
A Comprehensive Review of Pediatric Acute Encephalopathy



Abstract
1. Introduction
2. Classification of Acute Encephalopathy and Literature Search
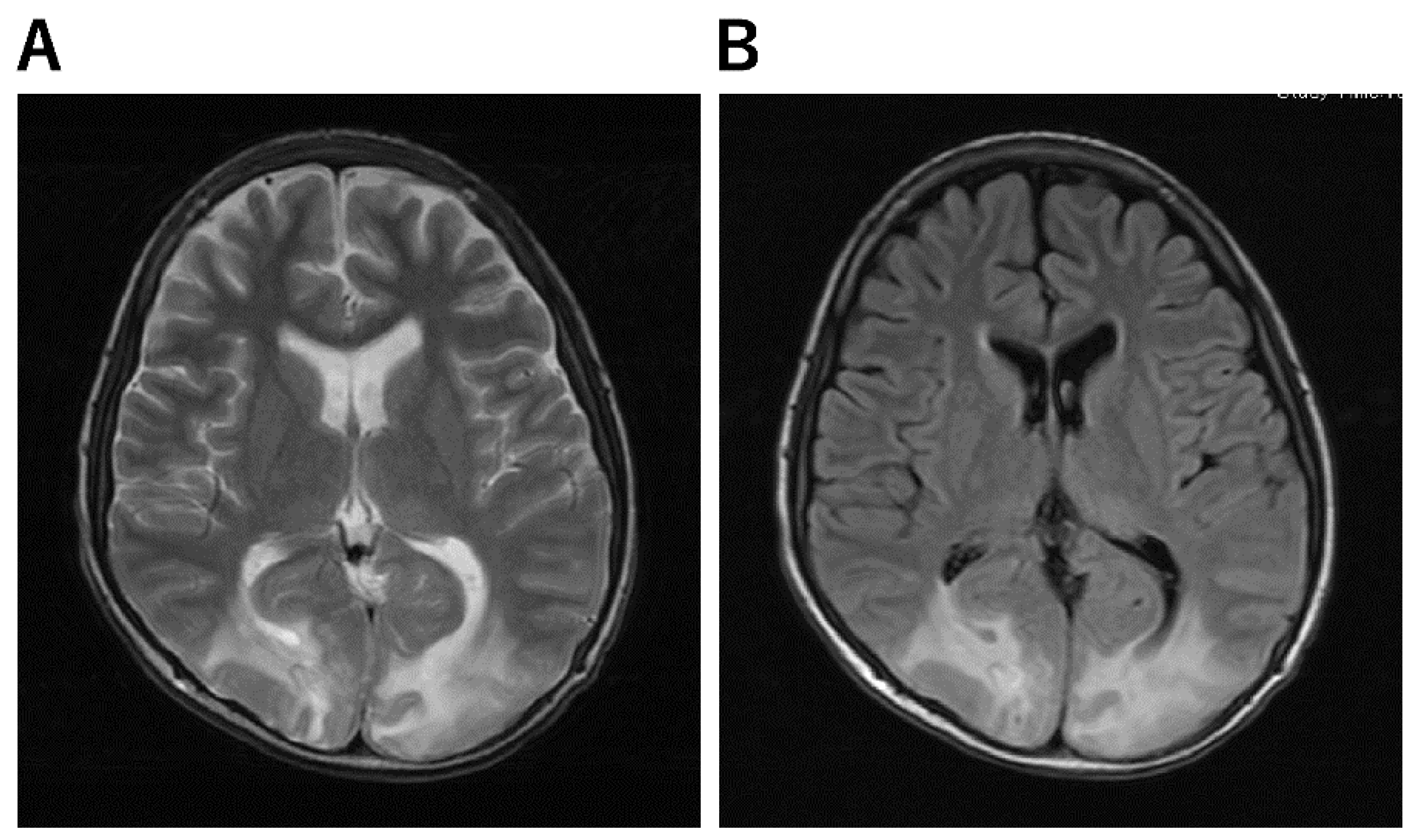
3. Clinical Presentation
4. Diagnosis
5. Management
6. Acute Encephalopathy in the COVID-19 Era
7. How to Evaluate Neurological Prognosis
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Slooter, A.J.C.; Otte, W.M.; Devlin, J.W.; Arora, R.C.; Bleck, T.P.; Claassen, J.; Duprey, M.S.; Ely, E.W.; Kaplan, P.W.; Latronico, N.; et al. Updated nomenclature of delirium and acute encephalopathy: Statement of ten Societies. Intensive Care Med. 2020, 46, 1020–1022. [Google Scholar] [CrossRef] [PubMed]
- Mizuguchi, M.; Ichiyama, T.; Imataka, G.; Okumura, A.; Goto, T.; Sakuma, H.; Takanashi, J.I.; Murayama, K.; Yamagata, T.; Yamanouchi, H.; et al. Guidelines for the diagnosis and treatment of acute encephalopathy in childhood. Brain Dev. 2021, 43, 2–31. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, A.; Saitoh, M.; Oka, A.; Okumura, A.; Kubota, M.; Saito, Y.; Takanashi, J.; Hirose, S.; Yamagata, T.; Yamanouchi, H.; et al. Epidemiology of acute encephalopathy in Japan, with emphasis on the association of viruses and syndromes. Brain Dev. 2012, 34, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Kasai, M.; Shibata, A.; Hoshino, A.; Maegaki, Y.; Yamanouchi, H.; Takanashi, J.I.; Yamagata, T.; Sakuma, H.; Okumura, A.; Nagase, H.; et al. Epidemiological changes of acute encephalopathy in Japan based on national sur-veillance for 2014–2017. Brain Dev. 2020, 42, 508–514. [Google Scholar] [CrossRef]
- Mizuguchi, M.; Yamanouchi, H.; Ichiyama, T.; Shiomi, M. Acute encephalopathy associated with influenza and other viral infections. Acta Neurol. Scand. 2007, 115, 45–56. [Google Scholar] [CrossRef]
- Shibata, A.; Kasai, M.; Hoshino, A.; Miyagawa, T.; Matsumoto, H.; Yamanaka, G.; Kikuchi, K.; Kuki, I.; Kumakura, A.; Hara, S.; et al. Thermolabile polymorphism of carnitine palmitoyltransferase 2: A genetic risk factor of overall acute encephalopathy. Brain Dev. 2019, 41, 862–869. [Google Scholar] [CrossRef]
- Shinohara, M.; Saitoh, M.; Nishizawa, D.; Miyagawa, T.; Matsumoto, H.; Yamanaka, G.; Kikuchi, K.; Kuki, I.; Kumakura, A.; Hara, S.; et al. ADORA2A polymorphism predisposes children to encephalopathy with febrile status epilepticus. Neurology 2013, 80, 1571–1576. [Google Scholar] [CrossRef]
- Shibata, A.; Kasai, M.; Terashima, H.; Hoshino, A.; Miyagawa, T.; Kikuchi, K.; Ishii, A.; Matsumoto, H.; Kubota, M.; Hirose, S.; et al. Case-control association study of rare nonsynonymous variants of SCN1A and KCNQ2 in acute encephalopathywith biphasic seizures and late reduced diffusion. J. Neurol. Sci. 2020, 414, 116808. [Google Scholar] [CrossRef]
- Kubota, M.; Chida, J.; Hoshino, H.; Ozawa, H.; Koide, A.; Kashii, H.; Koyama, A.; Mizuno, Y.; Hoshino, A.; Yamaguchi, M.; et al. Thermolabile CPT II variants and low blood ATP levels are closely related to severity of acute encephalopathy in Japanese children. Brain Dev. 2012, 34, 20–27. [Google Scholar] [CrossRef]
- Chen, Y.; Mizuguchi, H.; Yao, D.; Ide, M.; Kuroda, Y.; Shigematsu, Y.; Yamaguchi, S.; Kinoshita, M.; Kido, H. Thermolabile phenotype of carnitine palmitoyltransferase II variations as a predisposing factor for influenza-associated encephalopathy. FEBS Lett. 2005, 579, 2040–2044. [Google Scholar] [CrossRef]
- Saitoh, M.; Shinohara, M.; Ishii, A.; Ihara, Y.; Hirose, S.; Shiomi, M.; Kawawaki, H.; Kubota, M.; Yamagata, T.; Miyamoto, A.; et al. Clinical and genetic features of acute encephalopathy in children taking theophylline. Brain Dev. 2015, 37, 463–470. [Google Scholar] [CrossRef]
- Saitoh, M.; Ishii, A.; Ihara, Y.; Hoshino, A.; Terashima, H.; Kubota, M.; Kikuchi, K.; Yamanaka, G.; Amemiya, K.; Hirose, S.; et al. Missense mutations in sodium channel SCN1A and SCN2A predispose children to encepha-lopathy with severe febrile seizures. Epilepsy Res. 2015, 117, 1–6. [Google Scholar] [CrossRef]
- Fujita, Y.; Imataka, G.; Sakuma, H.; Takanashi, J.I.; Yoshihara, S. Multiple encephalopathy syndrome: A case of a novel radiological subtype of acute encephalopathy in childhood. Eur. Rev. Med. Pharmacol. Sci. 2022, 1, 11720. [Google Scholar]
- Hoshino, A.; Saitoh, M.; Miyagawa, T.; Kubota, M.; Takanashi, J.-I.; Miyamoto, A.; Tokunaga, K.; Oka, A.; Mizuguchi, M. Specific HLA genotypes confer susceptibility to acute necrotizing encephalopathy. Genes Immun. 2016, 17, 367–369. [Google Scholar] [CrossRef]
- Mohseni-Bod, H.; Drake, J.; Kukreti, V. Management of raised intracranial pressure in children with traumatic brain injury. J. Pediatr. Neurosci. 2014, 9, 207–215. [Google Scholar] [CrossRef]
- Lee, S.; Sanefuji, M.; Watanabe, K.; Uematsu, A.; Torisu, H.; Baba, H.; Kira, R.; Takada, Y.; Ishizaki, Y.; Toyoshima, M.; et al. Clinical and MRI characteristic of acute encephalopathy in children in congenital adrenal hyperplasia. J. Nurol. Sci. 2011, 306, 91–93. [Google Scholar] [CrossRef]
- Covanis, A. Epileptic encephalopathies (including severe epilepsy syndromes). Epilepsia 2012, 53, 114–126. [Google Scholar] [CrossRef]
- Scheffer, I.E.; Berkovic, S.; Capovilla, G.; Connolly, M.B.; French, J.; Guilhoto, L.; Hirsch, E.; Jain, S.; Mathern, G.W.; Moshe, S.; et al. ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017, 58, 512–521. [Google Scholar] [CrossRef]
- Markand, O.N. Lennox-Gastaut Syndrome (Childhood Epileptic Encephalopathy). J. Clin. Neurophysiol. 2003, 20, 426–441. [Google Scholar] [CrossRef]
- Hwang, S.-K.; Kwon, S. Early-onset epileptic encephalopathies and the diagnostic approach to underlying causes. Korean J. Pediatr. 2015, 58, 407–414. [Google Scholar] [CrossRef]
- Okumura, A.; Uematsu, M.; Imataka, G.; Tanaka, M.; Okanishi, T.; Kubota, T.; Sudo, A.; Tohyama, J.; Tsuji, M.; Ohmori, I.; et al. Acute encephalopathy in children with Dravet syndrome. Epilepsia 2012, 53, 79–86. [Google Scholar] [CrossRef]
- Imataka, G.; Wake, K.; Suzuki, M.; Yamanouchi, H.; Arisaka, O. Acute encephalopathy associated with hemolytic uremic syndrome caused by Escherichia coli O157: H7 and rotavirus infection. Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 1842–1844. [Google Scholar]
- Imataka, G.; Ishii, J.; Ando, Y.; Yoshihara, S.; Takagi, Y.; Nitta, A.; Arisaka, O.; Yoshihara, S. Long-term survival of a patient with acute neonatal-onset metabolic encephalopathy with carbamoyl phosphate synthetase 1 deficiency. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 10051–10053. [Google Scholar]
- Wake, K.; Imataka, G.; Ohnishi, T.; Kikuchi, J.; Hoshiyama, E.; Ono, K.; Yoshihara, S. A Case-Study of a Child with Reversible Posterior Leukoencephalopathy Syndrome (RPLS) Associated with Severe Burns throughout the Body. Iran. J. Public Health 2020, 49, 1390–1391. [Google Scholar] [CrossRef]
- Imataka, G.; Wake, K.; Yoshihara, S. A girl with acute encephalitis followed by acute cerebellitis/cerebellopathy. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 10708–10711. [Google Scholar]
- Miyake, N.; Fukai, R.; Ohba, C.; Chihara, T.; Miura, M.; Shimizu, H.; Kakita, A.; Imagawa, E.; Shiina, M.; Ogata, K.; et al. Biallelic TBCD Mutations Cause Early-Onset Neurodegenerative Encephalopathy. Am. J. Hum. Genet. 2016, 99, 950–961. [Google Scholar] [CrossRef]
- Abe, Y.; Sakai, T.; Okumura, A.; Akaboshi, S.; Fukuda, M.; Haginoya, K.; Hamano, S.; Hirano, K.; Kikuchi, K.; Kubota, M.; et al. Manifestations and characteristics of congenital adrenal hyperplasia-associated encephalopathy. Brain Dev. 2016, 38, 638–647. [Google Scholar] [CrossRef]
- National Institute of Neurological Disorders and Stroke. Encephalopathy Information Page. Available online: https://www.ninds.nih.gov/Disorders/All-Disorders/Encephalopathy-Information-Page. (accessed on 27 May 2022).
- Takahashi, A.; Kamei, E.; Sato, Y.; Shimada, S.; Tsubokawa, M.; Ohta, G.; Ohshima, Y.; Matsumine, A. Infant with right hemiplegia due to acute encephalopathy with biphasic seizures and late reduced diffusion (AESD): A case report. Medicine 2021, 100, e25468. [Google Scholar] [CrossRef]
- Venkatesan, A.; Tunkel, A.R.; Bloch, K.C.; Lauring, A.S.; Sejvar, J.; Bitnun, A.; Stahl, J.-P.; Mailles, A.; Drebot, M.; Rupprecht, C.E.; et al. Case Definitions, Diagnostic Algorithms, and Priorities in Encephalitis: Consensus Statement of the International Encephalitis Consortium. Clin. Infect. Dis. 2013, 57, 1114–1128. [Google Scholar] [CrossRef]
- Britton, P.N.; Eastwood, K.; Paterson, B.; Durrheim, D.N.; Dale, R.C.; Cheng, A.; Kenedi, C.; Brew, B.; Burrow, J.; Nagree, Y.; et al. Consensus guidelines for the investigation and management of encephalitis in adults and children in Australia and New Zealand. Intern. Med. J. 2015, 45, 563–576. [Google Scholar] [CrossRef]
- Lim, J.; Kwek, S.; How, C.; Chan, W. A clinical approach to encephalopathy in children. Singap. Med. J. 2020, 61, 626–632. [Google Scholar] [CrossRef] [PubMed]
- Erkkinen, M.G.; Berkowitz, A.L. A Clinical Approach to Diagnosing Encephalopathy. Am. J. Med. 2019, 132, 1142–1147. [Google Scholar] [CrossRef] [PubMed]
- Barbosa-Silva, M.C.; Lima, M.N.; Battaglini, D.; Robba, C.; Pelosi, P.; Rocco, P.R.M.; Maron-Gutierrez, T. Infectious disease-associated encephalopathies. Crit. Care 2021, 25, 236. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, S.S.; Soe, S.M.; Pillai, S.C.; Nosadini, M.; Barnes, E.H.; Gill, D.; Dale, R.C. Etiological associations and outcome predictors of acute electroencephalography in childhood encephalitis. Clin. Neurophysiol. 2016, 127, 3217–3224. [Google Scholar] [CrossRef]
- Maegaki, Y.; Kondo, A.; Okamoto, R.; Inoue, T.; Konishi, K.; Hayashi, A.; Tsuji, Y.; Fujii, S.; Ohno, K. Clinical Characteristics of Acute Encephalopathy of Obscure Origin: A Biphasic Clinical Course Is a Common Feature. Neuropediatrics 2006, 37, 269–277. [Google Scholar] [CrossRef]
- Okumura, A.; Suzuki, M.; Kidokoro, H.; Komatsu, M.; Shono, T.; Hayakawa, F.; Shimizu, T. The spectrum of acute encephalopathy with reduced diffusion in the unilateral hemisphere. Eur. J. Paediatr. Neurol. 2009, 13, 154–159. [Google Scholar] [CrossRef]
- Hosoya, M.; Ushiku, H.; Arakawa, H.; Morikawa, A. Low-voltage activity in EEG during acute phase of encephalitis predicts unfavorable neurological outcome. Brain Dev. 2002, 24, 161–165. [Google Scholar] [CrossRef]
- Okumura, A.; Komatsu, M.; Abe, S.; Kitamura, T.; Matsui, K.; Ikeno, M.; Shimizu, T. Amplitude-integrated electroencephalography in patients with acute encephalopathy with refractory, repetitive partial seizures. Brain Dev. 2011, 33, 77–82. [Google Scholar] [CrossRef]
- Accolla, E.A.; Kaplan, P.W.; Maeder-Ingvar, M.; Jukopila, S.; Rossetti, A. Clinical correlates of frontal intermittent rhythmic delta activity (FIRDA). Clin. Neurophysiol. 2011, 122, 27–31. [Google Scholar] [CrossRef]
- Ohno, A.; Okumura, A.; Fukasawa, T.; Nakata, T.; Suzuki, M.; Tanaka, M.; Okaia, Y.; Ito, Y.; Yamamoto, H.; Tsuji, T.; et al. Acute encephalopathy with biphasic seizures and late reduced diffusion: Predictive EEG findings. Brain Dev. 2022, 44, 221–228. [Google Scholar] [CrossRef]
- Solomon, T.; Hart, I.J.; Beeching, N.J. Viral encephalitis: A clinician’s guide. Pract. Neurol. 2007, 7, 288–305. [Google Scholar] [CrossRef]
- Kneen, R.; Solomon, T.; Appleton, R. The role of lumbar puncture in children with suspected central nervous system infection. BMC Pediatr. 2002, 2, 8. [Google Scholar] [CrossRef]
- Neeb, L.; Hoekstra, J.; Endres, M.; Siegerink, B.; Siebert, E.; Liman, T.G. Spectrum of cerebral spinal fluid findings in patients with posterior reversible enceph-alopathy syndrome. J. Neurol. 2016, 263, 30–34. [Google Scholar] [CrossRef]
- Okada, T.; Fujita, Y.; Imataka, G.; Takase, N.; Tada, H.; Sakuma, H.; Takanashi, J.-I. Increased cytokines/chemokines and hyponatremia as a possible cause of clinically mild encephalitis/encephalopathy with a reversible splenial lesion associated with acute focal bacterial nephritis. Brain Dev. 2022, 44, 30–35. [Google Scholar] [CrossRef]
- Imataka, G.; Yoshihara, S. Immature ovarian teratoma with anti-NMDA-receptor encephalitis in a 13-year-old Japanese female patient. Med. J. Malays. 2021, 76, 436–437. [Google Scholar]
- Mizuguchi, M. Influenza encephalopathy and related neuropsychiatric syndromes. Influ. Other Respir. Viruses 2013, 7, 67–71. [Google Scholar] [CrossRef]
- Takanashi, J.; Tada, H.; Terada, H.; Barkovich, A.J. Excitotoxicity in acute encephalopathy with biphasic seizures and late reduced diffusion. Am. J. Neuroradiol. 2009, 30, 132–135. [Google Scholar] [CrossRef]
- Takanashi, J.; Oba, H.; Barkovich, A.J.; Tada, H.; Tanabe, Y.; Yamanouchi, H.; Fujimoto, S.; Kato, M.; Kawatani, M.; Sudo, A.; et al. Diffusion MRI abnormalities after prolonged febrile seizures with encephalopathy. Neurology 2006, 66, 1304–1309. [Google Scholar] [CrossRef]
- Takanashi, J.-I.; Mizuguchi, M.; Terai, M.; Barkovich, A.J. Disrupted glutamate-glutamine cycle in acute encephalopathy with biphasic seizures and late reduced diffusion. Neuroradiology 2015, 57, 1163–1168. [Google Scholar] [CrossRef]
- Yamanouchi, H.; Mizuguchi, M. Acute infantile encephalopathy predominantly affecting the frontal lobes (AIEF): A novel clinical category and its tentative diagnostic criteria. Epilepsy Res. 2006, 70, S263–S268. [Google Scholar] [CrossRef]
- Mizuguchi, M.; Hayashi, M.; Nakano, I.; Kuwashima, M.; Yoshida, K.; Nakai, Y.; Itoh, M.; Takashima, S. Concentric structure of thalamic lesions in acute necrotizing encephalopathy. Neuroradiology 2002, 44, 489–493. [Google Scholar] [CrossRef]
- Okumura, A.; Abe, S.; Kidokoro, H.; Mizuguchi, M. Acute necrotizing encephalopathy: A comparison between influenza and non-influenza cases. Microbiol. Immunol. 2009, 53, 277–280. [Google Scholar] [CrossRef]
- Takanashi, J.; Barkovich, A.; Shiihara, T.; Tada, H.; Kawatani, M.; Tsukahara, H.; Kikuchi, M.; Maeda, M. Widening Spectrum of a Reversible Splenial Lesion with Transiently Reduced Diffusion. Am. J. Neuroradiol. 2006, 27, 836–838. [Google Scholar]
- Imataka, G.; Yoshihara, S. Typical MRI Imaging with Clinically Mild Encephalitis/Encephalopathy of a Reversible Splenial Lesion (MERS) Caused by Influenza A Virus. Iran. J. Public Health 2020, 49, 191–192. [Google Scholar] [CrossRef]
- Anderson, R.-C.; Patel, V.; Sheikh-Bahaei, N.; Liu, C.S.J.; Rajamohan, A.G.; Shiroishi, M.S.; Kim, P.E.; Go, J.L.; Lerner, A.; Acharya, J. Posterior Reversible Encephalopathy Syndrome (PRES): Pathophysiology and Neuro-Imaging. Front. Neurol. 2020, 11, 463. [Google Scholar] [CrossRef]
- Tsukahara, H.; Fujii, Y.; Matsubara, K.; Yamada, M.; Nagaoka, Y.; Saito, Y.; Yashiro, M.; Tsuge, M.; Goto, S.; Kitamura, T.; et al. Prognostic value of brain injury biomarkers in acute encephalitis/encephalopathy. Pediatr. Int. 2013, 55, 461–464. [Google Scholar] [CrossRef]
- Kasai, M.; Omae, Y.; Kawai, Y.; Shibata, A.; Hoshino, A.; Mizuguchi, M.; Tokunaga, K. GWAS identifies candidate susceptibility loci and microRNA biomarkers for acute encephalopathy with biphasic seizures and late reduced diffusion. Sci. Rep. 2022, 12, 1332. [Google Scholar] [CrossRef]
- Okumura, A.; Mizuguchi, M.; Kidokoro, H.; Tanaka, M.; Abe, S.; Hosoya, M.; Aiba, H.; Maegaki, Y.; Yamamoto, H.; Tanabe, T.; et al. Outcome of acute necrotizing encephalopathy in relation to treatment with corticosteroids and gammaglobulin. Brain Dev. 2009, 31, 221–227. [Google Scholar] [CrossRef]
- Esen, F.; Ozcan, P.E.; Tuzun, E.; Boone, M.D. Mechanisms of action of intravenous immunoglobulin in septic encephalopathy. Rev. Neurosci. 2018, 29, 417–423. [Google Scholar] [CrossRef]
- Shima, T.; Okumura, A.; Kurahashi, H.; Numoto, S.; Abe, S.; Ikeno, M.; Shimizu, T. A nationwide survey of norovirus-associated encephalitis/encephalopathy in Japan. Brain Dev. 2019, 41, 263–270. [Google Scholar] [CrossRef]
- Tanuma, N.; Miyata, R.; Nakajima, K.; Okumura, A.; Kubota, M.; Hamano, S.-I.; Hayashi, M. Changes in Cerebrospinal Fluid Biomarkers in Human Herpesvirus-6-Associated Acute Encephalopathy/Febrile Seizures. Mediat. Inflamm. 2014, 2014, 564091. [Google Scholar] [CrossRef] [PubMed]
- Gwer, S.; Gatakaa, H.; Mwai, L.; Idro, R.; Newton, C.R. The role for osmotic agents in children with acute encephalopathies: A systematic review. BMC Pediatr. 2010, 10, 23. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, M.; Tanaka, T.; Fujita, K.; Maruyama, A.; Nagase, H. Targeted temperature management of acute encephalopathy without AST elevation. Brain Dev. 2015, 37, 328–333. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, I.; Okubo, Y.; Nariai, H.; Michihata, N.; Matsui, H.; Fushimi, K.; Yasunaga, H. Recent treatment patterns and variations for pediatric acute encephalopathy in Japan. Brain Dev. 2020, 42, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Ishida, Y.; Nishiyama, M.; Yamaguchi, H.; Tomioka, K.; Takeda, H.; Tokumoto, S.; Toyoshima, D.; Maruyama, A.; Seino, Y.; Aoki, K.; et al. Early steroid pulse therapy for children with suspected acute encephalopathy. Medicine 2021, 100, e26660. [Google Scholar] [CrossRef] [PubMed]
- Neal, E.G.; Chaffe, H.M.; Schwartz, R.H.; Lawson, M.S.; Edwards, N.; Fitzsimmons, G.; Whitney, A.; Cross, J.H. A randomized controlled trial of classical and medium chain triglyceride ketogenic diets in the treatment of childhood epilepsy. Epilepsia 2008, 49, 9. [Google Scholar]
- Brophy, G.M.; Bell, R.; Claassen, J.; Alldredge, B.; Bleck, T.P.; Glauser, T.; Laroche, S.M.; Riviello, J.J.J.; Shutter, L.; Sperling, M.R.; et al. Guidelines for the evaluation and management of status epilepticus. Neurocrit. Care 2012, 7, 3–23. [Google Scholar] [CrossRef]
- Cereda, C.; Berger, M.M.; Rossetti, A.O. Bowel Ischemia: A Rare Complication of Thiopental Treatment for Status Epilepticus. Neurocritical Care 2009, 10, 355–358. [Google Scholar] [CrossRef]
- Nakagawa, T.; Fujita, K.; Saji, Y.; Maruyama, A.; Nagase, H. Induced hypothermia normothermia with general anesthesia prevents neurological damage in febrile refractory status epilepticus in children. No Hattatsu 2011, 43, 459–463. [Google Scholar]
- Imataka, G.; Osamu, A. Brain hypothermia therapy for childhood acute encephalopathy based on clinical evidence. Exp. Ther. Med. 2015, 10, 1624–1626. [Google Scholar] [CrossRef]
- Imataka, G.; Tsuboi, Y.; Kano, Y.; Ogino, K.; Tsuchioka, T.; Ohnishi, T.; Kaji, Y.; Wake, K.; Ichikawa, G.; Suzumura, H.; et al. Treatment with mild brain hypothermia for cardiopulmonary resuscitation after myoclonic seizures in infant with robertsonian type of trisomy 13. Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 2852–2855. [Google Scholar]
- Imataka, G.; Wake, K.; Yamanouchi, H.; Ono, K.; Arisaka, O. Brain hypothermia therapy for status epilepticus in childhood. Eur. Rev. Med. Pharmacol. Sci. 2014, 18, 1883–1888. [Google Scholar]
- Hoshide, M.; Yasudo, H.; Inoue, H.; Matsushige, T.; Sakakibara, A.; Nawata, Y.; Hidaka, I.; Kobayashi, H.; Kohno, F.; Ichiyama, T.; et al. Efficacy of hypothermia therapy in patients with acute encephalopathy with biphasic seizures and late reduced diffusion. Brain Dev. 2020, 42, 515–522. [Google Scholar] [CrossRef]
- Fujita, Y.; Imataka, G.; Kikuchi, J.; Yoshihara, S. Successful mild brain hypothermia therapy followed by targeted temperature man-agement for pediatric hemorrhagic shock and encephalopathy syndrome. Eur. Rev. Med. Pharmacol. Sci. 2021, 25, 3002–3006. [Google Scholar]
- Krishnan, V.; Kumar, V.; Shankaran, S.; Thayyil, S. Rise and Fall of Therapeutic Hypothermia in Low-Resource Settings: Lessons from the HELIX Trial. Indian J. Pediatr. 2021. Epub ahead of print. [Google Scholar] [CrossRef]
- Imataka, G.; Arisaka, O. An infant with steroid-refractory cytomegalovirus-associated ADEM who responded to immuno-globulin therapy. Eur. Rev. Med. Pharmacol. Sci. 2014, 18, 2148–2151. [Google Scholar]
- Yamaguchi, H.; Nishiyama, M.; Tokumoto, S.; Ishida, Y.; Tomioka, K.; Aoki, K.; Seino, Y.; Toyoshima, D.; Takeda, H.; Kurosawa, H.; et al. Detailed characteristics of acute encephalopathy with biphasic seizures and late reduced diffusion: 18-year data of a single-center consecutive cohort. J. Neurol. Sci. 2020, 411, 116684. [Google Scholar] [CrossRef]
- Watanabe, Y.; Motoi, H.; Oyama, Y.; Ichikawa, K.; Takeshita, S.; Mori, M.; Nezu, A.; Yokota, S. Cyclosporine for acute encephalopathy with biphasic seizures and late reduced dif-fusion. Pediatr. Int. 2014, 56, 577–582. [Google Scholar] [CrossRef]
- Tada, H.; Takanashi, J.-I.; Okuno, H.; Kubota, M.; Yamagata, T.; Kawano, G.; Shiihara, T.; Hamano, S.-I.; Hirose, S.; Hayashi, T.; et al. Predictive score for early diagnosis of acute encephalopathy with biphasic seizures and late reduced diffusion (AESD). J. Neurol. Sci. 2015, 358, 62–65. [Google Scholar] [CrossRef]
- Yokochi, T.; Takeuchi, T.; Mukai, J.; Akita, Y.; Nagai, K.; Obu, K.; Kakuma, T.; Matsuishi, T. Prediction of acute encephalopathy with biphasic seizures and late reduced diffusion in patients with febrile status epilepticus. Brain Dev. 2016, 38, 217–224. [Google Scholar] [CrossRef]
- Wong, A.; Simon, E.; Zimmerman, R.; Wang, H.-S.; Toh, C.-H.; Ng, S.-H. Acute Necrotizing Encephalopathy of Childhood: Correlation of MR Findings and Clinical Outcome. Am. J. Neuroradiol. 2006, 27, 1919–1923. [Google Scholar]
- Ormitti, F.; Ventura, E.; Summa, A.; Picetti, E.; Crisi, G. Acute Necrotizing Encephalopathy in a Child during the 2009 Influenza A(H1N1) Pandemia: MR Imaging in Diagnosis and Follow-Up. Am. J. Neuroradiol. 2010, 31, 396–400. [Google Scholar] [CrossRef]
- Vargas, W.S.; Merchant, S.; Solomo, G. Favorable outcomes in acute necrotizing encephalopathy in a child treated with hypo-thermia. Pediatr. Neurol. 2012, 46, 87–389. [Google Scholar] [CrossRef]
- Yoshihara, S.; Fujita, Y.; Miyamoto, K.; Imataka, G.; Yoshihara, S. Kawasaki Disease with Mild Encephalitis/Encephalopathy with Reversible Splenial Lesion in a 2-Year-Old Girl. Indian J. Pediatr. 2021, 88, 718. [Google Scholar] [CrossRef]
- Imataka, G.; Fujita, Y.; Kano, Y.; Yoshihara, S. Abnormal predominantly frontal high voltage and slow EEG findings with acute en-cephalopathy associated with acute focal bacterial nephritis. J. Biol. Regul. Homeost. Agents 2020, 34, 1131–1133. [Google Scholar]
- Imataka, G.; Yamaguchi, T.; Ishii, J.; Ogino, K.; Okamoto, K.; Tsuchioka, T.; Yoshihara, S. MERS associated with bacterial translocation in a pediatric patient with congenital portal vein hypoplasia: A case report. Exp. Ther. Med. 2018, 16, 2831–2834. [Google Scholar] [CrossRef]
- Serino, D.; Santarone, M.E.; Caputo, D.; Fusco, L. Febrile infection-related epilepsy syndrome (FIRES): Prevalence, impact and management strategies. Neuropsychiatr. Dis. Treat 2019, 15, 1897–1903. [Google Scholar] [CrossRef]
- Lee, Y.-J. Febrile Infection-Related Epilepsy Syndrome: Refractory Status Epilepticus and Management Strategies. Ann. Child Neurol. 2020, 28, 8–15. [Google Scholar] [CrossRef]
- Montalvan, V.; Lee, J.; Bueso, T.; De Toledo, J.; Rivas, K. Neurological manifestations of COVID-19 and other coronavirus infections: A systematic review. Clin. Neurol. Neurosurg. 2020, 194, 105921. [Google Scholar] [CrossRef]
- Mun, C.; Wong, A.; Toh, C.H. Spectrum of neuroimaging mimics in children with COVID-19 infection. Biomed. J. 2021, 45, 50–62. [Google Scholar]
- Laçinel Gürlevik, S.; Günbey, C.; Ozsurekci, Y.; Oygar, P.D.; Kesici, S.; Gocmen, R.; Aydin, O.; Temucin, Ç.; Tufan, E.; Terzi, K.; et al. Neurologic manifestations in children with COVID-19 from a tertiary center in Turkey and literature review. Eur. J. Paediatr. Neurol. 2022, 37, 139–154. [Google Scholar] [CrossRef] [PubMed]
- Nepal, G.; Shrestha, G.S.; Rehrig, J.H.; Gajurel, B.P.; Ojha, R.; Agrawal, A.; Panthi, S.; Khatri, B.; Adhikari, I. Neurological Manifestations of COVID-19 Associated Multi-system Inflammatory Syndrome in Children: A Systematic Review and Meta-analysis. J. Nepal Health Res. Counc. 2021, 19, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Ngo, B.; Lapp, S.A.; Siegel, B.; Patel, V.; Hussaini, L.; Bora, S.; Philbrook, B.; Weinschenk, K.; Wright, L.; Anderson, E.J.; et al. Cerebrospinal fluid cytokine, chemokine, and SARS-CoV-2 antibody profiles in children with neuropsychiatric symptoms associated with COVID-19. Mult. Scler. Relat. Disord. 2021, 55, 103169. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.E.; Asfour, A.; Sewell, T.B.; Hooe, B.; Pryce, P.; Earley, C.; Shen, M.Y.; Kerner-Rossi, M.; Thakur, K.T.; Vargas, W.S.; et al. Neurological issues in children with COVID-19. Neurosci. Lett. 2020, 743, 135567. [Google Scholar] [CrossRef] [PubMed]
- Toubiana, J.; Poirault, C.; Corsia, A.; Bajolle, F.; Fourgeaud, J.; Angoulvant, F.; Debray, A.; Basmaci, R.; Salvador, E.; Biscardi, S.; et al. Kawasaki-like multisystem inflammatory syndrome in children during the COVID-19 pandemic in Paris, France: Prospective observational study. BMJ 2020, 369, m2094. [Google Scholar] [CrossRef] [PubMed]
- Panariello, A.; Bassetti, R.; Radice, A.; Rossotti, R.; Puoti, M.; Corradin, M.; Moreno, M.; Percudani, M. Anti-NMDA receptor encephalitis in a psychiatric COVID-19 patient: A case report. Brain Behav. Immun. 2020, 87, 179–181. [Google Scholar] [CrossRef] [PubMed]
- Monti, G.; Giovannini, G.; Marudi, A.; Bedin, R.; Melegari, A.; Simone, A.M.; Santangelo, M.; Pignatti, A.; Bertellini, E.; Trenti, T.; et al. Anti-NMDA receptor encephalitis presenting as new onset refractory status epilepticus in COVID-19. Seizure 2020, 81, 18–20. [Google Scholar] [CrossRef] [PubMed]
- Siracusa, L.; Cascio, A.; Giordano, S.; Medaglia, A.A.; Restivo, G.A.; Pirrone, I.; Saia, G.F.; Collura, F.; Colomba, C. Neurological complications in pediatric patients with SARS-CoV-2 infection: A sys-tematic review of the literature. Ital. J. Pediatr. 2021, 47, 123. [Google Scholar] [CrossRef]
- Urso, L.; Distefano, M.G.; Cambula, G.; Colomba, A.I.; Nuzzo, D.; Picone, P.; Giacomazza, D.; Sicurella, L. The case of encephalitis in a COVID-19 pediatric patient. Neurol. Sci. 2022, 43, 105–112. [Google Scholar] [CrossRef]
- Nishiyama, M.; Nagase, H.; Tanaka, T.; Fujita, K.; Kusumoto, M.; Kajihara, S.; Yamaguchi, Y.; Maruyama, A.; Takeda, H.; Uetani, Y.; et al. Short and long-term outcomes in children with suspected acute encepha-lopathy. Brain Dev. 2016, 38, 731–737. [Google Scholar] [CrossRef]
- Oba, C.; Kashiwagi, M.; Tanabe, T.; Nomura, S.; Ogino, M.; Matsuda, T.; Murata, S.; Nakamura, M.; Shirasu, A.; Inoue, K.; et al. Prognostic factors in the early phase of acute encepha-lopathy. Pediatr. Int. 2018, 60, 270–275. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Microbiological classification | • Influenza-associated encephalopathy • Human herpesvirus (HHV)-6/7 encephalopathy • Rotavirus encephalopathy • Respiratory syncytial virus encephalopathy • Herpes simplex virus encephalopathy • Varicella-zoster virus encephalopathy • Progressive multifocal leukoencephalopathies (PML) associated with the HIV virus such as subacute sclerosing panencephalitis (SSPE) caused by the measles virus and subacute encephalitis caused by the rubella virus. Bacterial infection-associated encephalopathy (Acute encephalopathy associated with hemolytic uremic syndrome (HUS) caused by E. coli O157:H7 and rotavirus infection and salmonella infection) [22] • Encephalopathy caused Bacillus cereulide-producing Bacillus cereus. • Mycoplasma infection-associated encephalopathy • Acute disseminated encephalomyelitis (ADEM) • Others |
| Metabolic errors | • Classic Reye syndrome • Encephalopathy secondary to inherited metabolic disorders (acute metabolic encephalopathy with carbamoyl phosphate synthetase 1 deficiency) [23] |
| Cytokine storm | • Encephalopathy with diffuse brain swelling Rey-like syndrome, sepsis-like encephalopathy) • Hemorrhagic shock and encephalopathy syndrome (HSES) • Acute necrotizing encephalopathy (ANE) • Non-herpetic limbic encephalitis (NHLE) |
| Excitotoxicity | • Acute encephalopathy with biphasic seizures and late reduced diffusion (AESD) • Acute infantile encephalopathy predominantly affects the frontal lobes (AIEF) • Hemiconvulsion–hemiplegiaepilepsy syndrome (HHE) • Anti-N-methyl-D-aspartate receptor encephalitis |
| Unknown or others | • Mild encephalitis/encephalopathy with a reversible splenial lesion (MERS) • Posterior reversible leukoencephalopathy syndrome (PRES or RPLS) [24] • Febrile infection-related epilepsy syndrome (FIRES) synonym: acute encephalitis with refractory, repetitive partial seizures (AERRPS) • Acute cerebellitis/cerebellopathy [25] • Epileptic encephalopathies with child onset • Acute encephalopathy with a background of genetic abnormalities in the early neonatal period (NEXMIF gene abnormality, Biallelic TBCD Mutations, mutations in ARX genes) [20,26] • Dravet syndrome • Acute encephalopathy associated with congenital adrenal hyperplasia (CAH) • Unclassified encephalopathy |
| Acute Encephalopathy Syndrome | Imaging Characteristics |
|---|---|
| Acute encephalopathy with biphasic seizures and late reduced diffusion (AESD) [29,48,49,50] | a. No abnormal lesion within 2 days. b. Subcortical white matter lesions between days 3 and 9, which are most obvious on DWI (bright tree appearance). The lesions are predominantly frontal or frontoparietal in a location with sparing of the peri-Rolandic region (central sparing). c. After 9 days, the bright tree appearance on DWI has disappeared, and T2WI or FLAIR imaging show high-intensity lesions with central sparing, and cerebral atrophy. d. MRS shows acute Glu elevation (days 1–4), changing to subacute Gln elevation (days 4–12). e. Cranial MRI in the late phase shows cerebral cortical lesions of reduced diffusion, indicating cellular edema of the subcortical white matter. |
| Acute infantile encephalopathy (AIEF) [51] | a. Postictal edematous changes in the white matter and cortex of both frontal lobes. b. Reduced perfusion in the frontal lobes occasionally persisted and was linked with increasing bilateral atrophic alterations and prolonged high signal intensity on T2-weighted images. |
| Acute encephalopathy with febrile convulsive status epilepticus (AEFCSE) | a. Bright tree appearance of subcortical white matter on MRI/DWI with central sulcus being spared. b. Residual atrophy in the frontal region, or frontal and parietal combined region on CT or MR. |
| Acute necrotizing encephalopathy (ANE) [52,53] | a. Concentric structure of the thalamocerebral lesions; diffuse cerebral edema and symmetric and multifocal lesions in the thalamus and other CNS regions, including the posterior limb of the internal capsule, posterior putamen, cerebral and cerebellar deep white matter, and upper brainstem tegmentum. b. The thalamic lesions often show hemorrhagic degeneration and cystic change after 3 days, showing a high signal on T1WI and a low signal on T2WI or T2 star-weighted imaging. |
| Clinically mild encephalitis/encephalopathy with a reversible splenial lesion (MERS) [54,55] | a. MRI-DWI shows abnormal signals in the vast portion of the corpus callosum. b. Abnormal intensities in the splenium on ADC-map, FLAIR, T1, and T2-weighted images. |
| Posterior reversible encephalopathy syndrome (PRES) [24,56] | a. Abnormal lesions in the occipital and parietal lobe areas. b. Extensive signal abnormality in the bilateral cerebellar hemispheres and within the thalami. c. T2-FLAIR images show signal abnormality within the midbrain, pons, and superior cerebellar peduncles. c. Fluid-sensitive MR sequences: parieto-occipital predominant white matter T2 hyperintensities |
| Markers Indicating Poor Outcomes | |
|---|---|
| Acute encephalopathy due to cytokine storm | Serum elevated aspartate aminotransferase Hyperglycemia Serum elevated TNF-α, cytochrome c Serum elevated IL-6, sTNFR1, IL- Serum elevated TIMP-1 Serum elevated soluble CD163 Serum elevated HMGB1 Hematuria or proteinuria |
| Acute encephalopathy due to excitotoxicity | Serum elevated MMP-9 Serum elevated MMP-9/TIMP-1 ratio CSF tau protein CSF S100B CSF VILIP-1 CSF IL-6 hsa-mir-34c hsa-mir-449b hsa-mir-449c serine/threonine kinase 39 gene |
| Anti-seizure medication treatment for status epilepticus (A) Midazolam (0.5 mg/kg) administered via the nasal cavity or cheek mucosa (B) Midazolam (0.15 mg/kg) intravenously (i.v.) administered (up to two doses possible) (C) Between ages 0 and 2 years: intravenous phenobarbital, between 15 and 20 mg/kg (10 min i.v.), ≥2 years: fosphenytoin 22.5 mg/kg (10 min i.v.) (D) Sodium thiopental between 3 and 5 mg/kg (slow i.v.) |
| Brain hypothermia therapy |
| This protocol applies to infants weighing ≥7.5 kg, and aged ≥6 months. |
| Introductory period 1. Status epilepticus/acute encephalopathy admission: ICU (request for admission), contact brainwave dept/radiology (brain and chest CT) 2. Check vital signs, establish a peripheral line 3. Establish central venous line: establish double/triple lumen catheter + arterial line 4. Fluid infusion between 80 and 100 mL/kg/day: under whole-body management, fluid control must not be reduced more than necessary to maintain blood pressure and cerebral circulation. Blood pressure is evaluated using an arterial pressure monitor. Maintenance fluids comprise an acetic acid preparation maintenance fluid and a lactic acid preparation. Vitamins are administered. When theophylline is administered, vitamin B6 is measured (light-shielding blood collection tube: administer vitamin B6 for theophylline-related seizures. Take care not to induce cardiac arrest by sudden administration of B6). 5. Management of blood count, electrolytes, blood sugars, albumin, and clotting value. Ferritin, IL-2R, β2MG, procalcitonin, immune globulin, etc. submitted. 6. Mannitol 3–5 mL/kg × 4–6 times/day (administered over 1 h). 7. Harvest spinal fluid (after the first administration of mannitol). General spinal fluid + various cytokines (IL-6, IL-1β, TNF-α), Tau protein, submitted. Freeze and store the remaining fluid at −80 °C. 8. If possible time-wise, implement MRI (DWI/ADC-map). 9. Intratracheal intubation (if difficult, use muscle relaxant or inhalation anesthetic) 10. Artificial ventilation: PCO2 at 35 to 40 mmHg (do not over-ventilate). PEEP kept slightly low considering brain hypertension. Raise head by 10°. If brain hypertension occurs: request placement of cerebral pressure monitor by a neurosurgeon. 11. Steroid pulse therapy: methylprednisolone 30 mg/kg for >2 h for 3 days, during which heparin or fragmin therapy is continued ≥APTT 1.5. 12. Administer famotidine 0.5 mg/kg twice, or omeprazole. 13. Brain hypothermia therapy: use a whole-body blanket-cooling method to induce target body temperature (direct intestine/bladder temperature of 34.0 to 35.0 °C) within 6 h of onset. If necessary, cool the head or wash the stomach with a normal saline solution while taking care not to cause electrolyte abnormalities, or use chilled fluid infusion. 14. Anti-seizure medication: sodium thiopental, 5–10 mg/kg/h (if this cannot be used, consider midazolam, 0.3 to 0.9 mg/kg/h). 15. Sedation depth should be confirmed by portable electroencephalograph/paperless electroencephalograph (Makin2) as reaching suppression burst within 6 h of beginning therapy. Cooling period 16. Target temperature to be maintained for 48 h (or a maximum of 72 h). Confirm the BIS monitor value at suppression burst (aim for 40 or below) and adjust the sodium thiopental dose administered based on the BIS value as appropriate. Cases achieving a positive sedation depth should have their sodium thiopental dose reduced prior to rewarming at a BIS value between 60 and 70 and at body temperature of 35.0 °C. Caution: If spikes remain with suppression bursts, consider complete suppression (pupils will constrict to mydriasis, and response to light is lost: BIS value of 20 or lower). 17. Use INVOSTM at an appropriate time to check oxygen saturation at the left and right front scalp to evaluate brain circulation. 18. Blood pressure maintenance: appropriate dose of dopamine hydrochloride (5 µg/kg/min = 0.3 mg/kg/h = 0.015 mL/kg/h), manage electrolyte abnormalities and blood glucose. Heart rate will fall to bradycardia with falling body temperature. 19. Administer antibacterial as appropriate. In applicable conditions, cerebroprotective edaravone, sivelestat Na as a neutrophil elastase inhibitor, and acyclovir. Rewarming period 20. Rewarming is implemented at a pace of 0.5 °C per 12 h. Care should be taken to avoid pneumonia in line with increased sputum secretions. Aim to remove the patient from artificial respiration on the fifth to seventh day. For cases in which laryngitis is likely, intravenous dexamethasone or epinephrine should be administered prior to removal of the tube. 21. For cases in which critical complications are envisaged, TRH therapy should be initiated at an early stage. 22. Including rehabilitation, aim to discharge the patient one month after onset. 23. Prior to discharge, evaluate brain waves, implement neuroradiological images/nuclear medicine tests and assess development. Where necessary, anti-seizure mediation should be periodically administered for preventative purposes. |
| Subtype Description | Incidence in Japan | Clinical Manifestations | Diagnosis | Treatment |
|---|---|---|---|---|
| ADEM [2,77] | ||||
| An immune-mediated inflammatory demyelinating condition that predominately affects the white matter of the brain and spinal cord. | 0.40 per 100,000 | Polyfocal neurologic deficits and is typically self-limiting. | Based on clinical features and findings on neuroimaging and laboratory investigations. ADEM lacks a specific identified biological marker rendering a reliable laboratory diagnosis, long-term follow-up is important as there are instances where an illness initially diagnosed as ADEM is ultimately replaced with a diagnosis of MS | Combination of intravenous corticosteroids and IVIG, (2) cyclosporin, (3) cyclophosphamide, or (4) plasma exchange/plasmapheresis |
| AESD [78,79] | ||||
| Biphasic seizure and altered consciousness during the acute phase, followed by restricted diffusion in bilateral cerebral parenchyma on MRI during the subacute stage | Incidence is higher in Asian countries, especially Japan, and the genetic background may be a possible higher incidence | A prolonged febrile seizure is the first symptom. Brief seizures may be present in mild cases. Involuntary movements may act prognostic factor AESD prediction score by Tada et al. [80] Level of consciousness 12–24 h after seizure GCS 14–13 (JCS 1–3) and GCS 12–9 (JCS 10–30) were scored 2, GCS 8–3 (JCS 100–300) scored 3, age below 1.5 years scored 1, duration of ES above 40 min scored 1, mechanical intubation scored 1, AST on admission above 40 mEq/l scored 1, blood glucose on admission above 200 mg/dl scored 1, and Cr on admission above 0.35 scored 1 AESD prediction score by Yokochi et al. [81] pH < 7.014 1 point ALT ≥ 28 2 points Glu ≥ 228 2 points Time to awakening ≥ 11 h 2 points Cre ≥ 0.3 1 point Ammonia ≥ 125 2 points 4 points or more: High-risk group for AESD for both scores [77,78] | High signal intensity on DWI for the anterior area Severe acidosis is the most common lab finding. EEG may usefully differentiate AESD from PFS | Cyclosporine, methylprednisolone pulse therapy, intravenous immunoglobulin, and other therapies that suppress inflammatory cytokines. TTM may be considered but need to differentiate AESD from FSE at the early acute stage. TTM during the prolonged convulsive phase, prior to the diagnosis of AESD, could prevent the patient from developing a second phase of convulsions and thus prevent the patient from developing AESD. |
| AEFCSE | ||||
| Develops with prolonged febrile convulsion, followed by mild unconsciousness, then subsequently provoking a cluster of convulsions (late seizures) with a comatose state. | Incidence is higher in Asian countries, especially Japan | Pyrexia followed by partial seizures and subsequently hemiconvulsions. Transient neurological symptoms and Intellectual disability, attention deficit | EEG findings showed slow waves predominantly on the right hemisphere and a high signal in the subcortical white matter as with the cerebral lobar distribution pattern. Acute infantile encephalopathy predominantly affects the frontal lobes (AIEF). Thermolabile genotype of CPT II variations consisting of three single nucleotide polymorphisms in exons 4 (1055T > G/F352C and 1102G > A/V368I) and 5 (1939A > G/M647V) | TTM |
| ANE [82,83,84] | ||||
| Multiple bilateral brain lesions, primarily involving the thalami, but also involving the putamina, internal and external capsules, cerebellar white matter, and the brainstem tegmentum. | Many cases have been reported in Asia as well as in a number of Western countries. | Dramatic neurological deficits/symptoms. Neurological deficits may be preceded by a viral prodrome. | Associated thalamic, putamina, cerebral, cerebellar, and brainstem abnormalities are hypodense on CT. Bilateral symmetrical thalamic involvement. Abnormal signals on MRI are hypointense on T1 and hyperintense on T2. Restricted diffusion of the involved regions. ANE of childhood can be distinguished from ADEM clinically by the onset of encephalitic features shortly after the prodromal illness, whereas in ADEM, they may take 1 to 2 weeks to develop. | Immunomodulatory therapy such as corticosteroids or intravenous immunoglobulin is often used. TTM has also been used. TTM is critical to the outcome of children with ANE, especially if started within 12 h of onset. |
| MERS [85,86,87] | ||||
| An infection-associated encephalitis/encephalopathy syndrome that is predominately caused by a virus or a variety of pathological conditions, including MIS-C. There have been scattered reports of MERS associated with AFBN and Kawasaki disease. AFBN + MERS, a urinary tract infection in children, is particularly noteworthy | Fever, headache, neck rigidity, and Kerning sign (+) | serum VCA IgG (+), EBNA-1 IgG (−), EBV IgM (−), and inflammation in the analysis of CSF Cranial MRI+C showed that the blood vessels on the surface of the brain were increasing and thickening, and diffuse slow waves were detected on the EEG The high-signal intensity in the splenium of the corpus callosum on T2W Splenial hyperintensity as a “boomerang sign” on DWI and reduced diffusion on ADC | Supplementation, steroids and IVIG, acyclovir, and prescribed oral sodium, but some cases improve with the natural course of the disease. | |
| PRES [56] | ||||
| A clinical–radiological syndrome characterized by a headache, seizures, altered mental status, visual loss and white matter vasogenic edema affecting the posterior occipital and parietal lobes of the brain predominantly. | Visual disturbance can vary from blurred vision and homonymous hemianopsia to cortical blindness. Altered consciousness may vary from mild confusion or agitation to coma. | The bilateral occipital, parietal, frontal cortex, and subcortical white matter T2/fluid-attenuated inversion recovery hyperintensities | Intravenous fluids, antibiotics, antiepileptics | |
| FIRES [88,89] | ||||
| A subtype of NORSE that requires a prior febrile infection between 2 weeks and 24 h before the onset of refractory status epilepticus with or without fever at the onset of status epilepticus. | Very rare, affects approximately 1 in a million children | Focal seizures with impaired awareness and bilateral tonic–clonic seizures. Seizures progress to continuous or nearly continuous seizures. | EEG Leptomeningeal enhancement, bilateral claustrum hyperintensity or progressive mesial temporal lobe atrophy on MRI. | Benzodiazepines, Barbiturates, Ketamine, Lidocaine, Magnesium, Ketogenic diet, Cannabidiol |
| Score | Category | Description |
|---|---|---|
| 1 | Normal | At an age-appropriate level; school-age children attend regular school |
| 2 | Mild disability | Conscious, alert, able to interact at age-appropriate level; regular school, but grades perhaps not age-appropriate, possibility of mild neurologic deficit |
| 3 | Moderate disability | Conscious, age-appropriate independent activities of daily life; special education classroom and/or learning deficit present |
| 4 | Severe disability | Conscious, dependent on others for daily support because of impaired brain function |
| 5 | Coma or vegetative state | Any degree of coma, unaware, even if awake in appearance, without interaction with the environment; no evidence of cortex function; possibility for some reflexive response, spontaneous eye-opening, sleep–wake cycles |
| 6 | Brain death/death | Brain death, death |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Imataka, G.; Kuwashima, S.; Yoshihara, S. A Comprehensive Review of Pediatric Acute Encephalopathy. J. Clin. Med. 2022, 11, 5921. https://doi.org/10.3390/jcm11195921
Imataka G, Kuwashima S, Yoshihara S. A Comprehensive Review of Pediatric Acute Encephalopathy. Journal of Clinical Medicine. 2022; 11(19):5921. https://doi.org/10.3390/jcm11195921
Chicago/Turabian StyleImataka, George, Shigeko Kuwashima, and Shigemi Yoshihara. 2022. "A Comprehensive Review of Pediatric Acute Encephalopathy" Journal of Clinical Medicine 11, no. 19: 5921. https://doi.org/10.3390/jcm11195921
APA StyleImataka, G., Kuwashima, S., & Yoshihara, S. (2022). A Comprehensive Review of Pediatric Acute Encephalopathy. Journal of Clinical Medicine, 11(19), 5921. https://doi.org/10.3390/jcm11195921