recurrent macrophage activation syndrome. Nat. Genet. 2014, 46, 1140–1146. [Google Scholar] [CrossRef] [Green Version]
- Teachey, D.T.; Rheingold, S.R.; Maude, S.L.; Zugmaier, G.; Barrett, D.M.; Seif, A.E.; Nichols, K.E.; Suppa, E.K.; Kalos, M.; Berg, R.A.; et al. Cytokine release syndrome after blinatumomab treatment related to abnormal macrophage activation and ameliorated with cytokine-directed therapy. Blood 2013, 121, 5154–5157. [Google Scholar] [CrossRef]
- Morris, E.C.; Neelapu, S.S.; Giavridis, T.; Sadelain, M. Cytokine release syndrome and associated neurotoxicity in cancer immunotherapy. Nat. Rev. Immunol. 2021, 22, 85–96. [Google Scholar] [CrossRef]
- A Misbah, S.; Weeratunga, P. Immunoglobulin replacement and quality of life after CAR T-cell therapy. Lancet Oncol. 2020, 21, e6. [Google Scholar] [CrossRef] [Green Version]
- Hill, J.A.; Seo, S.K. How I prevent infections in patients receiving CD19-targeted chimeric antigen receptor T cells for B-cell malignancies. Blood 2020, 136, 925–935. [Google Scholar] [CrossRef]
- Rabilloud, T.; Potier, D.; Pankaew, S.; Nozais, M.; Loosveld, M.; Payet-Bornet, D. Single-cell profiling identifies pre-existing CD19-negative subclones in a B-ALL patient with CD19-negative relapse after CAR-T therapy. Nat. Commun. 2021, 12, 865. [Google Scholar] [CrossRef]
- Weiland, J.; Pal, D.; Case, M.; Irving, J.; Ponthan, F.; Koschmieder, S.; Heidenreich, O.; Von Stackelberg, A.; Eckert, C.; Vormoor, J.; et al. BCP-ALL blasts are not dependent on CD19 expression for leukaemic maintenance. Leukemia 2016, 30, 1920–1923. [Google Scholar] [CrossRef] [PubMed]
- Dourthe, M.-E.; Rabian, F.; Yakouben, K.; Chevillon, F.; Cabannes-Hamy, A.; Méchinaud, F.; Grain, A.; Chaillou, D.; Rahal, I.; Caillat-Zucman, S.; et al. Determinants of CD19-positive vs CD19-negative relapse after tisagenlecleucel for B-cell acute lymphoblastic leukemia. Leukemia 2021, 35, 3383–3393. [Google Scholar] [CrossRef] [PubMed]
- Sotillo, E.; Barrett, D.M.; Black, K.L.; Bagashev, A.; Oldridge, D.; Wu, G.; Sussman, R.; LaNauze, C.; Ruella, M.; Gazzara, M.R.; et al. Convergence of Acquired Mutations and Alternative Splicing of CD19 Enables Resistance to CART-19 Immunotherapy. Cancer Discov. 2015, 5, 1282–1295. [Google Scholar] [CrossRef] [Green Version]
- Gardner, R.; Wu, D.; Cherian, S.; Fang, M.; Hanafi, L.-A.; Finney, O.; Smithers, H.; Jensen, M.C.; Riddell, S.R.; Maloney, D.G.; et al. Acquisition of a CD19-negative myeloid phenotype allows immune escape of MLL-rearranged B-ALL from CD19 CAR-T-cell therapy. Blood 2016, 127, 2406–2410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; You, M.J.; Young, K.H.; Lin, P.; Lu, G.; Medeiros, L.J.; Bueso-Ramos, C.E. Advances in the molecular pathobiology of B-lymphoblastic leukemia. Hum. Pathol. 2012, 43, 1347–1362. [Google Scholar] [CrossRef]
- Salvaris, R.; Fedele, P. Targeted Therapy in Acute Lymphoblastic Leukaemia. J. Pers. Med. 2021, 11, 715. [Google Scholar] [CrossRef]
- Zhang, W.-Y.; Liu, Y.; Wang, Y.; Wang, C.-M.; Yang, Q.-M.; Zhu, H.-L.; Han, W.-D. Long-term safety and efficacy of CART-20 cells in patients with refractory or relapsed B-cell non-Hodgkin lymphoma: 5-years follow-up results of the phase I and IIa trials. Signal Transduct. Target. Ther. 2017, 2, 17054. [Google Scholar] [CrossRef] [Green Version]
- Liang, A.; Ye, S.; Li, P.; Huang, J.; Zhu, S.; Yao, X.; Zhou, L.; Xu, Y.; Zhu, J.; Zheng, C.; et al. Safety and efficacy of a novel anti-CD20 chimeric antigen receptor (CAR)-T cell therapy in relapsed/refractory (r/r) B-cell non-Hodgkin lymphoma (B-NHL) patients after failing CD19 CAR-T therapy. J. Clin. Oncol. 2021, 39, 2508. [Google Scholar] [CrossRef]
- Shadman, M.; Yeung, C.; Redman, M.; Lee, S.Y.; Lee, D.H.; Ra, S.; Ujjani, C.S.; Dezube, B.J.; Poh, C.; Warren, E.H.; et al. Safety and Efficacy of Third Generation CD20 Targeted CAR-T (MB-106) for Treatment of Relapsed/Refractory B-NHL and CLL. Blood 2021, 138, 3872. [Google Scholar] [CrossRef]
- Schultz, L.M.; Muffly, L.S.; Spiegel, J.Y.; Ramakrishna, S.; Hossain, N.; Baggott, C.; Sahaf, B.; Patel, S.; Craig, J.; Yoon, J.; et al. Phase I Trial Using CD19/CD22 Bispecific CAR T Cells in Pediatric and Adult Acute Lymphoblastic Leukemia (ALL). Blood 2019, 134, 744. [Google Scholar] [CrossRef]
- Shalabi, H.; Yates, B.; Shahani, S.; Qin, H.; HIghfill, S.L.; Panch, S.; Tran, M.; Stroncek, D.; Hoffman, L.; Little, L.; et al. Abstract CT051: Safety and efficacy of CD19/CD22 CAR T cells in children and young adults with relapsed/refractory ALL. Cancer Res. 2020, 80, CT051. [Google Scholar]
- Shah, N.N.; Johnson, B.D.; Schneider, D.; Zhu, F.; Szabo, A.; Keever-Taylor, C.A.; Krueger, W.; Worden, A.A.; Kadan, M.J.; Yim, S.; et al. Bispecific anti-CD20, anti-CD19 CAR T cells for relapsed B cell malignancies: A phase 1 dose escalation and expansion trial. Nat. Med. 2020, 26, 1569–1575. [Google Scholar] [CrossRef] [PubMed]
- Köksal, H.; Dillard, P.; Josefsson, S.E.; Maggadottir, S.M.; Pollmann, S.; Fåne, A.; Blaker, Y.N.; Beiske, K.; Huse, K.; Kolstad, A.; et al. Preclinical development of CD37CAR T-cell therapy for treatment of B-cell lymphoma. Blood Adv. 2019, 3, 1230–1243. [Google Scholar] [CrossRef] [PubMed]
- Scarfò, I.; Ormhøj, M.; Frigault, M.J.; Castano, A.P.; Lorrey, S.; Bouffard, A.A.; Van Scoyk, A.; Rodig, S.J.; Shay, A.J.; Aster, J.C.; et al. Anti-CD37 chimeric antigen receptor T cells are active against B- and T-cell lymphomas. Blood 2018, 132, 1495–1506. [Google Scholar] [CrossRef] [PubMed]
- Patterson, J.D.; Henson, J.C.; Breese, R.O.; Bielamowicz, K.J.; Rodriguez, A. CAR T Cell Therapy for Pediatric Brain Tumors. Front. Oncol. 2020, 10, 1582. [Google Scholar] [CrossRef] [PubMed]
- Richards, R.M.; Sotillo, E.; Majzner, R.G. CAR T Cell Therapy for Neuroblastoma. Front. Immunol. 2018, 9, 2380. [Google Scholar] [CrossRef] [Green Version]
- Majzner, R.G.; Theruvath, J.L.; Nellan, A.; Heitzeneder, S.; Cui, Y.; Mount, C.W.; Rietberg, S.P.; Linde, M.H.; Xu, P.; Rota, C.; et al. CAR T Cells Targeting B7-H3, a Pan-Cancer Antigen, Demonstrate Potent Preclinical Activity Against Pediatric Solid Tumors and Brain Tumors. Clin. Cancer Res. 2019, 25, 2560–2574. [Google Scholar] [CrossRef]
- Raje, N.; Berdeja, J.; Lin, Y.; Siegel, D.; Jagannath, S.; Madduri, D.; Liedtke, M.; Rosenblatt, J.; Maus, M.V.; Turka, A.; et al. Anti-BCMA CAR T-Cell Therapy bb2121 in Relapsed or Refractory Multiple Myeloma. N. Engl. J. Med. 2019, 380, 1726–1737. [Google Scholar] [CrossRef]
- Qin, H.; Yang, L.; Chukinas, J.A.; Shah, N.N.; Tarun, S.; Pouzolles, M.; Chien, C.D.; Niswander, L.M.; Welch, A.R.; Taylor, N.A.; et al. Systematic preclinical evaluation of CD33-directed chimeric antigen receptor T cell immunotherapy for acute myeloid leukemia defines optimized construct design. J. Immunother. Cancer 2021, 9, e003149. [Google Scholar] [CrossRef]
- Wolf, P.; Alzubi, J.; Gratzke, C.; Cathomen, T. The potential of CAR T cell therapy for prostate cancer. Nat. Rev. Urol. 2021, 18, 556–571. [Google Scholar] [CrossRef]
- Akce, M.; Zaidi, M.Y.; Waller, E.K.; El-Rayes, B.F.; Lesinski, G.B. The Potential of CAR T Cell Therapy in Pancreatic Cancer. Front. Immunol. 2018, 9, 2166. [Google Scholar] [CrossRef] [PubMed]
- Bo, M.D.; De Mattia, E.; Baboci, L.; Mezzalira, S.; Cecchin, E.; Assaraf, Y.G.; Toffoli, G. New insights into the pharmacological, immunological, and CAR-T-cell approaches in the treatment of hepatocellular carcinoma. Drug Resist. Updat. 2020, 51, 100702. [Google Scholar]
- Kudo, K.; Imai, C.; Lorenzini, P.; Kamiya, T.; Kono, K.; Davidoff, A.M.; Chng, W.J.; Campana, D. T Lymphocytes Expressing a CD16 Signaling Receptor Exert Antibody-Dependent Cancer Cell Killing. Cancer Res. 2014, 74, 93–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rataj, F.; Jacobi, S.J.; Stoiber, S.; Asang, F.; Ogonek, J.; Tokarew, N.; Cadilha, B.; Van Puijenbroek, E.; Heise, C.; Duewell, P.; et al. High-affinity CD16-polymorphism and Fc-engineered antibodies enable activity of CD16-chimeric antigen receptor-modified T cells for cancer therapy. Br. J. Cancer 2019, 120, 79–87. [Google Scholar] [CrossRef] [Green Version]
- Tamada, K.; Geng, D.; Sakoda, Y.; Bansal, N.; Srivastava, R.; Li, Z.; Davila, E. Redirecting Gene-Modified T Cells toward Various Cancer Types Using Tagged Antibodies. Clin. Cancer Res. 2012, 18, 6436–6445. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.S.Y.; Kim, J.Y.; Kazane, S.A.; Choi, S.-H.; Yun, H.Y.; Kim, M.S.; Rodgers, D.T.; Pugh, H.M.; Singer, O.; Sun, S.B.; et al. Versatile strategy for controlling the specificity and activity of engineered T cells. Proc. Natl. Acad. Sci. USA 2016, 113, E450–E458. [Google Scholar] [CrossRef] [Green Version]
- Rodgers, D.T.; Mazagova, M.; Hampton, E.N.; Cao, Y.; Ramadoss, N.S.; Hardy, I.R.; Schulman, A.; Du, J.; Wang, F.; Singer, O.; et al. Switch-mediated activation and retargeting of CAR-T cells for B-cell malignancies. Proc. Natl. Acad. Sci. USA 2016, 113, E459–E468. [Google Scholar] [CrossRef] [Green Version]
- Bachmann, M. The UniCAR system: A modular CAR T cell approach to improve the safety of CAR T cells. Immunol. Lett. 2019, 211, 13–22. [Google Scholar] [CrossRef]
- Mu, J.; Edwards, J.; Zaritskaya, L.; Swers, J.; Gupta, A.; LaFleur, D.; Hilbert, D.; Richman, L. Selective targeting of HER2-overexpressing solid tumors with a next-generation CAR-T cell therapy. J. Clin. Oncol. 2020, 38, 3041. [Google Scholar] [CrossRef]
- Grote, S.; Mittelstaet, J.; Baden, C.; Chan, K.C.; Seitz, C.; Schlegel, P.; Kaiser, A.; Handgretinger, R.; Schleicher, S. Adapter chimeric antigen receptor (AdCAR)-engineered NK-92 cells: An off-the-shelf cellular therapeutic for universal tumor targeting. Oncoimmunology 2020, 9, 1825177. [Google Scholar] [CrossRef]
- Urbanska, K.; Lanitis, E.; Poussin, M.; Lynn, R.C.; Gavin, B.P.; Kelderman, S.; Yu, J.; Scholler, N.; Powell, D.J., Jr. A Universal Strategy for Adoptive Immunotherapy of Cancer through Use of a Novel T-cell Antigen Receptor. Cancer Res. 2012, 72, 1844–1852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, J.H.; Collins, J.J.; Wong, W.W. Universal Chimeric Antigen Receptors for Multiplexed and Logical Control of T Cell Responses. Cell 2018, 173, 1426–1438.e11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.S.; Ma, J.S.Y.; Yun, H.; Cao, Y.; Kim, J.Y.; Chi, V.; Wang, D.; Woods, A.; Sherwood, L.; Caballero, D.; et al. Redirection of Genetically Engineered CAR-T Cells Using Bifunctional Small Molecules. J. Am. Chem. Soc. 2015, 137, 2832–2835. [Google Scholar] [CrossRef] [PubMed]
- Ryman, J.T.; Meibohm, B. Pharmacokinetics of Monoclonal Antibodies. CPT Pharmacomet. Syst. Pharmacol. 2017, 6, 576–588. [Google Scholar] [CrossRef]
- Waldmann, T.A.; Strober, W. Metabolism of Immunoglobulins. Prog. Allergy 1969, 13, 891–903. [Google Scholar]
- Knauf, M.J.; Bell, D.P.; Hirtzer, P.; Luo, Z.P.; Young, J.D.; Katre, N.V. Relationship of effective molecular size to systemic clearance in rats of recombinant interleukin-2 chemically modified with water-soluble polymers. J. Biol. Chem. 1988, 263, 15064–15070. [Google Scholar] [CrossRef]
- Lampson, L.A. Monoclonal antibodies in neuro-oncology: Getting past the blood-brain barrier. mAbs 2011, 3, 153–160. [Google Scholar] [CrossRef] [Green Version]
- Cavaco, M.; Gaspar, D.; Castanho, M.A.; Neves, V. Antibodies for the Treatment of Brain Metastases, a Dream or a Reality? Pharmaceutics 2020, 12, 62. [Google Scholar] [CrossRef] [Green Version]
- Cruz, E.; Kayser, V. Monoclonal antibody therapy of solid tumors: Clinical limitations and novel strategies to enhance treatment efficacy. Biol. Targets Ther. 2019, 13, 33–51. [Google Scholar] [CrossRef] [Green Version]
- Davis, K.L.; Mackall, C.L. Immunotherapy for acute lymphoblastic leukemia: From famine to feast. Blood Adv. 2016, 1, 265–269. [Google Scholar] [CrossRef] [Green Version]
- Lenk, L.; Alsadeq, A.; Schewe, D.M. Involvement of the central nervous system in acute lymphoblastic leukemia: Opinions on molecular mechanisms and clinical implications based on recent data. Cancer Metastasis Rev. 2020, 39, 173–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abid, H.; Watthanasuntorn, K.; Shah, O.; Gnanajothy, R. Efficacy of Pembrolizumab and Nivolumab in Crossing the Blood Brain Barrier. Cureus 2019, 11, e4446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cartellieri, M.; Feldmann, A.; Koristka, S.; Arndt, C.; Loff, S.; Ehninger, A.V.; Von Bonin, M.; Bejestani, E.P.; Ehninger, G.; Bachmann, M.P. Switching CAR T cells on and off: A novel modular platform for retargeting of T cells to AML blasts. Blood Cancer J. 2016, 6, e458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goebeler, M.-E.; Knop, S.; Viardot, A.; Kufer, P.; Topp, M.S.; Einsele, H.; Noppeney, R.; Hess, G.; Kallert, S.; Mackensen, A.; et al. Bispecific T-Cell Engager (BiTE) Antibody Construct Blinatumomab for the Treatment of Patients With Relapsed/Refractory Non-Hodgkin Lymphoma: Final Results From a Phase I Study. J. Clin. Oncol. 2016, 34, 1104–1111. [Google Scholar] [CrossRef]
- . Hong, W.X.; Haebe, S.; Lee, A.S.; Westphalen, C.B.; Norton, J.A.; Jiang, W.; Levy, R. Intratumoral Immunotherapy for Early-stage Solid Tumors. Clin. Cancer Res. 2020, 26, 3091–3099. [Google Scholar] [CrossRef] [Green Version]
- Adusumilli, P.S.; Zauderer, M.G.; Rivière, I.; Solomon, S.B.; Rusch, V.W.; O’Cearbhaill, R.E.; Zhu, A.; Cheema, W.; Chintala, N.K.; Halton, E.; et al. A Phase I Trial of Regional Mesothelin-Targeted CAR T-cell Therapy in Patients with Malignant Pleural Disease, in Combination with the Anti–PD-1 Agent Pembrolizumab. Cancer Discov. 2021, 11, 2748–2763. [Google Scholar] [CrossRef]
- Choi, B.D.; O’Rourke, D.M.; Maus, M.V. Engineering Chimeric Antigen Receptor T cells to Treat Glioblastoma. Int. J. Target. Ther. Cancer 2017, 6, 22–25. [Google Scholar]
- Schulz, H.; Pels, H.; Schmidt-Wolf, I.; Zeelen, U.; Germing, U.; Engert, A. Intraventricular treatment of relapsed central nervous system lymphoma with the anti-CD20 antibody rituximab. Haematology 2004, 89, 753–754. [Google Scholar]
- Sison, E.A.R.; Silverman, L.B. CNS prophylaxis in pediatric acute lymphoblastic leukemia. Hematology 2014, 2014, 198–201. [Google Scholar] [CrossRef] [Green Version]
- Ravandi, F.; Walter, R.B.; Subklewe, M.; Buecklein, V.; Jongen-Lavrencic, M.; Paschka, P.; Ossenkoppele, G.J.; Kantarjian, H.M.; Hindoyan, A.; Agarwal, S.K.; et al. Updated results from phase I dose-escalation study of AMG 330, a bispecific T-cell engager molecule, in patients with relapsed/refractory acute myeloid leukemia (R/R AML). J. Clin. Oncol. 2020, 38, 7508. [Google Scholar] [CrossRef]
- Krupka, C.; Kufer, P.; Kischel, R.; Zugmaier, G.; Bögeholz, J.; Köhnke, T.; Lichtenegger, F.S.; Schneider, S.; Metzeler, K.; Fiegl, M.; et al. CD33 target validation and sustained depletion of AML blasts in long-term cultures by the bispecific T-cell–engaging antibody AMG 330. Blood 2014, 123, 356–365. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chang, W.-C.; Wong, C.W.; Colcher, D.; Sherman, M.; Ostberg, J.R.; Forman, S.J.; Riddell, S.R.; Jensen, M.C. A transgene-encoded cell surface polypeptide for selection, in vivo tracking, and ablation of engineered cells. Blood 2011, 118, 1255–1263. [Google Scholar] [CrossRef] [PubMed]
- Di Stasi, A.; Tey, S.-K.; Dotti, G.; Fujita, Y.; Kennedy-Nasser, A.; Martinez, C.; Straathof, K.; Liu, E.; Durett, A.G.; Grilley, B.; et al. Inducible Apoptosis as a Safety Switch for Adoptive Cell Therapy. N. Engl. J. Med. 2011, 365, 1673–1683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Budde, L.E.; Berger, C.; Lin, Y.; Wang, J.; Lin, X.; Frayo, S.E.; Brouns, S.A.; Spencer, D.M.; Till, B.G.; Jensen, M.C.; et al. Combining a CD20 Chimeric Antigen Receptor and an Inducible Caspase 9 Suicide Switch to Improve the Efficacy and Safety of T Cell Adoptive Immunotherapy for Lymphoma. PLoS ONE 2013, 8, e82742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sterner, R.M.; Sakemura, R.; Cox, M.J.; Yang, N.; Khadka, R.H.; Forsman, C.L.; Hansen, M.J.; Jin, F.; Ayasoufi, K.; Hefazi, M.; et al. GM-CSF inhibition reduces cytokine release syndrome and neuroinflammation but enhances CAR-T cell function in xenografts. Blood 2019, 133, 697–709. [Google Scholar] [CrossRef] [Green Version]
- Roybal, K.T.; Rupp, L.J.; Morsut, L.; Walker, W.J.; McNally, K.A.; Park, J.S.; Lim, W.A. Precision Tumor Recognition by T Cells With Combinatorial Antigen-Sensing Circuits. Cell 2016, 164, 770–779. [Google Scholar] [CrossRef] [Green Version]
- Kloss, C.C.; Condomines, M.; Cartellieri, M.; Bachmann, M.; Sadelain, M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat. Biotechnol. 2013, 31, 71–75. [Google Scholar] [CrossRef]
- Zajc, C.U.; Dobersberger, M.; Schaffner, I.; Mlynek, G.; Pühringer, D.; Salzer, B.; Djinović-Carugo, K.; Steinberger, P.; Linhares, A.D.S.; Yang, N.J.; et al. A conformation-specific ON-switch for controlling CAR T cells with an orally available drug. Proc. Natl. Acad. Sci. USA 2020, 117, 14926–14935. [Google Scholar] [CrossRef]
- Park, S.; Pascua, E.; Lindquist, K.C.; Kimberlin, C.; Deng, X.; Mak, Y.S.L.; Melton, Z.; Johnson, T.O.; Lin, R.; Boldajipour, B.; et al. Direct control of CAR T cells through small molecule-regulated antibodies. Nat. Commun. 2021, 12, 710. [Google Scholar] [CrossRef]
- Weber, E.W.; Lynn, R.C.; Sotillo, E.; Lattin, J.; Xu, P.; Mackall, C.L. Pharmacologic control of CAR-T cell function using dasatinib. Blood Adv. 2019, 3, 711–717. [Google Scholar] [CrossRef]
- Mestermann, K.; Giavridis, T.; Weber, J.; Rydzek, J.; Frenz, S.; Nerreter, T.; Mades, A.; Sadelain, M.; Einsele, H.; Hudecek, M. The tyrosine kinase inhibitor dasatinib acts as a pharmacologic on/off switch for CAR T cells. Sci. Transl. Med. 2019, 11, eaau5907. [Google Scholar] [CrossRef] [PubMed]
- Deng, Q.; Han, G.; Puebla-Osorio, N.; Ma, M.C.J.; Strati, P.; Chasen, B.; Dai, E.; Dang, M.; Jain, N.; Yang, H.; et al. Characteristics of anti-CD19 CAR T cell infusion products associated with efficacy and toxicity in patients with large B cell lymphomas. Nat. Med. 2020, 26, 1878–1887. [Google Scholar] [CrossRef] [PubMed]
- Li-Weber, M.; Krammer, P.H. Regulation of IL4 gene expression by T cells and therapeutic perspectives. Nat. Rev. Immunol. 2003, 3, 534–543. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, S.; Sukumaran, S.; Bajgain, P.; Watanabe, N.; Heslop, H.E.; Rooney, C.M.; Brenner, M.K.; Fisher, W.E.; Leen, A.M.; Vera, J.F. Improving Chimeric Antigen Receptor-Modified T Cell Function by Reversing the Immunosuppressive Tumor Microenvironment of Pancreatic Cancer. Mol. Ther. 2017, 25, 249–258. [Google Scholar] [CrossRef] [Green Version]
- Kloss, C.C.; Lee, J.; Zhang, A.; Chen, F.; Melenhorst, J.J.; Lacey, S.F.; Maus, M.V.; Fraietta, J.A.; Zhao, Y.; June, C.H. Dominant-Negative TGF-beta Receptor Enhances PSMA-Targeted Human CAR T Cell Proliferation And Augments Prostate Cancer Eradication. Mol. Ther. 2018, 26, 1855–1866. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Ranganathan, R.; Jiang, S.; Fang, C.; Sun, J.; Kim, S.; Newick, K.; Lo, A.; June, C.H.; Zhao, Y.; et al. A Chimeric Switch-Receptor Targeting PD1 Augments the Efficacy of Second-Generation CAR T Cells in Advanced Solid Tumors. Cancer Res. 2016, 76, 1578–1590. [Google Scholar] [CrossRef] [Green Version]
- Rafiq, S.; Yeku, O.O.; Jackson, H.J.; Purdon, T.J.; Van Leeuwen, D.G.; Drakes, D.J.; Song, M.; Miele, M.M.; Li, Z.; Wang, P.; et al. Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances anti-tumor efficacy in vivo. Nat. Biotechnol. 2018, 36, 847–856. [Google Scholar] [CrossRef]
- Suarez, E.R.; de Chang, K.; Sun, J.; Sui, J.; Freeman, G.J.; Signoretti, S.; Zhu, Q.; Marasco, W.A. Chimeric antigen receptor T cells secreting anti-PD-L1 antibodies more effectively regress renal cell carcinoma in a humanized mouse model. Oncotarget 2016, 7, 34341–34355. [Google Scholar] [CrossRef] [Green Version]
- Cherkassky, L.; Morello, A.; Villena-Vargas, J.; Feng, Y.; Dimitrov, D.S.; Jones, D.R.; Sadelain, M.; Adusumilli, P.S. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J. Clin. Investig. 2016, 126, 3130–3144. [Google Scholar] [CrossRef] [Green Version]
- Rupp, L.J.; Schumann, K.; Roybal, K.T.; Gate, R.E.; Ye, C.J.; Lim, W.A.; Marson, A. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Sci. Rep. 2017, 7, 737. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Li, N.; Feng, K.; Chen, M.; Zhang, Y.; Liu, Y.; Yang, Q.; Nie, J.; Tang, N.; Zhang, X.; et al. Phase I study of CAR-T cells with PD-1 and TCR disruption in mesothelin-positive solid tumors. Cell. Mol. Immunol. 2021, 18, 2188–2198. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.D.; Yu, X.; Castano, A.P.; Darr, H.; Henderson, D.B.; Bouffard, A.A.; Larson, R.C.; Scarfò, I.; Bailey, S.R.; Gerhard, G.M.; et al. CRISPR-Cas9 disruption of PD-1 enhances activity of universal EGFRvIII CAR T cells in a preclinical model of human glioblastoma. J. Immunother. Cancer 2019, 7, 304. [Google Scholar] [CrossRef] [PubMed]
- Poirot, L.; Philip, B.; Schiffer-Mannioui, C.; Le Clerre, D.; Chion-Sotinel, I.; Derniame, S.; Potrel, P.; Bas, C.; Lemaire, L.; Galetto, R.; et al. Multiplex Genome-Edited T-cell Manufacturing Platform for “Off-the-Shelf” Adoptive T-cell Immunotherapies. Cancer Res. 2015, 75, 3853–3864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riley, J.L. PD-1 signaling in primary T cells. Immunol. Rev. 2009, 229, 114–125. [Google Scholar] [CrossRef]
- Stenger, D.; Stief, T.A.; Kaeuferle, T.; Willier, S.; Rataj, F.; Schober, K.; Vick, B.; Lotfi, R.; Wagner, B.; Grünewald, T.G.P.; et al. Endogenous TCR promotes in vivo persistence of CD19-CAR-T cells compared to a CRISPR/Cas9-mediated TCR knockout CAR. Blood 2020, 136, 1407–1418. [Google Scholar] [CrossRef]
- Roth, T.L.; Puig-Saus, C.; Yu, R.; Shifrut, E.; Carnevale, J.; Li, P.J.; Hiatt, J.; Saco, J.; Krystofinski, P.; Li, H.; et al. Reprogramming human T cell function and specificity with non-viral genome targeting. Nature 2018, 559, 405–409. [Google Scholar] [CrossRef]
- MacLeod, D.T.; Antony, J.; Martin, A.J.; Moser, R.J.; Hekele, A.; Wetzel, K.J.; Brown, A.E.; Triggiano, M.A.; Hux, J.A.; Pham, C.D.; et al. Integration of a CD19 CAR into the TCR Alpha Chain Locus Streamlines Production of Allogeneic Gene-Edited CAR T Cells. Mol. Ther. 2017, 25, 949–961. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Liu, G.; Wang, J.; Zheng, Z.-Y.; Jia, L.; Rui, W.; Huang, D.; Zhou, Z.-X.; Zhou, L.; Wu, X.; et al. Chimeric STAR receptors using TCR machinery mediate robust responses against solid tumors. Sci. Transl. Med. 2021, 13, abb5191. [Google Scholar] [CrossRef]
- Baeuerle, P.A.; Ding, J.; Patel, E.; Thorausch, N.; Horton, H.; Gierut, J.; Scarfo, I.; Choudhary, R.; Kiner, O.; Krishnamurthy, J.; et al. Synthetic TRuC receptors engaging the complete T cell receptor for potent anti-tumor response. Nat. Commun. 2019, 10, 2087. [Google Scholar] [CrossRef] [Green Version]
- Koneru, M.; Purdon, T.J.; Spriggs, D.; Koneru, S.; Brentjens, R.J. IL-12 secreting tumor-targeted chimeric antigen receptor T cells eradicate ovarian tumors in vivo. OncoImmunology 2015, 4, e994446. [Google Scholar] [CrossRef] [Green Version]
- Hu, B.; Ren, J.; Luo, Y.; Keith, B.; Young, R.M.; Scholler, J.; Zhao, Y.; June, C.H. Augmentation of Antitumor Immunity by Human and Mouse CAR T Cells Secreting IL-18. Cell Rep. 2017, 20, 3025–3033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurton, L.V.; Singh, H.; Najjar, A.M.; Switzer, K.C.; Mi, T.; Maiti, S.; Olivares, S.; Rabinovich, B.; Huls, H.; Forget, M.-A.; et al. Tethered IL-15 augments antitumor activity and promotes a stem-cell memory subset in tumor-specific T cells. Proc. Natl. Acad. Sci. USA 2016, 113, E7788–E7797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majzner, R.G.; Mackall, C.L. Clinical lessons learned from the first leg of the CAR T cell journey. Nat. Med. 2019, 25, 1341–1355. [Google Scholar] [CrossRef] [PubMed]
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef]
- Yang, J.; He, J.; Zhang, X.; Wang, Z.; Zhang, Y.; Cai, S.; Sun, Z.; Ye, X.; He, Y.; Shen, L.; et al. A Feasibility and Safety Study of a New CD19-Directed Fast CAR-T Therapy for Refractory and Relapsed B Cell Acute Lymphoblastic Leukemia. Blood 2019, 134, 825. [Google Scholar] [CrossRef]
- Whittington, M.D.; McQueen, R.B.; Ollendorf, D.A.; Kumar, V.M.; Chapman, R.H.; Tice, J.; Pearson, S.D.; Campbell, J.D. Long-term Survival and Value of Chimeric Antigen Receptor T-Cell Therapy for Pediatric Patients With Relapsed or Refractory Leukemia. JAMA Pediatr. 2018, 172, 1161–1168. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, R.R.; Gloude, N.J.; Schiff, D.; Murphy, J.D. Cost-Effectiveness of Chimeric Antigen Receptor T-Cell Therapy in Pediatric Relapsed/Refractory B-Cell Acute Lymphoblastic Leukemia. JNCI J. Natl. Cancer Inst. 2019, 111, 719–726. [Google Scholar] [CrossRef]
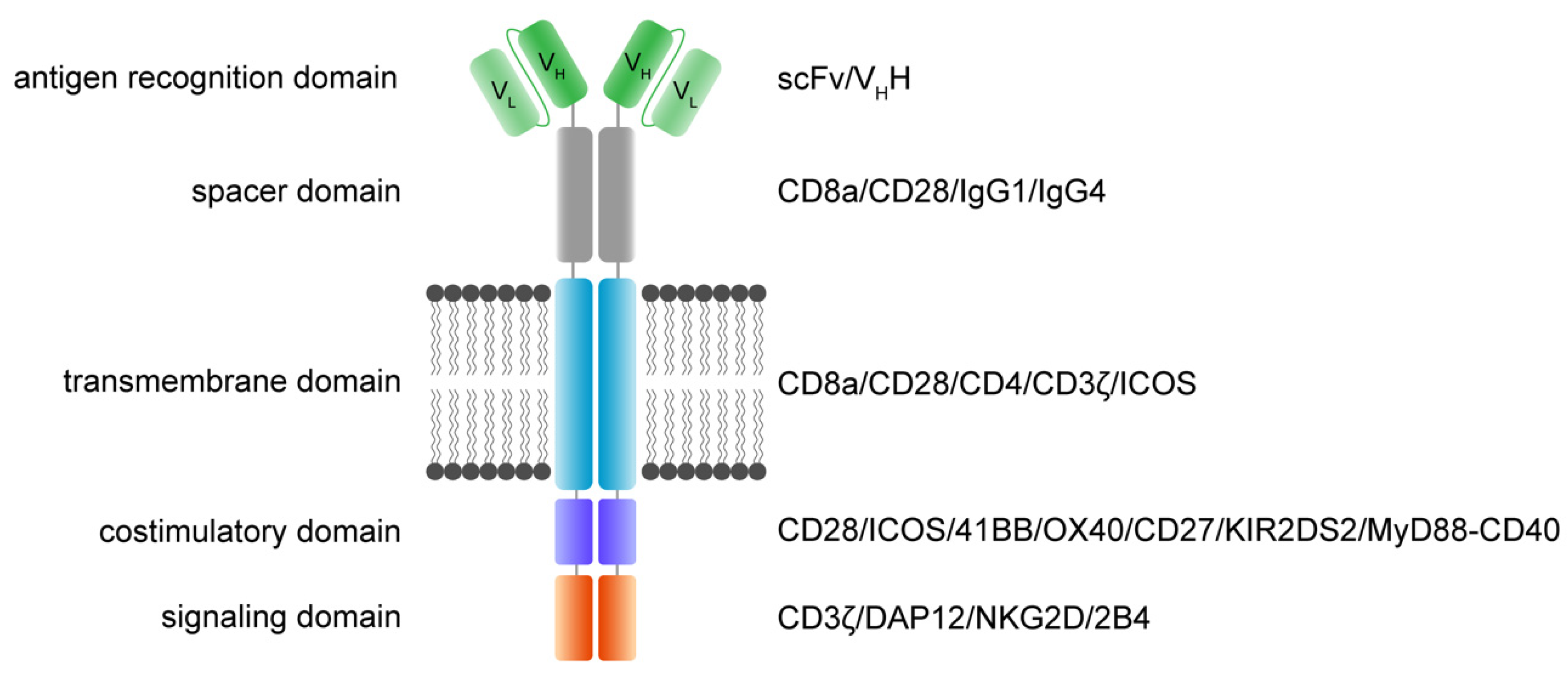
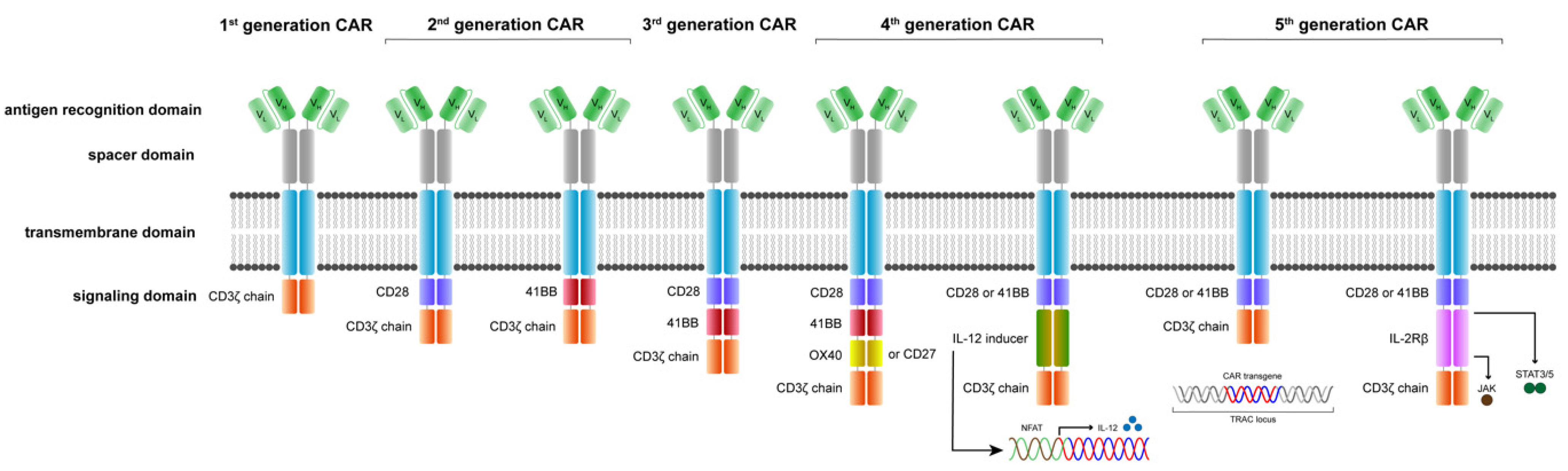
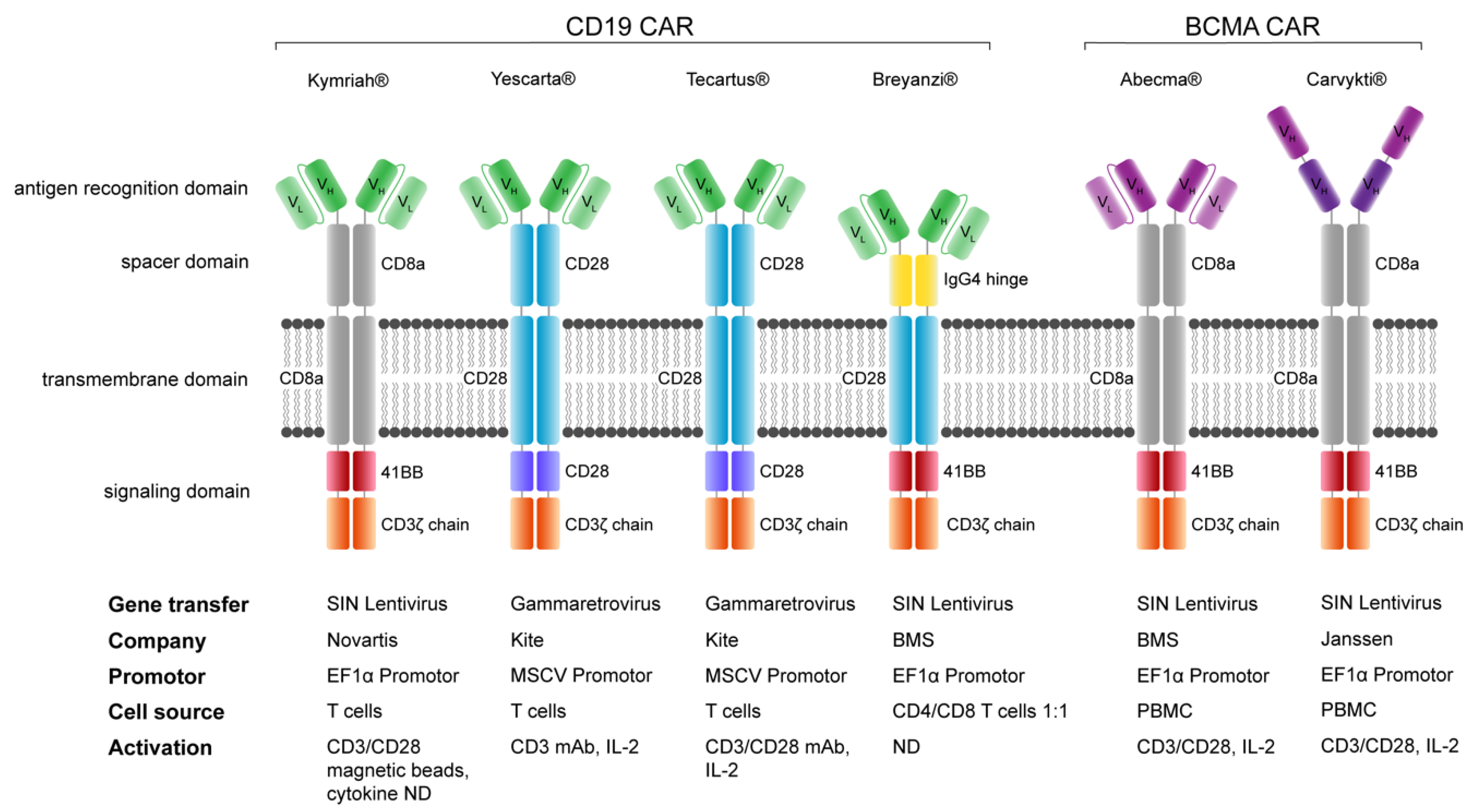
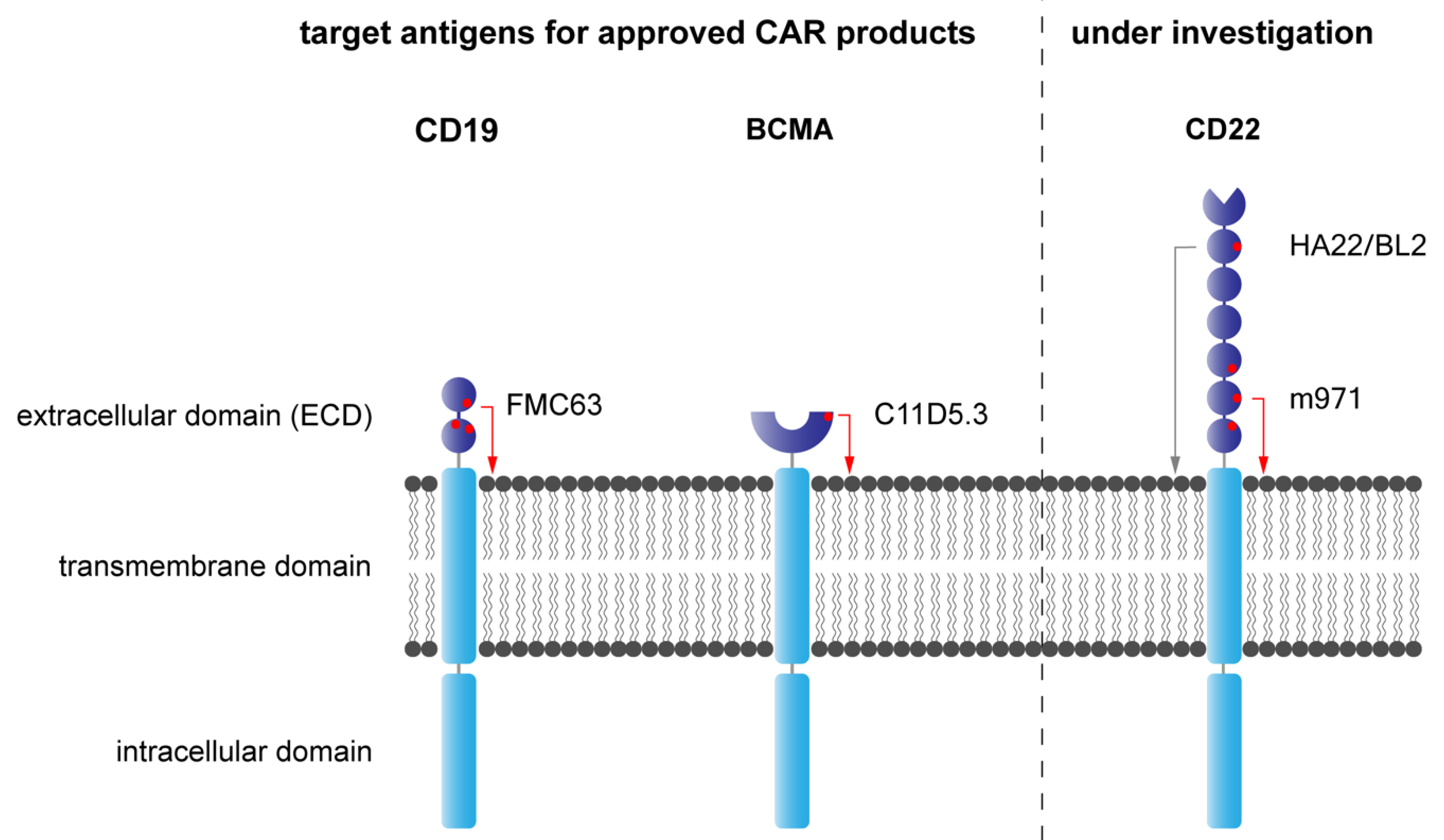

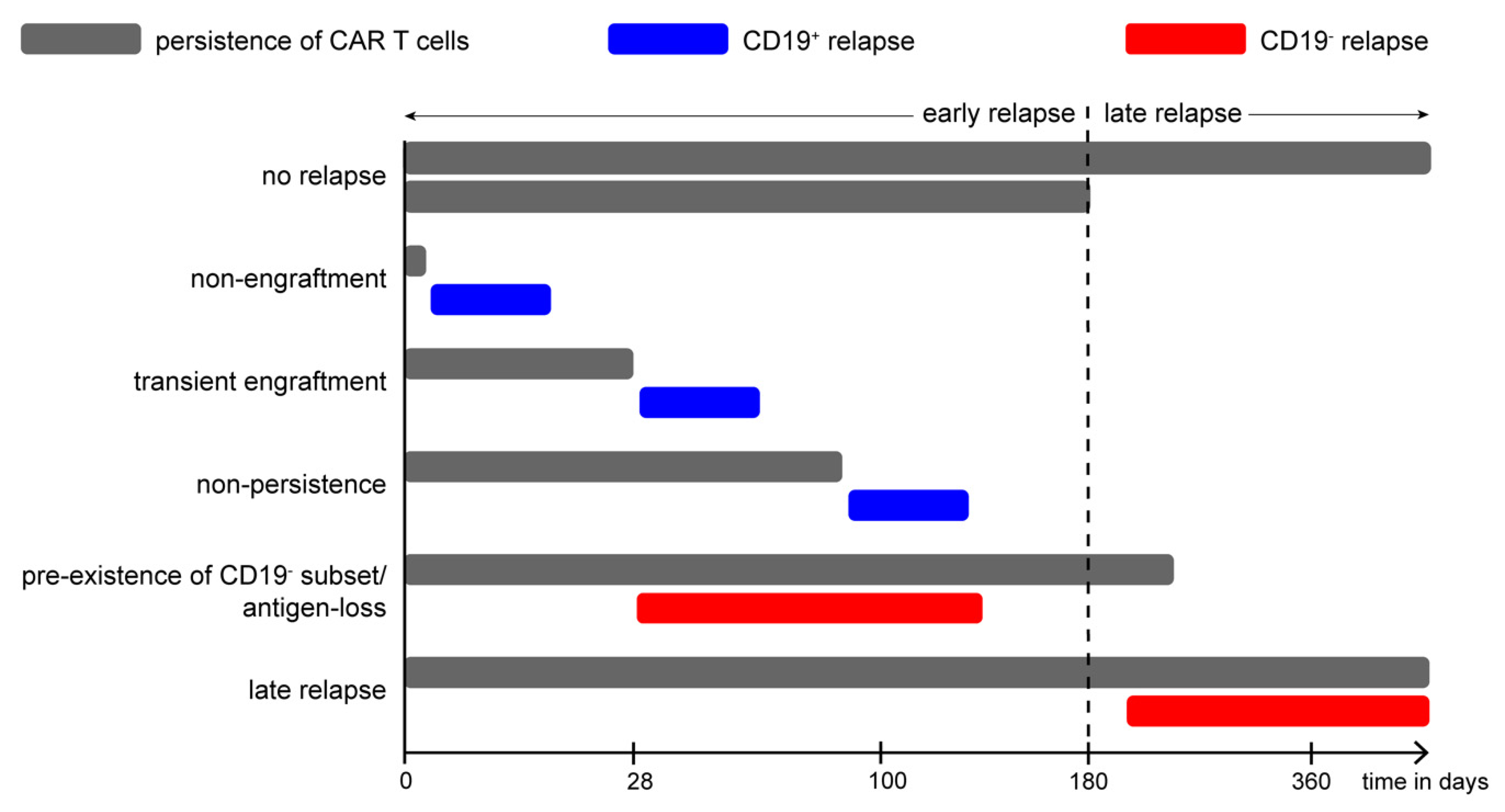


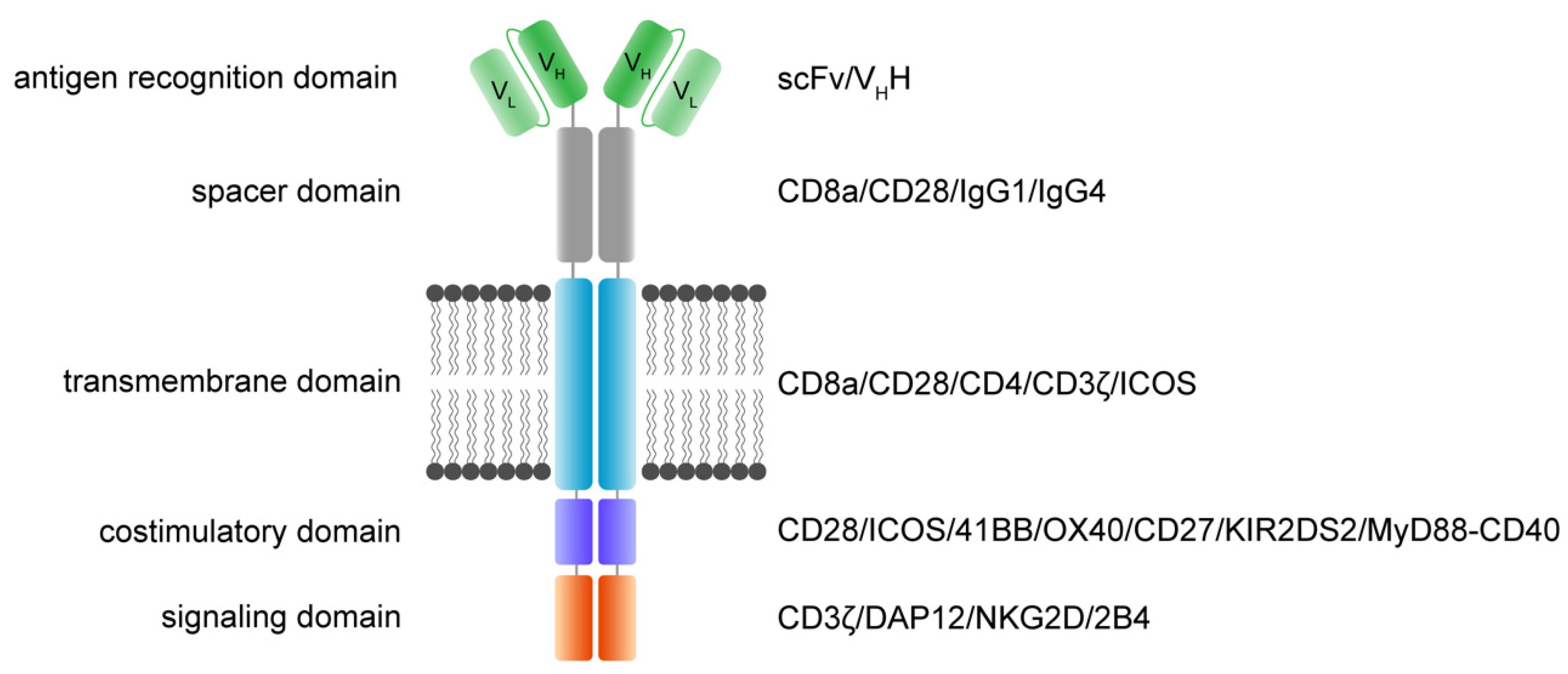{kind=link}
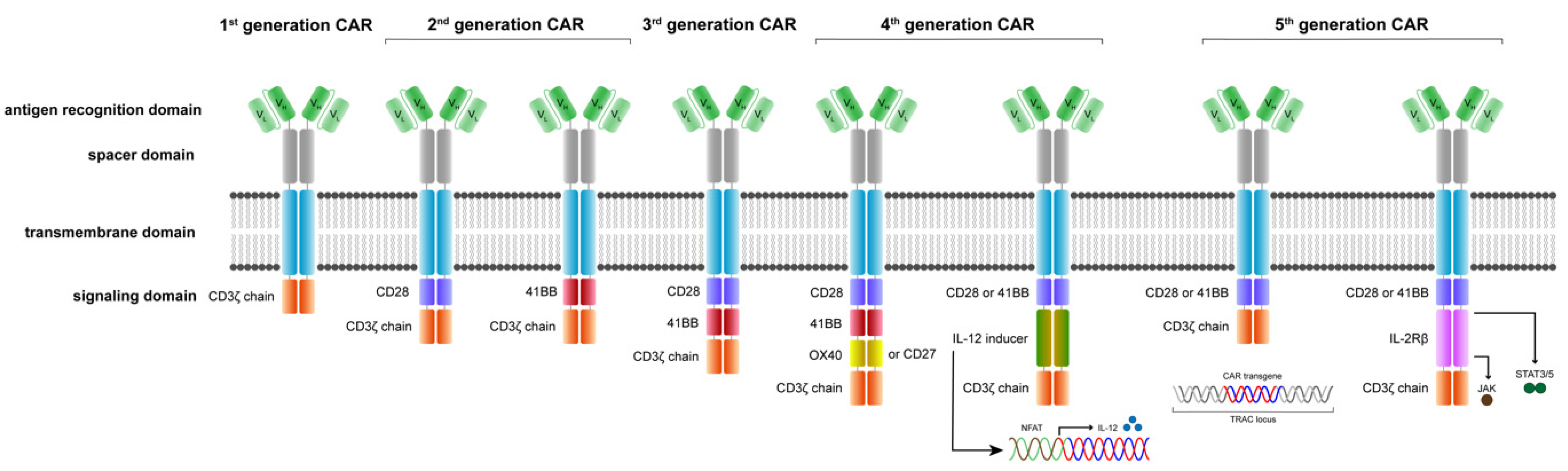{kind=link}
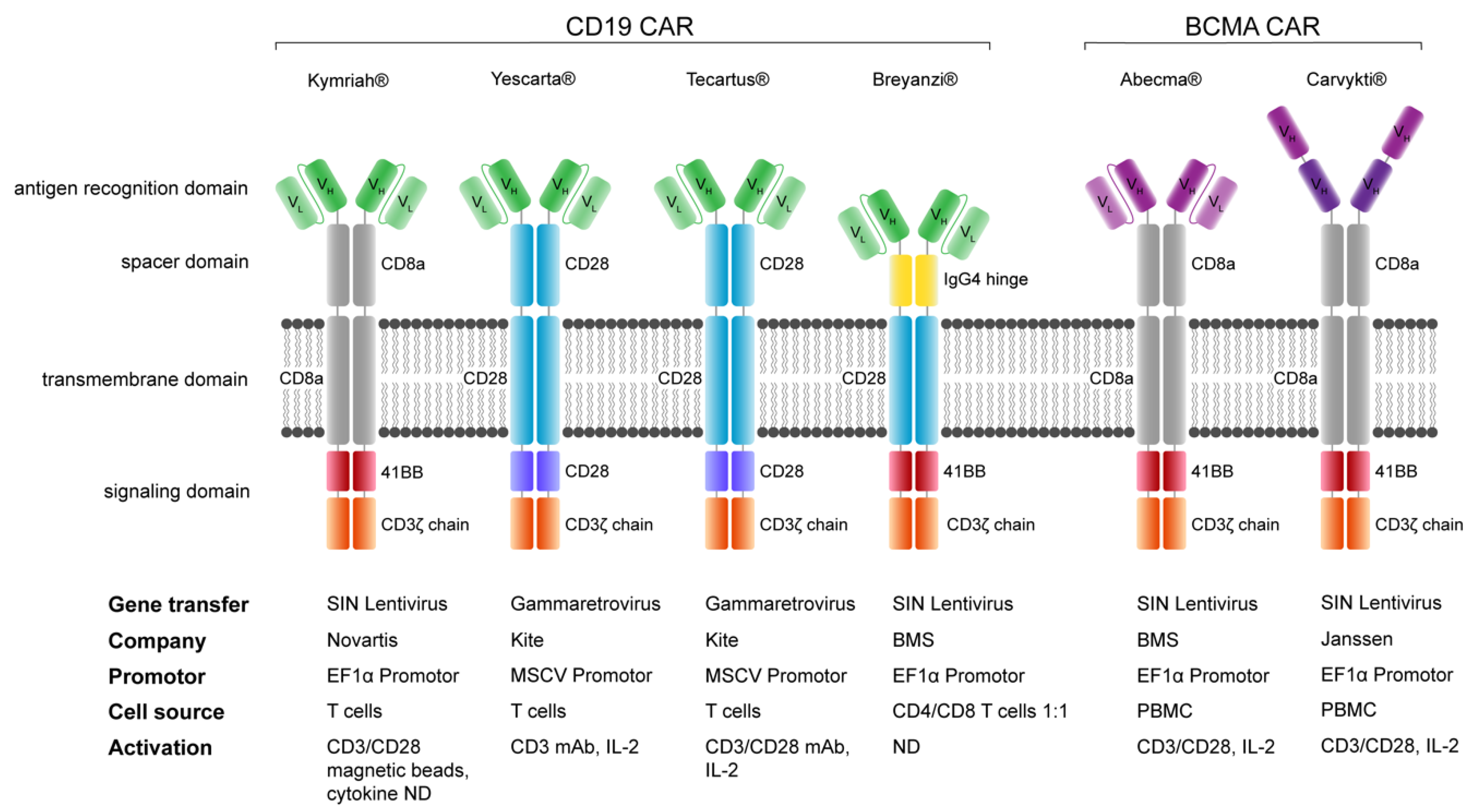{kind=link}
{kind=link}
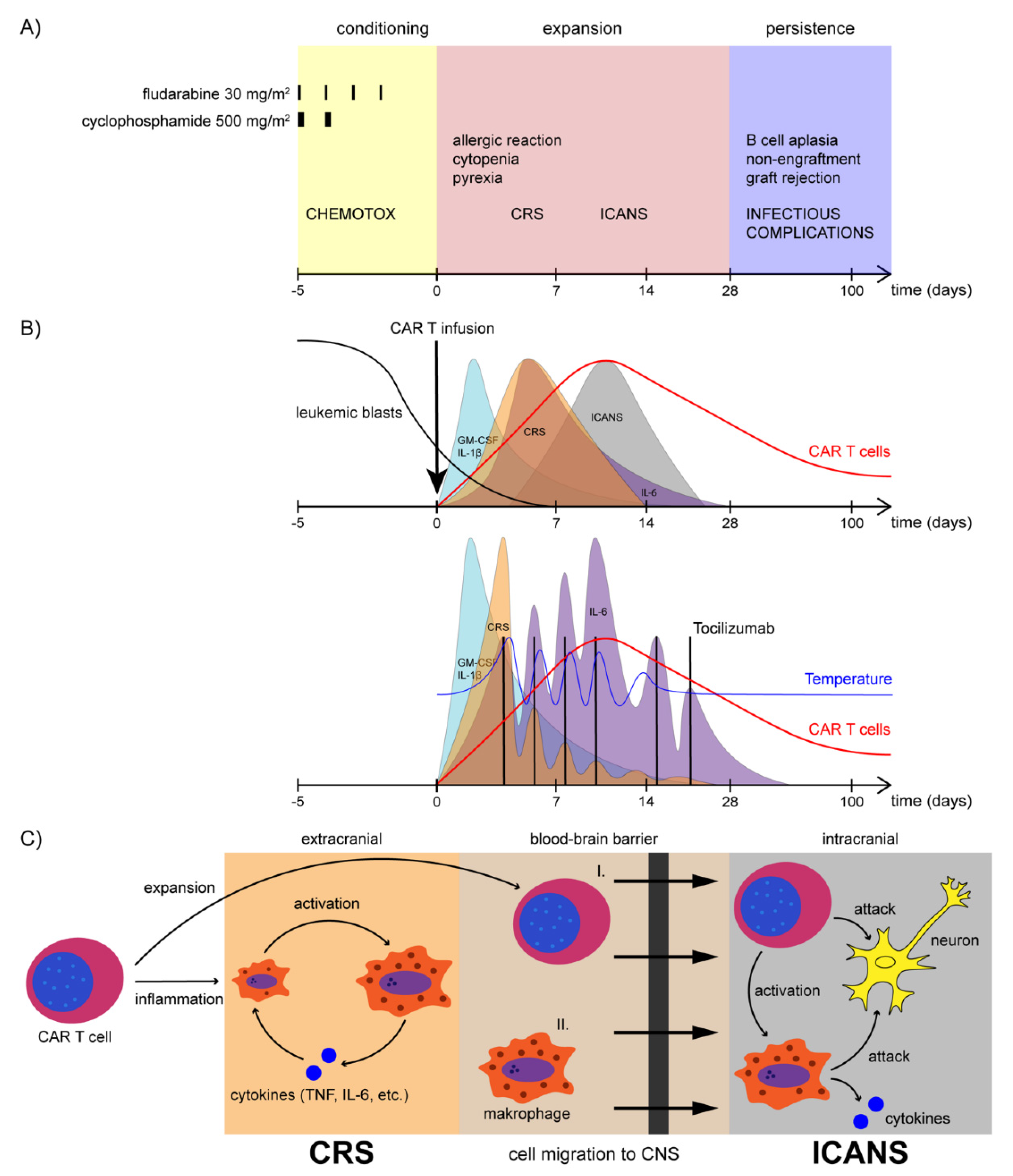{kind=link}
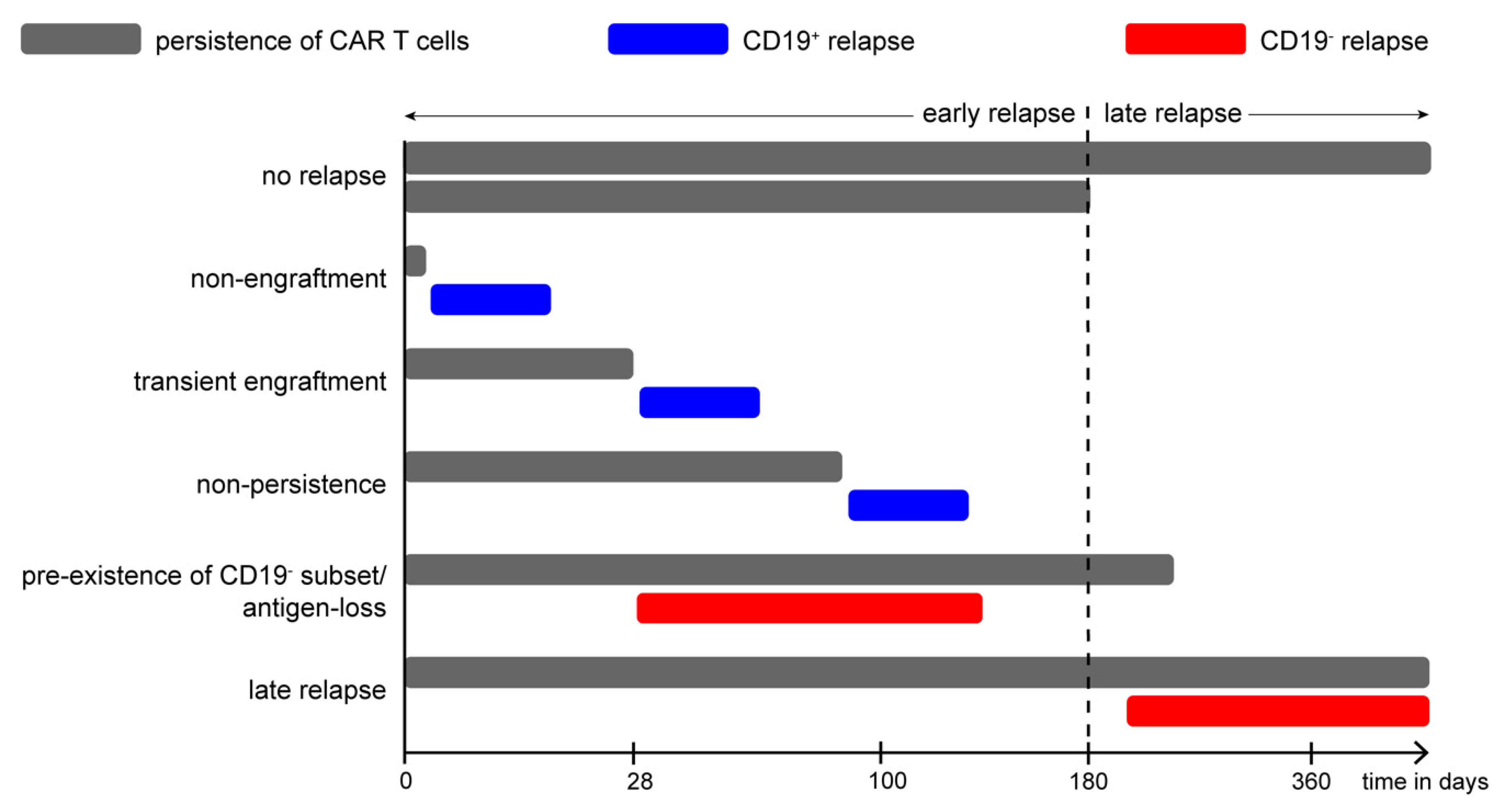{kind=link}
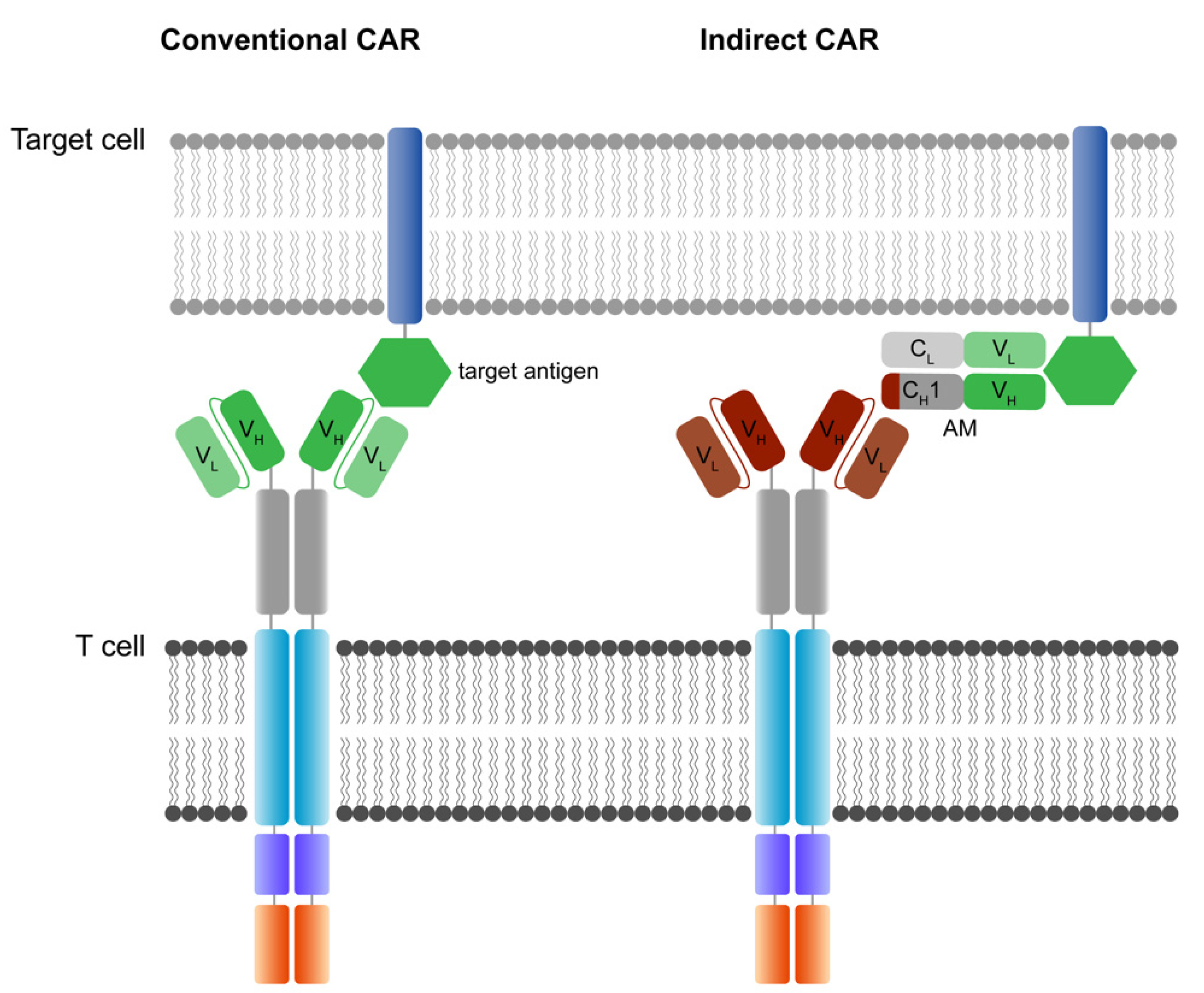{kind=link}
| Name | Target Antigen | Brand | FDA Approval | Indications |
|---|---|---|---|---|
| Tisagenlecleucel | CD19 | Kymriah | August 2017 May 2018 | r/r B-cell precursor ALL, r/r large B-cell lymphoma |
| Axicabtagene ciloleucel | CD19 | Yescarta | October 2017 March 2021 | r/r large B-cell lymphoma r/r follicular lymphoma |
| Brexucabtagene autoleucel | CD19 | Tecartus | July 2020 October 2021 | r/r MCL (July 2020) r/r B-cell precursor ALL (Oct 2021) |
| Lisocabtagene maraleucel | CD19 | Breyanzi | February 2021 | r/r large B-cell lymphoma |
| Idecabtagene vicleucel | BCMA | Abecma | March 2021 | r/r MM |
| Ciltacabtagene autoleucel | BCMA | Carvykti | February 2022 | r/r MM |
| CAR Target | Condition Treated | Eligible Age | Status | ClinicalTrials.gov ID |
|---|---|---|---|---|
| CD20 | B-cell Non-Hodgkin’s lymphomas | ≥18 | Recruiting | NCT03277729 |
| CD20 | B-cell lymphoma r/r to anti-CD19-CAR-Ttherapy | 14 to 70 | Unknown | NCT04036019 |
| CD20 | Lymphomas r/r to chemotherapy | ≥18, <90 | Unknown | NCT01735604 |
| CD22 | r/r B-cell lymphoma/leukemia | 3 to 39 | Recruiting | NCT02315612 |
| CD22 | B-ALL | 1–30 | Recruiting | NCT04088864 |
| CD22 | B-ALL | 1–24 | Recruiting | NCT02650414 |
| CD22 | r/r B-ALL | 15–70 | Recruiting | NCT04150497 |
| CD19, CD20 | r/r B-cell lymphoma/leukemia | 16–70 | Completed | NCT03097770 |
| CD19, CD20 | r/r B-cell lymphoma/leukemia | 18–70 | Active, not recruiting | NCT03019055 |
| CD19, CD20 | r/r B-cell lymphoma/leukemia | 18–70 | Recruiting | NCT04007029 |
| CD19, CD20 | r/r B-ALL | 1–39 | Recruiting | NCT04049383 |
| CD19, CD22 | r/r B-cell lymphoma/leukemia | ≥18 | Recruiting | NCT03233854 |
| CD19, CD22 | r/r B-cell lymphoma/leukemia | 3–39 | Recruiting | NCT03448393 |
| CD19, CD22 | r/r B-cell lymphoma/leukemia | 6 months to 70 | Recruiting | NCT04029038 |
| CD19, CD22 | B-cell lymphoma/leukemia | ≤30 | Recruiting | NCT03330691 |
| CD19, CD22 | r/r B-ALL | 1–30 | Recruiting | NCT03241940 |
| CD37 | B and T cell lymphoma/leukemia | ≥18 | Recruiting | NCT04136275 |
| CD79B | r/r B-ALL, B-cell NHL | No age limit | Not yet recruiting | NCT04609241 |
| CD33 | AML | 1–35 | Recruiting | NCT03971799 |
| CD123 | AML | ≥12 | Recruiting | NCT02159495 |
| CD123 | AML | 18–65 | Recruiting | NCT03190278 |
| CD123 | AML | 18–70 | Recruiting | NCT04014881 |
| CD123 | AML | ≥18 | Active, not recruiting | NCT03766126 |
| CD33, CLL-1, CD123 | AML | 6 months to 75 | Recruiting | NCT04010877 |
| CLL-1 | AML | ≤75 | Recruiting | NCT04219163 |
| CD38 | AML | 6–65 | Recruiting | NCT04351022 |
| CD33, CLL-1 | AML/MDS/MPN/CML | No age limit | Recruiting | NCT03795779 |
| CAR Target | Condition Treated | Eligible Age | Status | ClinicalTrials.gov ID |
|---|---|---|---|---|
| B7-H3 | Pediatric CNS tumors | 1–26 | Recruiting | NCT04185038 |
| B7-H3 | Pediatric solid tumors | ≤26 | Recruiting | NCT04483778 |
| B7-H3 | Solid tumors | 1–75 | Recruiting | NCT04432649 |
| GD2 | DIPG/high grade glioma | 12 months to 18 | Recruiting | NCT04099797 |
| GD2 | DIPG/DMG | 2–30 | Recruiting | NCT04196413 |
| GD2 | Osteosarcoma, neuroblastoma | ≤35 | Recruiting | NCT04539366 |
| GD2 | Neuroblastoma | 12 months to 25 | Recruiting | NCT03373097 |
| GD2 | Neuroblastoma, sarcoma | 1–74 | Recruiting | NCT03635632 |
| GD2 | Osteosarcoma, neuroblastoma | 18 months to 18 | Recruiting | NCT03721068 |
| EGFR | Pediatric CNS tumors | ≥15 and ≤26 | Recruiting | NCT03638167 |
| EGFR | Pediatric solid tumors | 1–30 | Recruiting | NCT03618381 |
| EGFRvIII | Hematological and solid tumors | 4–70 | Recruiting | NCT03638206 |
| HER2 | Pediatric CNS tumors | 1–26 | Recruiting | NCT03500991 |
| HER2 | CNS tumors | ≥3 | Recruiting | NCT02442297 |
| IL13Ra2 | Pediatric CNS tumors | 4–35 | Recruiting | NCT04510051 |
| IL13Ra2 | Glioma | 12–75 | Recruiting | NCT02208362 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boettcher, M.; Joechner, A.; Li, Z.; Yang, S.F.; Schlegel, P. Development of CAR T Cell Therapy in Children—A Comprehensive Overview. J. Clin. Med. 2022, 11, 2158. https://doi.org/10.3390/jcm11082158
Boettcher M, Joechner A, Li Z, Yang SF, Schlegel P. Development of CAR T Cell Therapy in Children—A Comprehensive Overview. Journal of Clinical Medicine. 2022; 11(8):2158. https://doi.org/10.3390/jcm11082158
Chicago/Turabian StyleBoettcher, Michael, Alexander Joechner, Ziduo Li, Sile Fiona Yang, and Patrick Schlegel. 2022. "Development of CAR T Cell Therapy in Children—A Comprehensive Overview" Journal of Clinical Medicine 11, no. 8: 2158. https://doi.org/10.3390/jcm11082158
APA StyleBoettcher, M., Joechner, A., Li, Z., Yang, S. F., & Schlegel, P. (2022). Development of CAR T Cell Therapy in Children—A Comprehensive Overview. Journal of Clinical Medicine, 11(8), 2158. https://doi.org/10.3390/jcm11082158