
Effectiveness of Different Rituximab Doses Combined with Leflunomide in the Treatment or Retreatment of Rheumatoid Arthritis: Part 2 of a Randomized, Placebo-Controlled, Investigator-Initiated Clinical Trial (AMARA)

 , , , , , and
, , , , , and
Abstract
1. Introduction
2. Patients and Methods
2.1. Study Design
2.2. Outcomes
2.3. Statistical Analysis
3. Results
3.1. Patient Disposition
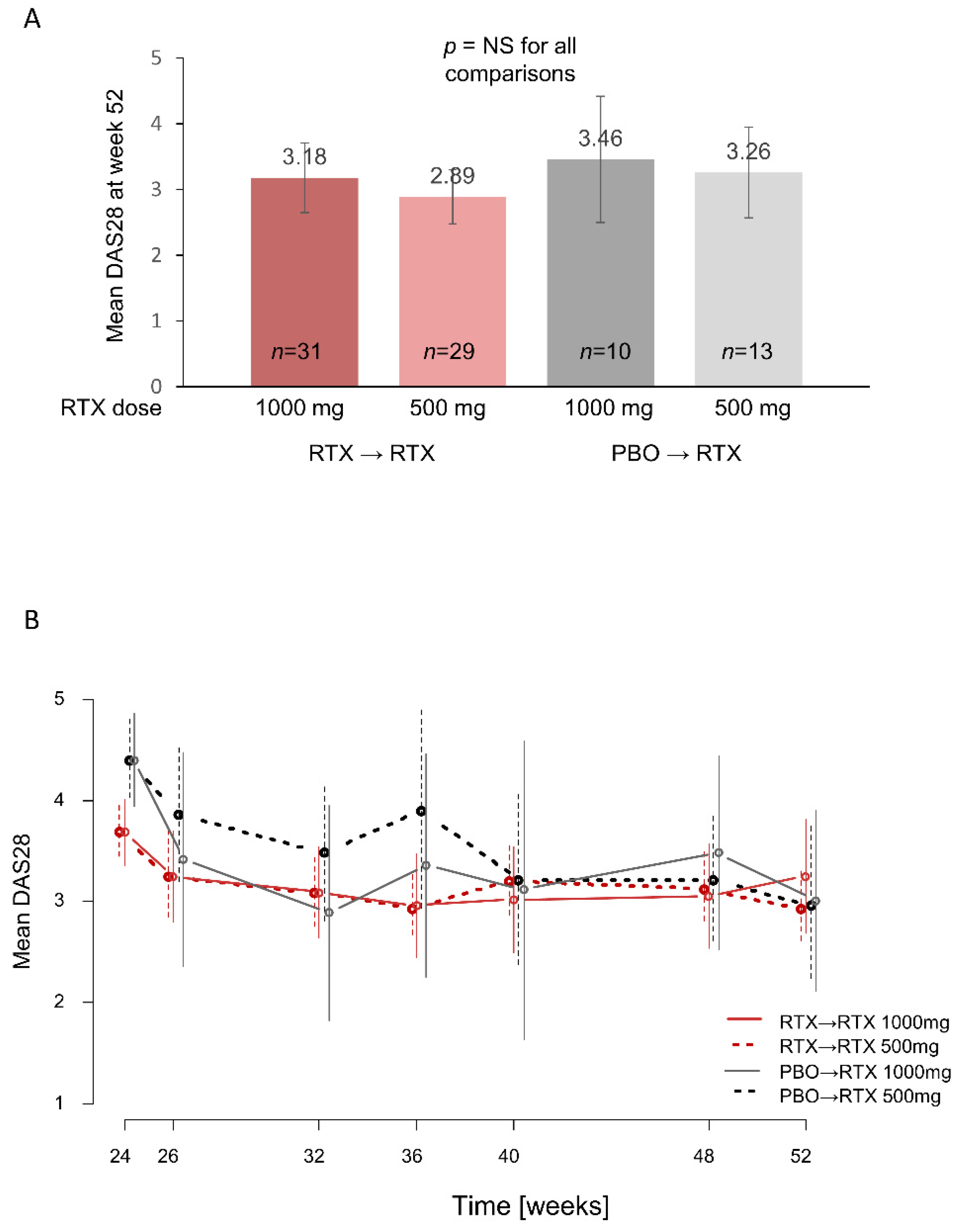
3.2. DAS28 Outcomes
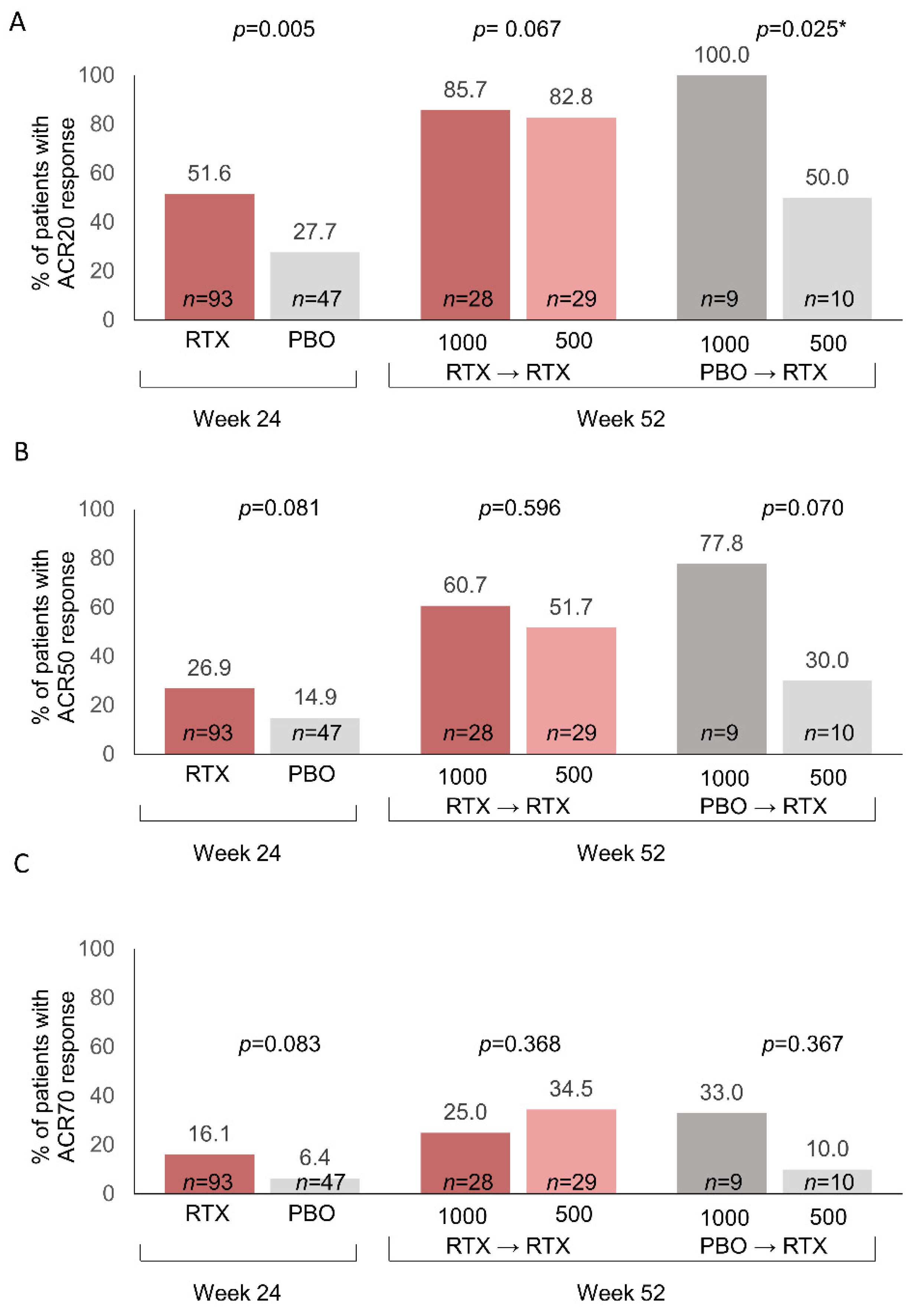
3.3. ACR Response Rates
3.4. Patient-reported Outcomes
3.5. Safety
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Edwards, J.C.; Szczepanski, L.; Szechinski, J.; Filipowicz-Sosnowska, A.; Emery, P.; Close, D.R.; Stevens, R.M.; Shaw, T. Efficacy of B-cell-targeted therapy with rituximab in patients with rheumatoid arthritis. N. Engl. J. Med. 2004, 350, 2572–2591. [Google Scholar] [CrossRef]
- Emery, P.; Fleischmann, R.; Filipowicz-Sosnowska, A.; Schechtman, J.; Szczepanski, L.; Kavanaugh, A.; Racewicz, A.J.; van Vollenhoven, R.F.; Li, N.F.; Agarwals, S.; et al. The efficacy and safety of rituximab in patients with active rheumatoid arthritis despite methotrexate treatment. Results of a phase IIb randomized, double-blind, placebo-controlled, dose-ranging trial. Arthritis Rheum. 2006, 54, 1390–1400. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.B.; Emery, P.; Greenwald, M.W.; Dougados, M.; Furie, R.A.; Genovese, M.C.; Keystone, E.C.; Loveless, J.E.; Burmster, G.R.; Cravets, M.W.; et al. Rituximab for rheumatoid arthritis refractory to anti-tumor necrosis factor therapy: Results of a multicenter, randomized, double-blind, placebo-controlled, phase III trial evaluating primary efficacy and safety at twenty-four weeks. Arthritis Rheum. 2006, 54, 2793–2806. [Google Scholar] [CrossRef] [PubMed]
- Roche Pharma, A.G. MabThera Summary of Product Characteristics. Available online: https://www.ema.europa.eu/en/documents/product-information/mabthera-epar-product-information_en.pdf (accessed on 2 June 2022).
- Genentech, Inc. RITUXAN® (rituximab) Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/103705s5467lbl.pdf (accessed on 14 July 2022).
- Einarsson, J.T.; Evert, M.; Geborek, P.; Saxne, T.; Lundgren, M.; Kapetanovic, M.C. Rituximab in clinical practice: Dosage, drug adherence, Ig levels, infections, and drug antibodies. Clin. Rheumatol. 2017, 36, 2743–2750. [Google Scholar] [CrossRef] [PubMed]
- Chatzidionysiou, K.; Lie, E.; Nasonov, E.; Lukina, G.; Hetland, M.L.; Tarp, U.; van Riel, P.L.; Nordström, D.C.; Gomez-Reino, J.; Pavelka, K.; et al. Effectiveness of two different doses of rituximab for the treatment of rheumatoid arthritis in an international cohort: Data from the CERERRA collaboration. Arthritis Res. Ther. 2016, 18, 50. [Google Scholar] [CrossRef] [PubMed]
- Emery, P.; Deodhar, A.; Rigby, W.F.; Isaacs, J.D.; Combe, B.; Racewicz, A.J.; Latinis, K.; Abud-Mendoza, C.; Szczepański, L.J.; Roschmann, R.A.; et al. Efficacy and safety of different doses and retreatment of rituximab: A randomized, placebo-controlled trial in patients who are biological naïve with active rheumatoid arthritis and an inadequate response to methotrexate (Study Evaluating Rituximab’s Efficacy in MTX iNadequate rEsponders (SERENE)). Ann. Rheum. Dis. 2010, 69, 1629–1635. [Google Scholar] [CrossRef] [PubMed]
- Mariette, X.; Rouanet, S.; Sibilia, J.; Combe, B.; Le Loët, X.; Tebib, J.; Jourdan, R.; Dougados, M. Evaluation of low-dose rituximab for the retreatment of patients with active rheumatoid arthritis: A non-inferiority randomised controlled trial. Ann. Rheum. Dis. 2014, 73, 1508–1514. [Google Scholar] [CrossRef] [PubMed]
- Bredemeier, M.; Campos, G.G.; de Oliveira, F.K. Updated systematic review and meta-analysis of randomized controlled trials comparing low- versus high-dose rituximab for rheumatoid arthritis. Clin. Rheumatol. 2015, 34, 1801–1805. [Google Scholar] [CrossRef] [PubMed]
- Henry, J.; Gottenberg, J.E.; Rouanet, S.; Pavy, S.; Sellam, J.; Tubach, F.; Belkhir, R.; Mariette, X.; Seror, R.; for the Auto-Immunity and Rituximab Investigators. Doses of rituximab for retreatment in rheumatoid arthritis: Influence on maintenance and risk of serious infection. Rheumatology 2018, 57, 538–547. [Google Scholar] [CrossRef] [PubMed]
- Verhoef, L.M.; den Broeder, N.; Thurlings, R.M.; van der Laan, W.H.; van der Weele, W.; Kok, M.R.; Bernelot Moens, H.J.; Woodworth, T.G.; van den Bemt, B.J.F.; van den Hoogen, F.H.J.; et al. Ultra-low doses of rituximab for continued treatment of rheumatoid arthritis (REDO study): A randomised controlled non-inferiority trial. Lancet Rheumatol. 2019, 1, e145–e153. [Google Scholar] [CrossRef]
- Rubbert-Roth, A.; Tak, P.P.; Zerbini, C.; Tremblay, J.L.; Carreño, L.; Armstrong, G.; Collinson, N.; Shaw, T.M.; on behalf of the MIRROR Trial Investigators. Efficacy and safety of various repeat treatment dosing regimens of rituximab in patients with active rheumatoid arthritis: Results of a phase III randomized study (MIRROR). Rheumatology 2010, 49, 1683–1693. [Google Scholar] [CrossRef] [PubMed]
- Buch, M.H.; Smolen, J.S.; Betteridge, N.; Breedveld, F.C.; Burmester, G.; Dörner, T.; Ferraccioli, G.; Gottenberg, J.E.; Isaacs, J.; Kvien, T.K.; et al. Updated consensus statement on the use of rituximab in patients with rheumatoid arthritis. Ann. Rheum. Dis. 2011, 70, 909–920. [Google Scholar] [CrossRef]
- Behrens, F.; Koehm, M.; Rossmanith, T.; Alten, R.; Aringer, M.; Backhaus, M.; Burmester, G.R.; Feist, E.; Herrmann, E.; Kellner, H.; et al. Rituximab plus leflunomide in rheumatoid arthritis: A randomized, placebo-controlled, investigator-initiated clinical trial (AMARA study). Rheumatology 2021, 60, 5318–5328. [Google Scholar] [CrossRef] [PubMed]
- Bellan, M.; Scotti, L.; Ferrante, D.; Calzaducca, E.; Manfredi, G.F.; Sainaghi, P.P.; Barone-Adesi, F. Risk of severe infection among rheumatoid arthritis patients on biological DMARDs: A population-based cohort study. J. Clin. Med. 2022, 11, 2955. [Google Scholar] [CrossRef] [PubMed]
- Riley, T.R.; George, M.D. Risk for infections with glucocorticoids and DMARDs in patients with rheumatoid arthritis. RMD Open 2021, 7, e001235. [Google Scholar] [CrossRef] [PubMed]
- Winthrop, K.L.; Saag, K.; Cascino, M.D.; Pei, J.; John, A.; Jahreis, A.; Haselkorn, T.; Furst, D.E. Long-term safety of rituximab in patients with rheumatoid arthritis: Results of a five-year observational study. Arthritis Care Res. 2019, 71, 993–1003. [Google Scholar] [CrossRef] [PubMed]
- Tak, P.P.; Rigby, W.; Rubbert-Roth, A.; Peterfy, C.; van Vollenhoven, R.F.; Stohl, W.; Healy, E.; Hessey, E.; Reynard, M.; Shaw, T. Sustained inhibition of progressive joint damage with rituximab plus methotrexate in early active rheumatoid arthritis: 2-year results from the randomised controlled trial IMAGE. Ann. Rheum. Dis. 2012, 71, 351–357. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | Rituximab→Rituximab | Placebo→Rituximab | ||
|---|---|---|---|---|
| Rituximab 1000 mg | Rituximab 500 mg | Rituximab 1000 mg | Rituximab 500 mg | |
| n | 31 | 29 | 10 | 13 |
| Baseline | ||||
| Age, years | 58.2 (11.3) | 57.1 (12.9) | 54.7 (6.3) | 55.3 (11.0) |
| BMI, kg/m2 | 26.8 (5.3) | 25.9 (6.1) | 29.8 (5.0) | 25.9 (6.0) |
| Females, n (%) | 25 (80.7%) | 19 (65.5%) | 7 (70.0%) | 9 (69.2%) |
| RF seropositive, n (%) | 21 (67.7%) | 19 (65.5%) | 4 (40.0%) | 7 (53.9%) |
| Anti-CCP seropositive,a n (%) | 22 (71.0%) | 18 (62.1%) | 4 (40.0%) | 7 (53.9%) |
| Previous use of conventional DMARDs (%) | 30 (96.8%) | 17 (93.8%) | 10 (100.0%) | 12 (94.6%) |
| Patients with at least one previous anti-TNF therapy, n (%) | 1 (3.2%) | 4 (13.8%) | 0 | 1 (7.7%) |
| Corticosteroid dose, b,c mean (SD) [median; Q1, Q3] | 6.3 (2.7) [5.0; 2.0, 10.0] | 5.3 (1.7) [5.0; 2.0, 10.0] | 6.7 (2.9) [5.0; 5.0, 10.0] | 5.5 (1.1) [5.0; 5.0, 7.5] |
| Leflunomide dose, c mean (SD) | 17.2 (4.5) | 19.6 (2.0) | 19.0 (3.2) | 18.3 (3.9) |
| 10 mg dose, n (%) | 8 (25.8%) | 1 (4.4%) | 1 (10.0%) | 2 (15.4%) |
| 20 mg dose, n (%) | 23 (74.2%) | 28 (96.6%) | 9 (90.0%) | 11 (84.6%) |
| Disease duration, years, mean (SD) [median; Q1, Q3] | 8.0 (9.7) [4.4; 1.9, 11.5] | 7.4 (7.6) [3.8; 2.0, 12.5] | 10.6 (12.8) [5.1; 4.1, 13.8] | 4.5 (3.7) [3.1; 2.2, 4.7] |
| DAS 28 | 5.7 (1.1) | 5.3 (1.0) | 5.9 (1.0) | 5.3 (1.0) |
| CDAI, mean (SD) [median; Q1, Q3] | 31.2 (11.8) [28.7; 23.7, 39.2] | 27.4 (8.7) [28.2; 21.0, 34.0] | 36.4 (12.7) [32.5; 27.2, 51.2] | 31.8 (10.5) [28.2; 24.4, 35.5] |
| Tender joint count (28 joints) | 10.5 (5.8) | 9.4 (4.2) | 14.0 (6.9) | 13.3 (6.3) |
| Swollen joint count (28 joints) | 8.7 (4.3) | 7.8 (3.5) | 10.5 (5.8) | 7.3 (2.9) |
| Tender joint count (68 joints) | 15.5 (10.4) | 13.9 (6.8) | 20.9 (12.9) | 19.0 (11.0) |
| Swollen joint count 66 joints) | 10.8 (5.3) | 9.7 (5.0) | 12.3 (6.9) | 8.4 (2.7) |
| C-reactive protein, mg/L, mean (SD) [median; Q1, Q3] | 10.8 (12.4) [7.4; 2.2, 13.3] | 9.5 (20.4) [3.6; 1.9, 7.6] | 9.2 (11.8) [5.9; 4.0, 8.0] | 5.1 (6.1) [3.2; 0.8, 5.5] |
| MDGA (10-cm VAS) | 58.4 (17.6) | 54.1 (18.0) | 57.0 (14.5) | 58.1 (18.7) |
| PtGA (10-cm VAS) | 60.6 (23.6) | 48.0 (23.8) | 62.2 (16.8) | 53.8 (27.7) |
| Week 24 | ||||
| DAS 28 | 3.4 (1.2) | 3.5 (1.5) | 4.2 (1.2) | 3.8 (1.4) |
| CDAI, mean (SD) [median; Q1, Q3] | 12.4 (9.6) [10.7; 5.1, 19.4] | 12.4 (10.7) [11.1; 3.4, 20.7] | 16.4 (10.8) [15.6; 6.8, 25.4] | 17.7 (12.8) [18.1; 8.6, 28.1] |
| Tender joint count (28 joints) | 3.4 (4.3) | 4.2 (5.6) | 6.5 (4.9) | 5.3 (6.7) |
| Swollen joint count (28 joints) | 3.8 (3.6) | 2.8 (3.3) | 4.8 (6.5) | 4.5 (4.6) |
| Tender joint count (68 joints) | 5.4 (8.1) | 7.4 (10.5) | 9.8 (9.3) | 7.2 (8.3) |
| Swollen joint count (66 joints) | 4.6 (4.6) | 3.9 (4.6) | 5.7 (8.0) | 6.2 (6.2) |
| CRP, mg/L, mean (SD) [median; Q1, Q3] | 5.0 (5.4) [3.3; 1.8, 5.6] | 6.0 (6.5) [2.7; 1.9, 8.5] | 13.2 (10.6) [10.1; 3.4, 20.6] | 3.6 (4.0) [2.3; 0.6, 3.6] |
| ACR20 response, n (%) | 19 (61.3%) | 19 (65.5%) | 7 (70.0%) | 4 (30.8%) |
| ACR50 response, n (%) | 10 (32.3%) | 11 (37.9%) | 3 (30.0%) | 2 (15.4%) |
| ACR70 response, n (%) | 6 (19.4%) | 7 (24.1%) | 1 (10.0%) | 1 (7.7%) |
| MDGA (100-mm VAS) | 26.8 (19.6) | 27.3 (22.1) | 23.7 (19.2) | 39.3 (25.9) |
| PtGA (100-mm VAS) | 25.5 (21.8) | 28.0 (24.3) | 27.1 (22.3) | 41.8 (27.4) |
| FACIT Fatigue | 73.8 (19.9) | 74.0 (18.9) | 64.4 (22.8) | 62.1 (22.8) |
| HAQ-DI | 1.0 (0.6) | 0.9 (0.48) | 1.1 (0.5) | 1.1 (0.3) |
| SF-36 Physical functioning Physical role limitations Pain General health perceptions Energy/vitality Social functioning Emotional role limitations Mental health | 68.2 (23.5) 63.8 (43.1) 58.3 (21.0) 56.8 (14.5) 62.2 (20.1) 81.3 (23.4) 72.4 (41.9) 67.4 (18.6) | 63.0 (27.4) 57.6 (44.9) 62.7 (23.5) 50.3 (21.1) 56.9 (20.2) 81.8 (19.9) 66.7 (42.4) 67.1 (19.1) | 52.0 (25.3) 59.4 (49.9) 55.9 (22.2) 52.5 (22.2) 51.5 (22.5) 71.3 (26.3) 50.0 (47.2) 63.2 (20.6) | 48.5 (19.2) 36.5 (42.8) 47.5 (20.4) 41.5 (19.9) 43.5 (17.5) 65.5 (20.4) 43.6 (49.8) 61.2 (18.2) |
| Adverse Events by SOC | Rituximab→Rituximab | Placebo→Rituximab | Total (n = 83) | ||
|---|---|---|---|---|---|
| Rituximab 1000 mg (n = 31) | Rituximab 500 mg (n = 29) | Rituximab 1000 mg (n = 10) | Rituximab 500 mg (n = 13) | ||
| AEs ≥5% in any subgroup | |||||
| Number of AEs | 38 | 33 | 7 | 17 | 95 |
| Number (%) of patients | 26 (83.9%) | 26 (89.7%) | 7 (70.0%) | 12 (92.3%) | 71 (74.7%) |
| Infections and infestations | 19 (61.3%) | 14 (48.3%) | 3 (30.0%) | 6 (35.3%) | 42 (50.6%) |
| Skin and subcutaneous tissue disorders | 3 (9.7%) | 5 (17.2%) | 1 (10.0%) | 1 (7.7%) | 10 (12.0%) |
| Musculoskeletal and connective tissue disorders | 2 (6.5%) | 2 (6.9%) | 1 (10.0%) | 2 (15.4%) | 7 (8.4%) |
| Investigations | 2 (6.5%) | 2 (6.9%) | 0 | 1 (7.7%) | 5 (6.0%) |
| Gastrointestinal disorders | 3 (9.7%) | 1 (3.4%) | 0 | 0 | 4 (4.8%) |
| General disorders and administration site conditions | 1 (3.2%) | 1 (3.4%) | 0 | 2 (15.4%) | 4 (4.8%) |
| Nervous system disorders | 2 (6.5%) | 1 (3.4%) | 0 | 1 (7.7%) | 4 (4.8%) |
| Ear and labyrinth disorders | 1 (3.2%) | 0 | 1 (10.0%) | 1 (7.7%) | 3 (3.6%) |
| Surgical and medical procedures | 0 | 2 (6.9%) | 0 | 1 (7.7%) | 3 (3.6%) |
| Metabolism and nutrition disorders | 0 | 1 (3.4%) | 1 (10.0%) | 0 | 2 (2.4%) |
| Vascular disorders | 1 (3.2%) | 0 | 0 | 1 (7.7%) | 2 (2.4%) |
| Immune system disorders | 0 | 0 | 0 | 1 (7.7%) | 1 (1.2%) |
| SAEs (all) | |||||
| Number of events | 6 | 7 | 1 | 3 | 17 |
| Number (%) of patients | 5 (16.1%) | 7 (24.1%) | 1 (10.0%) | 3 (23.1%) | 16 (19.3%) |
| Surgical and medical procedures a | 1 (3.2%) | 3 (10.3%) | 1 (7.7%) | 5 (6.0%) | |
| Infections and infestations b | 2 (6.5%) | 1 (3.4%) | 0 | 1 (7.7%) | 4 (4.8%) |
| Musculoskeletal and connective tissue disorders c | 1 (3.2%) | 2 (6.9%) | 1 (10.0%) | 0 | 4 (4.8%) |
| General disorders and administration site conditions d | 1 (3.2%) | 0 | 0 | 1 (7.7%) | 2 (2.4%) |
| Gastrointestinal disorders e | 1 (3.2%) | 0 | 0 | 0 | 1 (1.2%) |
| Vascular disorders f | 0 | 1 (3.4%) | 0 | 0 | 1 (1.2%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koehm, M.; Foldenauer, A.C.; Rossmanith, T.; Alten, R.; Aringer, M.; Backhaus, M.; Burmester, G.R.; Feist, E.; Kellner, H.; Krueger, K.; et al. Effectiveness of Different Rituximab Doses Combined with Leflunomide in the Treatment or Retreatment of Rheumatoid Arthritis: Part 2 of a Randomized, Placebo-Controlled, Investigator-Initiated Clinical Trial (AMARA). J. Clin. Med. 2022, 11, 7316. https://doi.org/10.3390/jcm11247316
Koehm M, Foldenauer AC, Rossmanith T, Alten R, Aringer M, Backhaus M, Burmester GR, Feist E, Kellner H, Krueger K, et al. Effectiveness of Different Rituximab Doses Combined with Leflunomide in the Treatment or Retreatment of Rheumatoid Arthritis: Part 2 of a Randomized, Placebo-Controlled, Investigator-Initiated Clinical Trial (AMARA). Journal of Clinical Medicine. 2022; 11(24):7316. https://doi.org/10.3390/jcm11247316
Chicago/Turabian StyleKoehm, Michaela, Ann C. Foldenauer, Tanja Rossmanith, Rieke Alten, Martin Aringer, Marina Backhaus, Gerd R. Burmester, Eugen Feist, Herbert Kellner, Klaus Krueger, and et al. 2022. "Effectiveness of Different Rituximab Doses Combined with Leflunomide in the Treatment or Retreatment of Rheumatoid Arthritis: Part 2 of a Randomized, Placebo-Controlled, Investigator-Initiated Clinical Trial (AMARA)" Journal of Clinical Medicine 11, no. 24: 7316. https://doi.org/10.3390/jcm11247316
APA StyleKoehm, M., Foldenauer, A. C., Rossmanith, T., Alten, R., Aringer, M., Backhaus, M., Burmester, G. R., Feist, E., Kellner, H., Krueger, K., Müller-Ladner, U., Rubbert-Roth, A., Tony, H.-P., Wassenberg, S., Burkhardt, H., & Behrens, F. (2022). Effectiveness of Different Rituximab Doses Combined with Leflunomide in the Treatment or Retreatment of Rheumatoid Arthritis: Part 2 of a Randomized, Placebo-Controlled, Investigator-Initiated Clinical Trial (AMARA). Journal of Clinical Medicine, 11(24), 7316. https://doi.org/10.3390/jcm11247316