
Assessment of Myocardial Diastolic Dysfunction as a Result of Myocardial Infarction and Extracellular Matrix Regulation Disorders in the Context of Mesenchymal Stem Cell Therapy

 , , , , ,
, , , , , 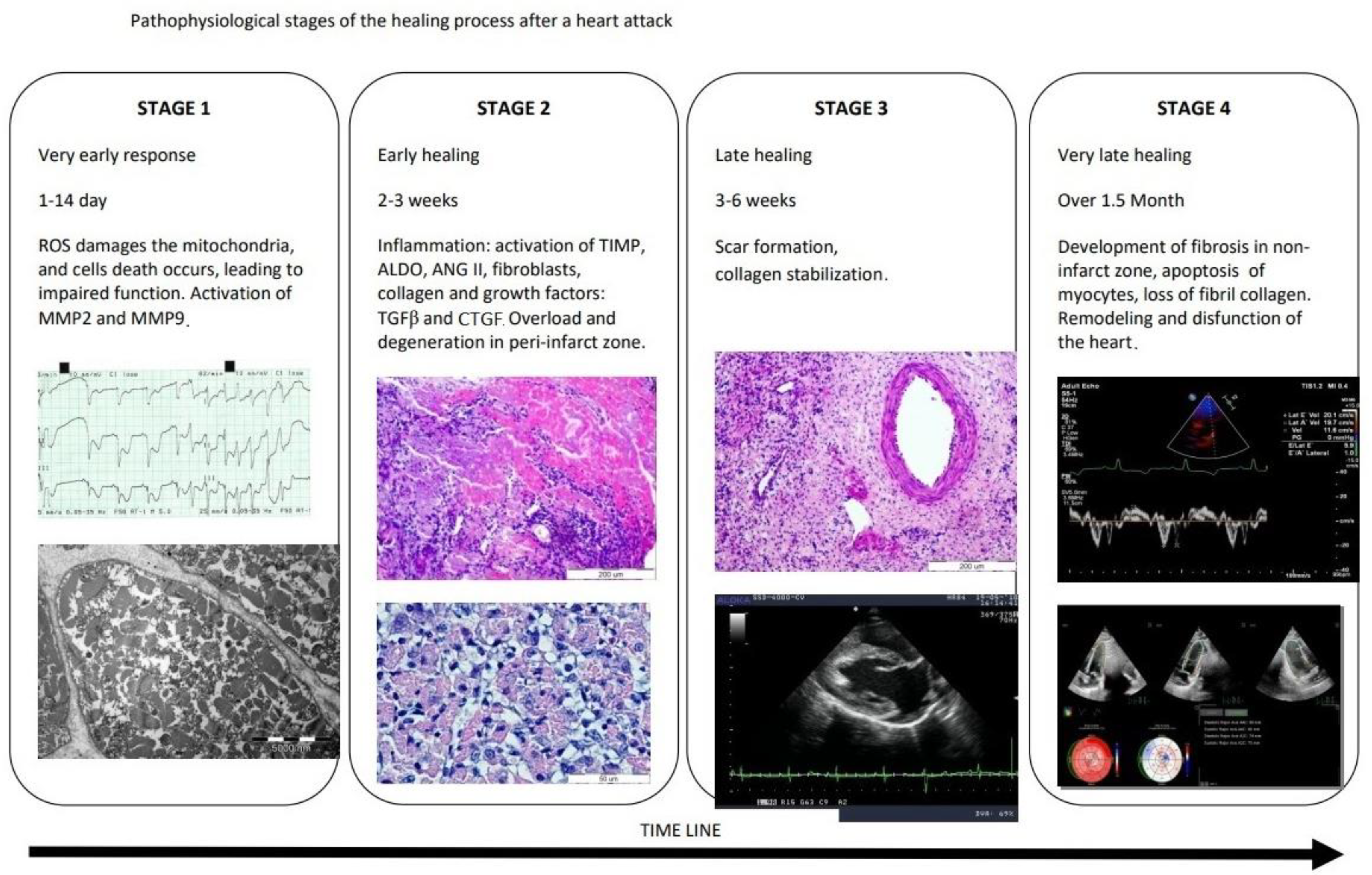{kind=link}
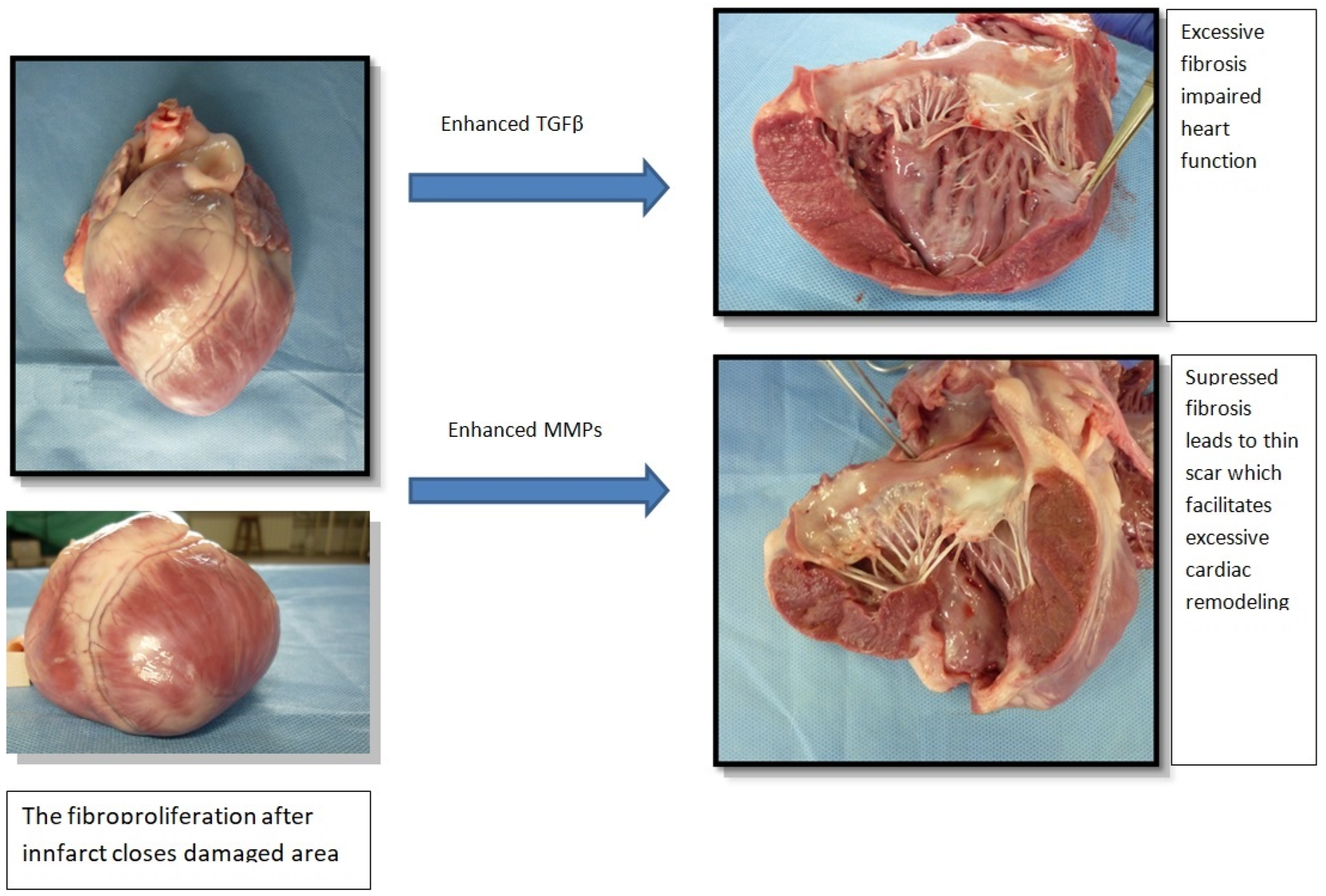{kind=link}
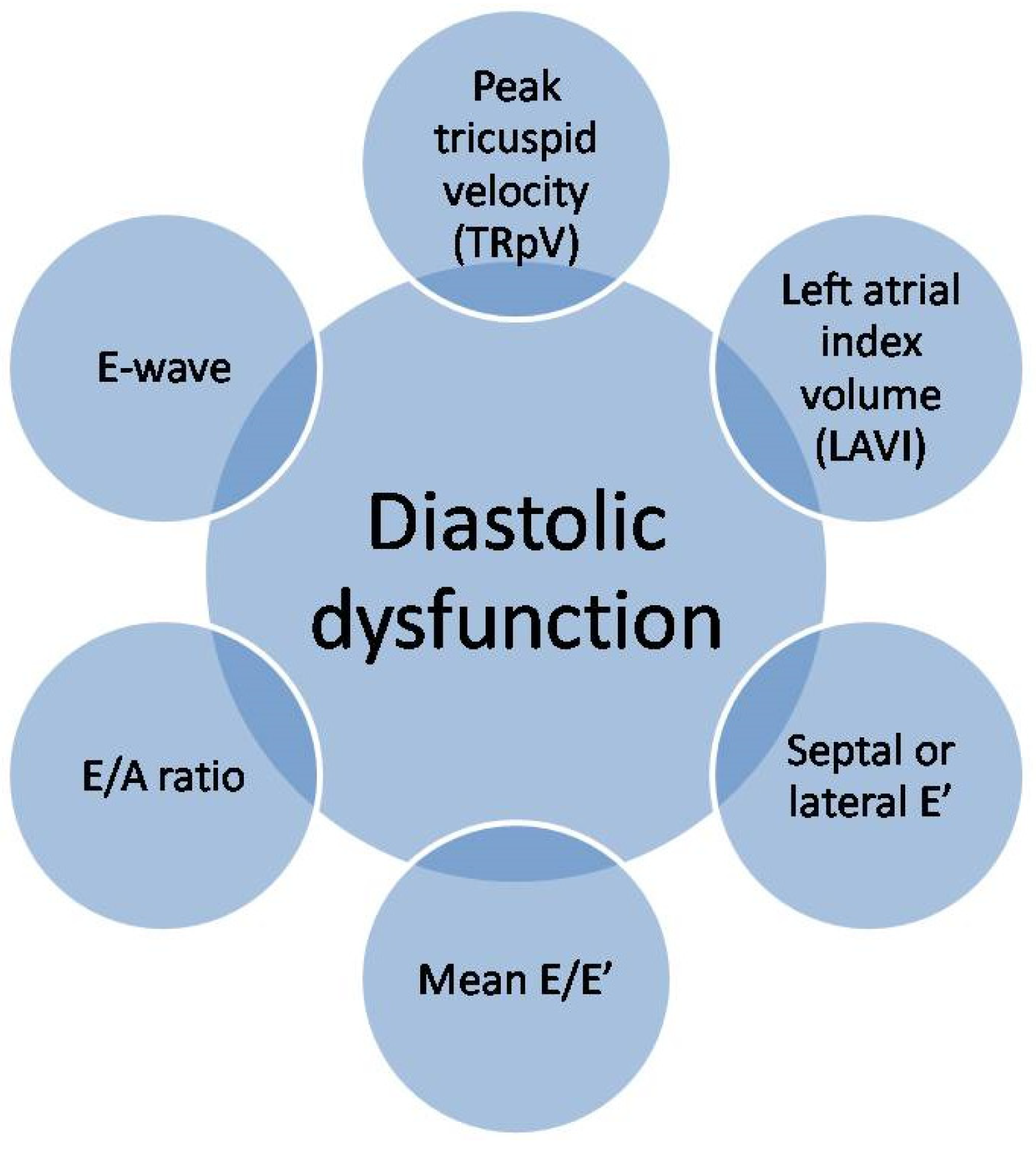{kind=link}
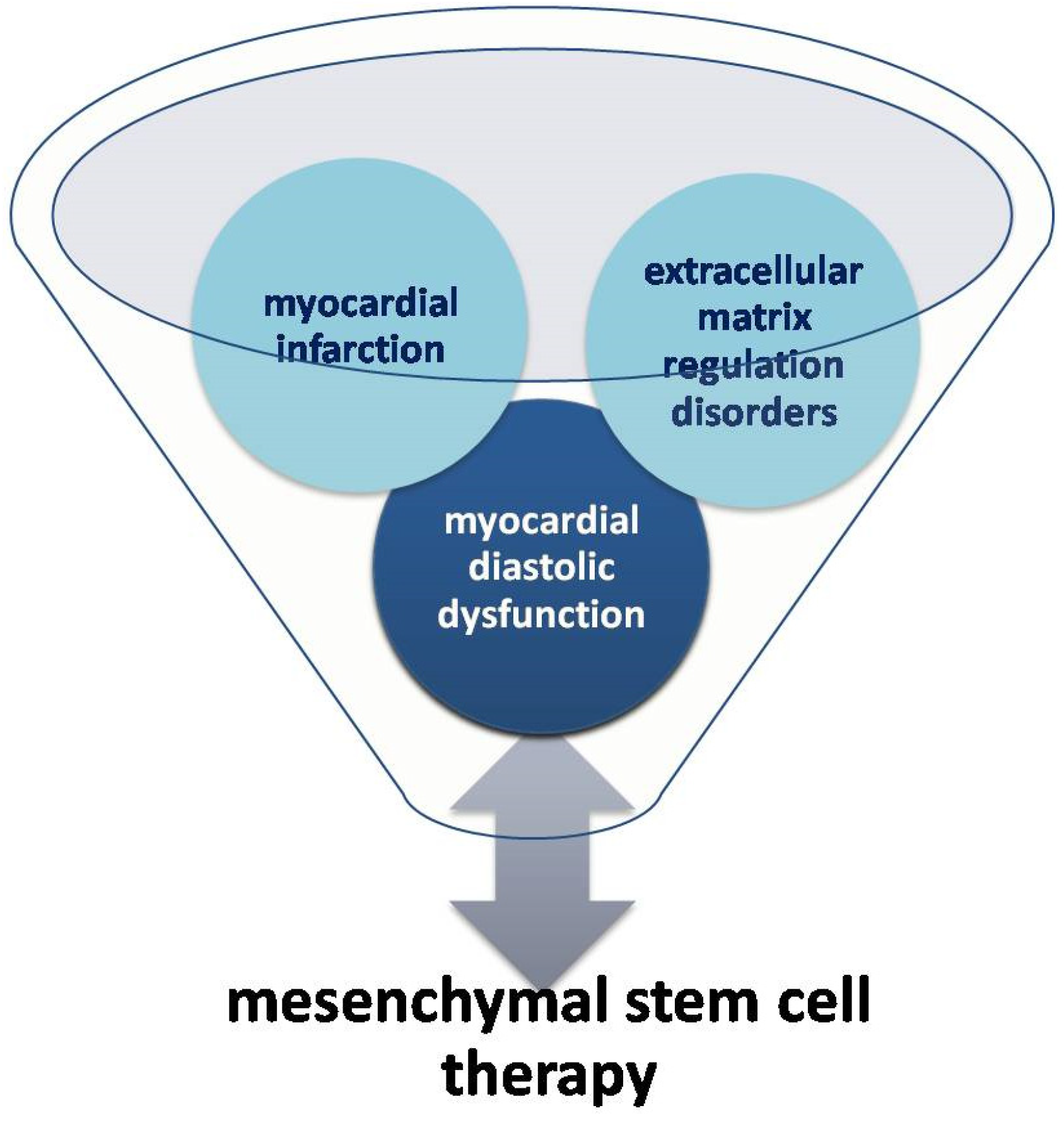{kind=link}
Abstract
1. Introduction
1.1. Mechanisms Underlying Diastolic Dysfunction after MI
1.2. Diagnosis of Diastolic Dysfunction
1.3. Stem Cell Therapy
1.4. Impact of Stem Cell Therapy on Diastolic Function of the Heart
1.5. Influence of Stem Cell Therapy on Cardiac Fibrosis
1.6. Application of Fibroblasts in Stem Cell Therapy
1.7. Post-Infarction Treatment of Diastolic Dysfunction
1.8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Cardiovascular Diseases (CVDs). Available online: http://www.who.int/mediacentre/factsheets/fs317/en/ (accessed on 1 May 2017).
- Budaj, A.; Beręsewicz, A. Ischemic Heart Disease; Interna Szczeklika Wyd. 10.; Gajewski, P., Szczeklik, A., Eds.; MedycynaPraktyczna: Kraków, Poland, 2019; pp. 168–226. ISBN 9788374305686. [Google Scholar]
- Gyöngyösi, M.; Winkler, J.; Ramos, I.; Do, Q.T.; Firat, H.; McDonald, K.; González, A.; Thum, T.; Díez, J.; Jaisser, F.; et al. Myocardial fibrosis: Biomedical research from bench to bedside. Eur. J. Heart Fail. 2019, 19, 177–191. [Google Scholar] [CrossRef]
- Weber, K.T.; Sun, Y.; Bhattacharya, S.K.; Ahokas, R.A.; Gerling, I.C. Myofibroblast- mediated mechanisms of pathological remodelling of the heart. Nat. Rev. Cardiol. 2013, 10, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Jugdutt, B.I. Remodeling of the myocardium and potential targets in the collagen degradation and synthesis pathways. Curr. Drug Targets Cardiovasc. Haematol. Disord. 2003, 3, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Weber, K.T.; Anversa, P.; Armstrong, P.W.; Brilla, C.G.; Burnett, J.C.; Cruickshank, J.M.; Devereux, R.B.; Giles, T.D.; Korsgaard, N.; Leier, C.V.; et al. Remodeling and reparation of the cardiovascular system. J. Am. Coll. Cardiol. 1992, 20, 3–16. [Google Scholar] [CrossRef]
- Jugdutt, B.I.; Joljart, M.J.; Khan, M.I. Rate of collagen deposition during healing after myocardial infarction in the rat and dog models: Mechanistic insights into ventricular remodeling. Circulation 1996, 94, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Weber, K.T.; Pick, R.; Jalil, J.E.; Janicki, J.S.; Carroll, E.P. Patterns of myocardial fibrosis. J. Mol. Cell Cardiol. 1989, 21, 121–131. [Google Scholar] [CrossRef]
- Karolko, B.; Przewłocka-Kosmala, M. Fibrosis markers in heart failure. Folia Cardiol. 2017, 12, 245–253. [Google Scholar]
- Kong, P.; Christia, P.; Frangogiannis, N. The pathogenesis of cardiac fibrosis. Cell Mol. Life Sci. 2014, 71, 549–574. [Google Scholar] [CrossRef]
- Li, A.H.; Liu, P.P.; Villarreal, F.J.; Garcia, R.A. Dynamic changes in myocardial matrix and relevance to disease: Translational perspectives. Circ. Res. 2014, 114, 916–927. [Google Scholar] [CrossRef]
- Olivetti, G.; Capasso, J.M.; Sonnenblick, E.H.; Anversa, P. Side-to-side slippage of myocytes participates in ventricular wall remodeling acutely after myocardial infarction in rats. Circ. Res. 1990, 67, 23–34. [Google Scholar] [CrossRef]
- Alberts, B.; Bray, D.; Lewis, J.; Raff, M.; Roberts, K.; Watson, J.D. Molecular Biology of the Cell, 3rd ed.; Garland Publishing: New York, NY, USA, 1994. [Google Scholar]
- Phillips, C.; Wenstrup, R.J. Biosynthetic and genetic disorders of collagen. In Wound Healing: Biochemical and Clinical Aspects; Cohen, I.K., Diegelmann, R.F., Lindblad, W.J., Eds.; WB Saunders Co.: Philadelphia, PA, USA, 1992; pp. 152–176. [Google Scholar]
- Factor, S.M.; Robinson, T.F.; Dominitz, R.; Cho, S.H. Alterations of the myocardial skeletal framework in acute myocardial infarction with and without ventricular rupture. Am. J. Cardiovasc. Pathol. 1987, 1, 91–97. [Google Scholar] [PubMed]
- Shoulders, M.D.; Raines, R.T. Collagenstructure and stability. Annu. Rev. Biochem. 2009, 78, 929–958. [Google Scholar] [CrossRef] [PubMed]
- Chimenti, C.; Kajstura, J.; Torella, D.; Urbanek, K.; Heleniak, H.; Colussi, C.; Di Meglio, F.; Nadal-Ginard, B.; Frustaci, A.; Leri, A.; et al. Senescence and death of primitive cells and myocytes lead to premature cardiac aging and heart failure. Circ. Res. 2003, 93, 604–613. [Google Scholar] [CrossRef] [PubMed]
- Camelliti, P.; Borg, T.K.; Kohl, P. Structural and functional characterisation of cardiac fibroblasts. Cardiovasc. Res. 2005, 65, 40–51. [Google Scholar] [CrossRef]
- Ceauşu, Z.; Popa, M.; Socea, B.; Gorecki, G.P.; Costache, M.; Ceauşu, M. Influence of the microenvironment dynamics on extracellular matrix evolution under hypoxic ischemic conditionns in the myocardium. Exp. Ther. Med. 2022, 23, 199. [Google Scholar] [CrossRef]
- Etoh, T.; Joffs, C.; Deschamps, A.M.; Davis, J.; Dowdy, K.; Hendrick, J.; Baicu, S.; Mukherjee, R.; Manhaini, M.; Spinale, F.G. Myocardial and interstitial matrix metalloproteinase activity after acute myocardial infarction in pigs. Am. J. Physiol. 2001, 281, H987–H994. [Google Scholar] [CrossRef]
- Chen, B.; Frangogiannis, N.G. The role of macrophages in nonischemic heart failure. JACC Basic Transl. Sci. 2018, 3, 245–248. [Google Scholar] [CrossRef]
- Murphy-Ullrich, J.E.; Sage, E.H. Revisiting the matricellular concept. Matrix Biol. 2014, 37, 1–14. [Google Scholar] [CrossRef]
- Dewald, O.; Ren, G.; Duerr, G.D.; Zoerlein, M.; Klemm, C.; Gersch, C.; Tincey, S.; Michael, L.H.; Entman, M.L.; Frangogiannis, N. Of mice and dogs: Species-specific differences in the inflammatory response following myocardial infarction. Am. J. Pathol. 2004, 164, 665–677. [Google Scholar] [CrossRef]
- Frangogiannis, N. The extracellular matrix in ischemic and nonischemicheart failure. Circ. Res. 2019, 125, 117–146. [Google Scholar] [CrossRef]
- Fu, X.; Khalil, H.; Kanisicak, O.; Boyer, J.G.; Vagnozzi, R.J.; Maliken, B.D.; Sargent, M.A.; Prasad, V.; Valiente-Alandi, I. Specialized fibroblast differentiated states underlie scar formation in the infarcted mouse heart. J. Clin. Investig. 2018, 128, 2127–2143. [Google Scholar] [CrossRef] [PubMed]
- Sager, H.B.; Hulsmans, M.; Lavine, K.J.; Moreira, M.B.; Heidt, T.; Courties, G.; Sun, Y.; Iwamoto, Y.; Tricot, B.; Khan, O.F.; et al. Proliferation and Recruitment Contribute to Myocardial Macrophage Expansion in Chronic Heart Failure. Circ. Res. 2016, 119, 853–864. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, D.; Sigusch, H.H.; Hensse, J.; Tyagi, S.C.; Körfer, R.; Figulla, H.R. Cardiac remodelling in end stage heart failure: Upregulation of matrix metalloproteinase (MMP) irrespective of the underlying disease, and evidence for a direct inhibitory effect of ACE inhibitors on MMP. Heart 2002, 88, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G.; Shimoni, S.; Chang, S.M.; Ren, G.; Dewald, O.; Gersch, C.; Shan, K.; Aggeli, C.; Reardon, M.; Letsou, G.V.; et al. Active interstitial remodeling: An important process in the hibernating human myocardium. J. Am. Coll. Cardiol. 2002, 39, 1468–1474. [Google Scholar] [CrossRef]
- Jugdutt, B.I. Prevention of ventricular remodelling post myocardial infarction: Timing and duration of therapy. Can. J. Cardiol. 1993, 9, 103–114. [Google Scholar]
- Mann, D.L. Inflammatory mediators and the failing heart: Past, present, and the foreseeable future. Circ. Res. 2002, 91, 988–998. [Google Scholar] [CrossRef]
- Duran, J.M.; Taghavi, S.; Berretta, R.M.; Makarewich, C.A.; Iii, T.S.; Starosta, T.; Udeshi, F.; George, J.C.; Kubo, H.; Houser, S.R. A characterization and targeting of the infarct border zone in a swine model of myocardial infarction. Clin. Transl. Sci. 2012, 5, 416–421. [Google Scholar] [CrossRef]
- Latini, R.; Maggioni, A.P.; Flather, M.; Sleight, P.; Tognoni, G. ACE inhibitor use in patients with myocardial infarction: Summary of evidence from clinical trials. Circulation 1995, 92, 3132–3137. [Google Scholar] [CrossRef]
- Gladden, J.D.; Linke, W.A.; Redfield, M.M. Heart failure with preserved ejection fraction. Pflug. Arch. Eur. J. Physiol. 2014, 466, 1037–1053. [Google Scholar] [CrossRef]
- Velagaleti, R.S.; Pencina, M.J.; Murabito, J.; Wang, T.; Parikh, N.I.; D’Agostino, R.B.; Levy, D.; Kannel, W.B.; Vasan, R.S. Long-term trends in the incidence of heart failure after myocardial infarction. Circulation 2008, 118, 2057–2262. [Google Scholar] [CrossRef]
- López, B.; González, A.; Ravassa, S.; Beaumont, J.; Moreno, M.U.; San José, G.; Querejeta, R.; Díez, J. Circulating biomarkers of myocardial fibrosis: The need for a reappraisal. J. Am. Coll. Cardiol. 2015, 65, 2449–2456. [Google Scholar] [CrossRef] [PubMed]
- Kossaify, A.; Nasr, M. Diastolic Dysfunction and the New Recommendations for Echocardiographic Assessment of Left Ventricular Diastolic Function: Summary of Guidelines and Novelties in Diagnosis and Grading. J. Diagn. Med. Sonogr. 2019, 35, 317–325. [Google Scholar] [CrossRef]
- Nagueh, S.F.; Smiseth, O.A.; Appleton, C.P. Recommendations for the evaluation of left ventricular diastolic function by echocardiography: An update from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. Eur. Heart J. Cardiovasc. Imaging 2016, 17, 1321–1360. [Google Scholar] [CrossRef]
- Nagueh, S.F.; Appleton, C.P.; Gillebert, T.C. Recommendations for the evaluation of left ventricular diastolic function by echocardiography. J. Am. Soc. Echocardiogr. 2009, 22, 107–133. [Google Scholar] [CrossRef] [PubMed]
- Shintani, S.; Murohara, T.; Ikeda, H.; Ueno, T.; Honma, T.; Katoh, A.; Sasaki, K.-I.; Shimada, T.; Oike, Y.; Imaizumi, T. Mobilization of endothelial progenitor cells in patients with acute myocardial infarction. Circulation 2001, 103, 2776–2779. [Google Scholar] [CrossRef]
- Huang, C.; Zhang, X.; Ramil, J.M.; Rikka, S.; Kim, L.; Lee, Y.; Gude, N.A.; Thistlethwaite, P.A.; Sussman, M.A.; Gottlieb, R.A.; et al. Juvenile exposure to anthracyclines impairs cardiac progenitor cell function and vascularisation resulting in greater susceptibility to stress-induced myocardial injury in adult mice. Circulation 2010, 121, 675–683. [Google Scholar] [CrossRef]
- Wojakowski, W.; Tendera, M.; Cybulski, W.; Zuba-Surma, E.K.; Szade, K.; Florczyk, U.; Kozakowska, M.; Szymula, A.; Krzych, Ł.; Paslawska, U.; et al. Effects of intracoronary delivery of allogenic bone marrow-derived stem cells expressing heme oxygenase-1 on the reperfusion injury in experimental myocardial infarction. Thromb. Hemost. 2012, 108, 464–475. [Google Scholar]
- Hatzistergos, K.E.; Takeuchi, L.M.; Saur, D.; Seidler, B.; Dymecki, S.M.; Mai, J.J.; White, I.A.; Balkan, W.; Kanashiro-Takeuchi, R.M.; Schally, A.V.; et al. cKit+ cardiac progenitors of neural crest origin. Proc. Natl. Acad. Sci. USA 2015, 112, 13051–13056. [Google Scholar] [CrossRef]
- Kim, M.C.; Kim, Y.S.; Kang, W.S.; Lee, K.H.; Cho, M.; Hong, M.H.; Lim, K.S.; Jeong, M.H.; Ahn, Y. Intramyocardial Injection of Stem Cells in Pig Myocardial Infarction Model: The First Trial in Korea. J. Korean Med. Sci. 2017, 32, 1708–1712. [Google Scholar] [CrossRef]
- Gómez-Heras, S.G.; Largo, C.; Larrea, J.L.; Vega-Clemente, L.; Flores, M.C.; Ruiz-Pérez, D.; García-Olmo, D.; García-Arranz, M. Main histological parameters to be evaluated in an experimental model of myocardial infarcttreated by stem cells on pigs. Peer J. 2019, 7, e7160. [Google Scholar] [CrossRef]
- Sopko, N.A.; Turturice, B.A.; Becker, M.E.; Brown, C.R.; Dong, F.; Popović, Z.B.; Penn, M.S. Bone marrow support of the heart in pressure overload is lost with aging. PLoS ONE 2010, 5, e15187. [Google Scholar] [CrossRef] [PubMed]
- Dimmler, S.; Zeiher, A.M. Wanted! The best cel for cardiac regeneration. Editorial comment. JACC 2004, 44, 2. [Google Scholar]
- Querejeta, R.; López, B.; González, A.; Sánchez, E.; Larman, M.; MartínezUbago, J.L.; Díez, J. Increased collagen type I synthesis in patients with heart failure of hypertensive origin: Relation to myocardialfibrosis. Circulation 2004, 110, 1263–1268. [Google Scholar] [CrossRef] [PubMed]
- Roberts, D.D.; Kaur, S.; Isenberg, J.S. Regulation of Cellular Redox Signaling by Matricellular Proteins in Vascular Biology, Immunology, and Cancer. Antioxid. Redox Signal. 2017, 27, 874–911. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.-N.; Cores, J.; Huang, K.; Cui, X.-L.; Luo, L.; Zhang, J.-Y.; Li, T.-S.; Qian, L.; Cheng, K. Concise Review: Is Cardiac Cell Therapy Dead? Embarrassing Trial Outcomes and New Directions for the Future. Stem. Cells Transl. Med. 2018, 7, 354–359. [Google Scholar] [CrossRef]
- Gnecchi, M.; Zhang, Z.; Ni, A.; Dzau, V.J. Paracrine mechanisms in adult stem cell signaling and therapy. Circ. Res. 2008, 103, 1204–1219. [Google Scholar] [CrossRef]
- Chimenti, I.; Smith, R.R.; Li, T.-S.; Gerstenblith, G.; Messina, E.; Giacomello, A.; Marbán, E. Relative roles of direct regeneration versus paracrine effects of human cardiosphere-derived cells transplanted into infarcted mice. Circ. Res. 2010, 106, 971–980. [Google Scholar] [CrossRef]
- Rota, M.; Padin-Iruegas, M.E.; Misao, Y.; De Angelis, A.; Maestroni, S.; Ferreira-Martins, J.; Fiumana, E.; Rastaldo, R.; Arcarese, M.L.; Mitchell, T.S.; et al. Local activation or implantation of cardiac progenitor cells rescues scarred infracted myocardium improving cardiac function. Circ. Res. 2008, 103, 107–116. [Google Scholar] [CrossRef]
- Lefer, D.J.; Marbán, E. Is Cardioprotection Dead? Circulation 2017, 136, 98–109. [Google Scholar] [CrossRef]
- Tang, X.-L.; Rokosh, G.; Sanganalmath, S.K.; Yuan, F.; Sato, H.; Mu, J.; Dai, S.; Li, C.; Chen, N.; Peng, Y.; et al. Intracoronary administration of cardiac progenitor cells alleviates left ventricular dysfunction in rats with a 30-day-old infarction. Circulation 2010, 121, 293–305. [Google Scholar] [CrossRef]
- Klarenbosch, B.R.; Chamuleau, S.A.J.; Teske, A.J. Deformation imaging to assess global and regional effects of cardiac regenerative therapy in ischaemic heart disease: A systematic review. J. Tissue Eng. Regen. Med. 2019, 13, 1872–1882. [Google Scholar] [CrossRef] [PubMed]
- Abushouk, A.I.; Salem, A.M.; Saad, A.; Afifi, A.M.; Afify, A.Y.; Afify, H.; Salem, H.S.; Ghanem, E.; Abdel-Daim, M.M. Mesenchymal Stem Cell Therapy for Doxorubicin-Induced Cardiomyopathy: Potential Mechanisms, Governing Factors, and Implications of the Heart Stem Cell Debate. Front. Pharmacol. 2019, 10, 635. [Google Scholar] [CrossRef]
- Traverse, J.H.; Henry, T.D.; Ellis, S.G.; Pepine, C.J.; Willerson, J.T.; Zhao, D.X.M.; Forder, J.R.; Byrne, B.J.; Hatzopoulos, A.K.; Penn, M.S.; et al. Effect of intracoronary delivery of autologous bone marrow mononuclear cells 2 to 3 weeks following acute myocardial infarction on left ventricular function: The late TIME randomized trial. JAMA 2011, 306, 2110–2119. [Google Scholar] [CrossRef] [PubMed]
- Perin, E.C.; Willerson, J.T.; Pepine, C.J.; Henry, T.D.; Ellis, S.G.; Zhao, D.X.M.; Silva, G.V.; Lai, D.; Thomas, J.D.; Kronenberg, M.W.; et al. Effect of transendocardial delivery of autologous bone marrow mononuclear cells on functional capacity, left ventricular function, and perfusion in chronic heart failure: The FOCUS-CCTRN trial. JAMA 2012, 307, 1717–1726. [Google Scholar] [CrossRef] [PubMed]
- Traverse, J.H.; Henry, T.D.; Pepine, C.J.; Willerson, J.T.; Zhao, D.X.M.; Ellis, S.G.; Forder, J.R.; Anderson, R.D.; Hatzopoulos, A.K.; Penn, M.S.; et al. Effect of the use and timing of bone marrow mononuclear cel delivery on left ventricular function after acute myocardial infarction: The tIME randomized trial. JAMA 2012, 308, 2380–2389. [Google Scholar] [CrossRef]
- Sürder, D.; Manka, R.; Cicero, V.L.; Moccetti, T.; Rufibach, K.; Soncin, S.; Turchetto, L.; Radrizzani, M.; Astori, G.; Schwitter, J.; et al. Intracoronary injection of bone marrow-derived mononuclear cells early or late after acute myocardial infarction. Circulation 2016, 127, 1968–1979. [Google Scholar] [CrossRef]
- Choudry, F.; Hamshere, S.; Saunders, N.; Veerapen, J.; Bavnbek, K.; Knight, C.; Pellerin, D.; Locca, D.; Westwood, M.; Rakhit, R.; et al. A randomized double-blind control study of early intra-coronary autologous bone marrow cell infusion in acute myocardial infarction: The REGENERATE-AMI clinical trial†. Eur. Heart J. 2016, 37, 256–263. [Google Scholar] [CrossRef]
- Quyyumi, A.A.; Vasquez, A.; Kereiakes, D.J.; Klapholz, M.; Schaer, G.L.; Abdel-Latif, A.; Frohwein, S.; Henry, T.D.; Schatz, R.A.; Dib, N.; et al. PreSERVE-AMI: A randomized, double-blind, placebocontrolled clinical trial of intracoronary administration of autologous CD34+ cells in patients with left ventricular dysfunction post STEMI. Circ. Res. 2017, 120, 324–331. [Google Scholar] [CrossRef] [PubMed]
- Wollert, K.C.; Meyer, G.P.; Müller-Ehmsen, J.; Tschöpe, C.; Bonarjee, V.; Larsen, A.I.; May, A.E.; Empen, K.; Chorianopoulos, E.; Tebbe, U.; et al. Intracoronary autologous bone marrow cell transfer after myocardial infarction: The BOOST-2 randomised placebo-controlled clinical trial. Eur. Heart J. 2017, 38, 2936–2943. [Google Scholar] [CrossRef]
- Fernández-Avilés, F.; Sanz-Ruiz, R.; Bogaert, J.; Plasencia, A.C.; Gilaberte, I.; Belmans, A.; Santos, M.E.F.; Charron, D.; Mulet, M.; Yotti, R.; et al. Safety and efficacy of intracoronary infusion of allogeneic human cardiac stem cells in patients with ST-segment elevation myocardial infarction and left ventricular dysfunction. Circ. Res. 2016, 123, 579–589. [Google Scholar] [CrossRef]
- Nicolau, J.C.; Furtado, R.H.; Silva, S.A.; Rochitte, C.E.; Rassi, A.; Moraes, J.B., Jr.; Quintella, E.; Costantini, C.R.; Korman, A.P.; Mattos, M.A.; et al. Stem-cell therapy in ST-segment elevation myocardial infarction with reduced ejection fraction: A multicenter, double-blind randomized trial. Clin. Cardiol. 2018, 41, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Bolli, R.; Mitrani, R.D.; Hare, J.M.; Pepine, C.J.; Perin, E.C.; Willerson, J.T.; Traverse, J.H.; Henry, T.D.; Yang, P.C.; Murphy, M.P.; et al. Cardiovascular Cell Therapy Research Network (CCTRN). Phase II study of autologous mesenchymal stromal cells and c-kit positive cardiac cells, singly or in combination, in patients with ischemic heart failure: The CCTRN CONCERT-HF study. Heart Fail. Eur. J. 2021, 23, 661–674. [Google Scholar] [CrossRef] [PubMed]
- Vagnozzi, R.J.; Maillet, M.; Sargent, M.A.; Khalil, H.; Johansen, A.K.; Schwanekamp, J.A.; York, A.J.; Huang, V.; Nahrendorf, M.; Sadayappan, S.; et al. An acute immune response underlies the benefits of heart stem cell therapy. Nature 2020, 577, 405–409. [Google Scholar] [CrossRef]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef]
- Huangfu, D.; Maehr, R.; Guo, W.; Eijkelenboom, A.; Snitow, M.; Chen, A.E.; Melton, D.A. Induction of pluripotent stem cells by defined factors is greatly improved by small-molecule compounds. Nat. Biotechnol. 2008, 26, 795–797. [Google Scholar] [CrossRef]
- Nakagawa, M.; Koyanagi, M.; Tanabe, K.; Takahashi, K.; Ichisaka, T.; Aoi, T.; Okita, K.; Mochiduki, Y.; Takizawa, N.; Yamanaka, S. Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat. Biotechnol. 2008, 26, 101–106. [Google Scholar] [CrossRef]
- O’Doherty, R.; Greiser, U.; Wang, W. Nonviral methods for inducing pluripotency to cells. Biomed. Res. Int. 2013, 2013, 705902. [Google Scholar] [CrossRef]
- Caulfield, J.B.; Borg, T.K. The collagen network of the heart. Lab Investig. 1979, 40, 364–372. [Google Scholar]
- Zannad, F.; Alla, F.; Dousset, B.; Perez, A.; Pitt, B. Limitation of excessive extracellular matrix turnover may contribute to survival benefit of spironolactone therapy in patients with congestive heart failure: Insights from the Randomized ALdactone Evaluation Study (RALES). Circulation 2000, 102, 2700–2706. [Google Scholar] [CrossRef]
- Cohn, J.N.; Tognoni, G. Valsartan Heart Failure Trial Investigators. A randomized trial of the angiotensin-receptor blocker valsartan in chronic heart failure. N. Engl. J. Med. 2001, 345, 1667–1675. [Google Scholar] [CrossRef]
- Abbate, A.; Kontos, M.C.; Grizzard, J.D.; Biondi-Zoccai, G.G.; Van Tassell, B.W.; Robati, R.; Roach, L.M.; Arena, R.A.; Roberts, C.S.; Varma, A.; et al. VCU-ART Investigators. Interleukin-1 blockade with anakinra to prevent adverse cardiac remodeling after acute myocardial infarction (Virginia Commonwealth University Anakinra Remodeling Trial [VCU-ART] Pilot study). Am. J. Cardiol. 2010, 105, 1371–1377.e1. [Google Scholar] [CrossRef] [PubMed]
- Biernacka, A.; Cavalera, M.; Wang, J.; Russo, I.; Shinde, A.; Kong, P.; Gonzalez-Quesada, C.; Rai, V.; Dobaczewski, M.; Lee, D.W.; et al. Smad3 Signaling Promotes Fibrosis While Preserving Cardiac and Aortic Geometry in Obese Diabetic Mice. Circ. Heart Fail. 2015, 8, 788–798. [Google Scholar] [CrossRef] [PubMed]
- Mishra, R.; Saha, P.; Datla, S.R.; Mellacheruvu, P.; Gunasekaran, M.; Guru, S.A.; Fu, X.; Chen, L.; Bolli, R.; Sharma, S.; et al. Transplanted allogeneic cardiac progenitor cells secrete GDF-15 and stimulate an active immune remodeling process in the ischemic myocardium. J. Transl. Med. 2022, 20, 323. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piątek-Matuszak, P.; Pasławski, R.; Pasławska, U.; Kiczak, L.; Płóciennik, M.; Janiszewski, A.; Michałek, M.; Gwizdała, A.; Kaźmierczak, J.; Gorący, J. Assessment of Myocardial Diastolic Dysfunction as a Result of Myocardial Infarction and Extracellular Matrix Regulation Disorders in the Context of Mesenchymal Stem Cell Therapy. J. Clin. Med. 2022, 11, 5430. https://doi.org/10.3390/jcm11185430
Piątek-Matuszak P, Pasławski R, Pasławska U, Kiczak L, Płóciennik M, Janiszewski A, Michałek M, Gwizdała A, Kaźmierczak J, Gorący J. Assessment of Myocardial Diastolic Dysfunction as a Result of Myocardial Infarction and Extracellular Matrix Regulation Disorders in the Context of Mesenchymal Stem Cell Therapy. Journal of Clinical Medicine. 2022; 11(18):5430. https://doi.org/10.3390/jcm11185430
Chicago/Turabian StylePiątek-Matuszak, Patrycja, Robert Pasławski, Urszula Pasławska, Liliana Kiczak, Michał Płóciennik, Adrian Janiszewski, Marcin Michałek, Adrian Gwizdała, Jarosław Kaźmierczak, and Jarosław Gorący. 2022. "Assessment of Myocardial Diastolic Dysfunction as a Result of Myocardial Infarction and Extracellular Matrix Regulation Disorders in the Context of Mesenchymal Stem Cell Therapy" Journal of Clinical Medicine 11, no. 18: 5430. https://doi.org/10.3390/jcm11185430
APA StylePiątek-Matuszak, P., Pasławski, R., Pasławska, U., Kiczak, L., Płóciennik, M., Janiszewski, A., Michałek, M., Gwizdała, A., Kaźmierczak, J., & Gorący, J. (2022). Assessment of Myocardial Diastolic Dysfunction as a Result of Myocardial Infarction and Extracellular Matrix Regulation Disorders in the Context of Mesenchymal Stem Cell Therapy. Journal of Clinical Medicine, 11(18), 5430. https://doi.org/10.3390/jcm11185430