
First and Second Level Haemoglobinopathies Diagnosis: Best Practices of the Italian Society of Thalassemia and Haemoglobinopathies (SITE)

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
- The ENERCA recommendations for preconception or antenatal screening, prenatal diagnosis and genetic counseling of hemoglobinopathies [5];
- The joint SOGC-CCMG Opinion for Reproductive Genetic Carrier Screening: An Update for All Canadian Providers of Maternity and Reproductive Healthcare in the Era of Direct-to-Consumer Testing [6];
- UK NHS sickle cell and thalassemia screening program, UK NHS sickle cell and thalassemia handbook for laboratories [7].
3. First Level Tests
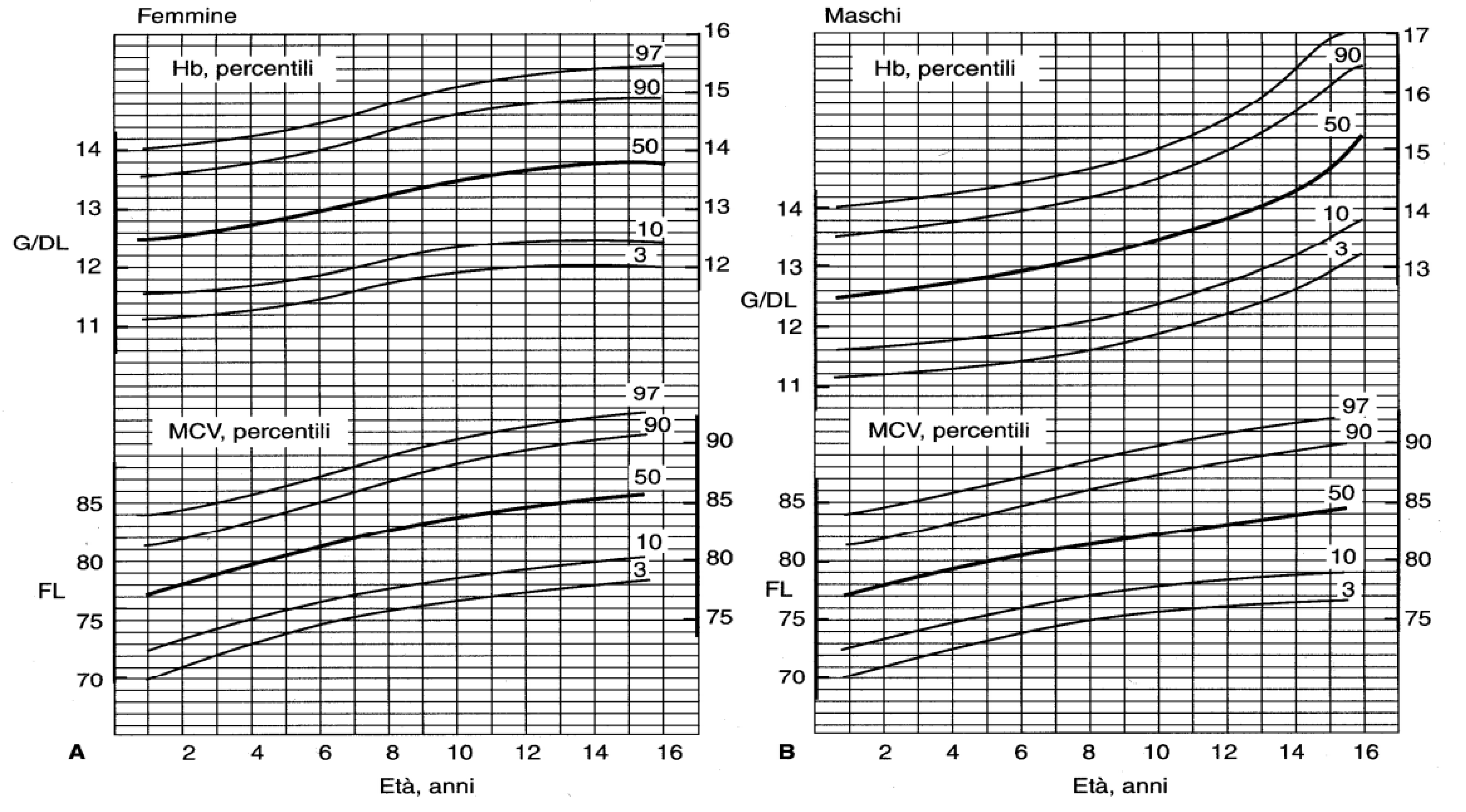
3.1. Complete Blood Count (CBC)
3.2. Separation of Hemoglobin
Hemoglobin A2
3.3. Fetal Hemoglobin
3.4. Hb Variants
3.5. Iron Parameters and Hemoglobinopathies
Recommendations
- Adequate counseling with a specialist with documented experience in the treatment of hemoglobinopathies (internist, hematologist, pediatrician, clinical geneticist) should be offered to all hemoglobinopathy carriers in order to explain the probabilities of disease transmission, the pathology, the state-of-the-art treatment and the possibilities of prenatal diagnosis [6].
- First level tests for hemoglobinopathies (CBC, iron parameters and HPLC and/or CE) should be delayed if the patient has received a red blood cell transfusion in the previous three months or in the case of iron deficiency.
- First level tests for diagnostic certainty of alpha+ thalassemia are not sufficient. (Diagnostic certainty can only be obtained by molecular analysis.)
- At birth, first level examinations for the diagnosis of heterozygous beta thalassemia, Hb Lepore, or delta beta thalassemia are not sufficient for diagnostic certainty.
- CBC and iron indices should be analyzed within 12/24 h of blood sampling.
- Hb pattern analysis can be carried out within seven days of blood sampling test in EDTA by storing the sample at 4 °C. In the case of a suspected unstable variant, the examination should be performed as soon as possible to avoid degradation of the variant.
- HPLC and/or CE are recommended for the screening of hemoglobinopathies. Both methods can be mutually used as confirmatory tests. To separate Hb fractions using HPLC, it is essential to use a cation exchange apparatus, with a double pump continuous linear gradient and with a variation coefficient of <5%.
- When interpreting HbA2, consider the presence of factors that might over- or underestimate it (see Table 3).
- The presence of HbF above normal values beyond the first year of life should be investigated with second level tests if associated with moderate microcytic anemia and/or splenomegaly.
4. Second Level Test
4.1. Molecular Analysis
- subjects with nondiriment first level tests to confirm or refute the diagnosis of a healthy carrier. To correctly identify α+-thalassemia carriers, molecular analysis is required [23]. Carriers of very mild or silent mutations of the β-globin gene, in which chain production is only minimally reduced, may have normal hematological parameters and are only recognizable by molecular analysis [17];
- suspected carriers of both α and β thalassemia to correctly define the thalassemia recurrence risk;
- the definition of the mutation responsible for the carrier status on the indication of a specialist in the field;
- sideropenic pregnant couples. It is preferable to search for alpha- and beta globin gene mutations if one partner is already known to be a beta thalassemia carrier before the normalization of iron levels in order to avoid the delay of a prenatal diagnosis if necessary [23].
- ○
- DNA can be extracted by a saliva sample from children for whom peripheral blood sampling is difficult;
- ○
- buccal swabs from bone marrow transplant recipients. (Note: a saliva sample might not allow a definitive report due to the presence of the leucocytes of the donor in the saliva).
Recommendations
- DNA analysis should always be performed in couples at risk (both partner carriers of hemoglobinopathies and/or triplication-quadruplication of α globin genes) in cases where the first level tests are not decisive or in couples where one partner is a carrier of alpha- and the other of beta thalassemia in order to exclude the possibility that the beta trait is masking an alpha trait, thus underestimating the risk of HbH in the fetus or vice versa.
- In the case of sideropenia in pregnant couples, it is preferable to start molecular analysis before iron level normalization to avoid a delay in a prenatal diagnosis if necessary [23].
- In the case of suspected α thalassemia, techniques such as reverse dot blot or GAP-PCR can be used as first level screening for recurrent α globin gene mutations. In negative cases with strong clinical suspicion of α thalassemia carrier, it is recommended to proceed with second level tests such as direct sequencing or MLPA.
- Direct sequencing of the HBB gene is recommended.
- In laboratories equipped with NGS, this can be considered as an alternative to Sanger sequencing, following the recommendations in force for NGS genetic diagnosis.
- For subjects with HbA2 borderline levels, it is recommended to rule out β+ mutations and α globin gene triplication/quadruplication.
- Genotype–phenotype correspondence should always be verified.
- During counseling, it should be clearly specified that first level tests can only exclude the risk of transfusion-dependent thalassemia or sickle cell syndrome, conditions for which prenatal diagnosis is indicated (Table 12).
4.2. Prenatal Diagnosis (PND)
4.3. Cell-Free Fetal DNA (cffDNA)
4.4. Preimplantation Genetic Testing (PGT)
4.5. Gamete Donation
4.5.1. Recommendations
- At-risk couples should receive counseling with a specialist (internist, hematologist, pediatrician, clinical geneticist) in the treatment of hemoglobinopathies.
- Couples for whom prenatal counseling has not been possible should be informed of the risk of recurrence and of PND possibilities for future pregnancies [6].
- It is preferable to perform PND through CVS in the first trimester of gestation.
- Before PND, the genotype of both parents must be determined.
- If the phenotype of the father is not available, PND limits should be clearly explained (e.g., possible failure to detect a second mutation of the β-globin gene because of technical limitations).
- Even if PND has been performed, it is preferable to perform a carrier test in the child, ideally in the first year of life, in the case of an unaffected fetus; in the case of an affected fetus, it is recommended to examine the patient within 3 months of life.
- In all at-risk couples in whom a prenatal diagnosis is not made, it is mandatory to perform a carrier test in the infant within 3–6 months of life in order to ensure appropriate treatment.
- In the case of gamete donation/preimplantation genetic testing, specific advice from an experienced center is recommended.
- With the techniques currently available in diagnostics, the analysis of cell-free fetal DNA (cffDNA) is not sufficient to diagnose fetal hemoglobinopathies.
4.5.2. Informed Consent to Investigations
4.5.3. Medical Reporting
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kattamis, A.; Forni, G.L.; Aydinok, Y.; Viprakasit, V. Changing patterns in the epidemiology of β-thalassemia. Eur. J. Haematol. 2020, 105, 692–703. [Google Scholar] [CrossRef] [PubMed]
- Schünemann, H.J.; Wiercioch, W.; Brozek, J.; Etxeandia-Ikobaltzeta, I.; Mustafa, R.A.; Manja, V.; Brignardello-Petersen, R.; Neumann, I.; Falavigna, M.; Alhazzani, W.; et al. GRADE Evidence to Decision (EtD) frameworks for adoption, adaptation, and de novo development of trustworthy recommendations: GRADE-ADOLOPMENT. J. Clin. Epidemiol. 2017, 81, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Traeger-Synodinos, J.; Harteveld, C.L.; Old, J.M.; Petrou, M.; Galanello, R.; Giordano, P.; Angastioniotis, M.; De la Salle, B.; Henderson, S.; May, A.; et al. EMQN Best Practice Guidelines for molecular and haematology methods for carrier identification and prenatal diagnosis of the haemoglobinopathies. Eur. J. Hum. Genet. 2015, 23, 426–437. [Google Scholar] [CrossRef] [PubMed]
- Ryan, K.; Bain, B.J.; Worthington, D.; James, J.; Plews, D.; Mason, A.; Roper, D.; Rees, D.C.; de la Salle, B.; Streetly, A.; et al. Significant haemoglobinopathies: Guidelines for screening and diagnosis. Br. J. Haematol. 2010, 149, 35–49. [Google Scholar] [CrossRef] [PubMed]
- Recommendations for Preconceptional or Antenatal Screening, Prenatal Diagnosis and Genetic Counselling of Haemoglobinopathies. Available online: http://www.enerca.org (accessed on 1 June 2021).
- Wilson, R.D.; De Bie, I.; Armour, C.M.; Brown, R.N.; Campagnolo, C.; Carroll, J.C.; Okun, N.; Nelson, T.; Zwingerman, R. Joint SOGC–CCMG Opinion for Reproductive Genetic Carrier Screening: An Update for All Canadian Providers of Maternity and Reproductive Healthcare in the Era of Direct-to-Consumer Testing. J. Obstet. Gynaecol. Can. 2016, 38, 742–762.e3. [Google Scholar] [CrossRef]
- Sickle Cell and Thalassaemia Screening Programme: Standards. Available online: https://www.gov.uk/topic/population-screening-programmes/sickle-cell-thalassaemia (accessed on 1 June 2021).
- Cousens, N.E.; Gaff, C.L.; Metcalfe, S.A.; Delatycki, M.B. Carrier screening for Beta-thalassaemia: A review of international practice. Eur J. Hum. Genet. 2010, 18, 1077–1083. [Google Scholar] [CrossRef]
- American College of Obstetricians and Gynecologists. Committee Opinion No. 691: Carrier Screening for Genetic Conditions. Obstet. Gynecol. 2017, 129, e41–e55. [Google Scholar] [CrossRef]
- Old, J.M. Screening and genetic diagnosis of haemoglobinopathies. Scand. J. Clin. Lab. Investig. 2007, 67, 71–86. [Google Scholar] [CrossRef]
- Lees, C.M.; Davies, S.; Dezateux, C. Neonatal screening for sickle cell disease. Cochrane Database Syst. Rev. 2000, 2000, CD001913. [Google Scholar] [CrossRef]
- Baronciani, D.; Casale, M.; De Franceschi, L.; Graziadei, G.; Longo, F.; Origa, R.; Rigano, P.; Pinto, V.; Marchetti, M.; Gigante, A.; et al. Selecting β-thalassemia Patients for Gene Therapy: A Decision-making Algorithm. HemaSphere 2021, 5, e555. [Google Scholar] [CrossRef]
- Lichtman, M.A.; Murphy, M.S.; Adamson, J.W. Detection of mutant hemoglobins with altered affinity for oxygen. A simplified technique. Ann. Intern. Med. 1976, 84, 517–520. [Google Scholar] [CrossRef] [PubMed]
- Forni, G.L.; Barella, S.; Cappellini, M.D.; Maggio, A.; Piga, A. Architettura Della Rete Italiana Talassemie Ed Emoglobinopatie. 2018. Available online: http://www.siteitalia.org/forza_download.php?file=Architettura_Rete_Italiana.pdf (accessed on 2 May 2021).
- Saarinen, U.M.; Silmes, M.A. Developmental changes in red blood cell counts and indices of infants after exclusion of iron deficiency by laboratory criteria and continuous iron supplementation. J. Pediatr. 1978, 92, 414. [Google Scholar] [CrossRef]
- Dallman, P.R.; Siimes, M.A. Percentile curves for hemoglobin and red cellvolume in infnacy and childhood. J. Pediatr. 1979, 94, 26–31. [Google Scholar] [CrossRef]
- Brancaleoni, V.; Di Pierro, E.; Motta, I.; Cappellini, M.D. Laboratory diagnosis of thalassemia. Int. Jnl. Lab. Hem. 2016, 38, 32–40. [Google Scholar] [CrossRef]
- Buch, A.C.; Karve, P.P.; Panicker, N.K.; Singru, S.A.; Gupta, S.C. Role of red cell distribution width in classifying microcytic hypochromic anaemia. J. Indian Med. Assoc. 2011, 109, 297–299. [Google Scholar]
- Trent, R.J.; Webster, B.; Bowden, D.K.; Gilbert, A.; Joy Ho, P.; Lindeman, R.; Lammi, A.; Rowell, J.; Hinchcliffe, M.; Colley, A.; et al. Complex phenotypes in the haemoglobinopathies: Recommendations on screening and DNA testing. Pathology 2006, 38, 507–519. [Google Scholar] [CrossRef]
- Galanello, R.; De Virgiliis, S.; Addis, M.; Paglietti, E.; Ruggeri, R.; Cao, A. Haematological characteristics of the beta 0 thalassaemia trait in Sardinian children. J. Clin. Pathol. 1980, 33, 946–948. [Google Scholar] [CrossRef]
- Iolascon, A.; Pinto, L.; Nobili, B.; Cutillo, S. Developmental changes in HbA2 and HbF on neocytes and gerocytes in normal infants during the first year of life. Acta Haematol. 1983, 70, 278–279. [Google Scholar] [CrossRef]
- Brugnara, C.; Schiller, B.; Moran, J. Reticulocyte hemoglobin equivalent (Ret He) and assessment of iron-deficient states. Clin. Lab Haematol. 2006, 28, 303–308. [Google Scholar] [CrossRef]
- Lee, S.Y.; Yap, E.S.; Lee, E.Y.; Goh, J.H.; Liu, T.C.; Yip, C. Evaluation of Thalassaemia Screening Tests in the Antenatal and Non-Antenatal Populations in Singapore. Ann. Acad Med. Singap 2019, 48, 5–15. [Google Scholar] [CrossRef]
- Traeger-Synodinos, J.; Harteveld, C.L. Preconception carrier screening and prenatal diagnosis in thalassemia and hemoglobinopathies: Challenges and future perspectives. Expert Rev. Mol. Diagn. 2017, 17, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Li, D.-Z.; Yang, Y.-D. Invasive prenatal diagnosis of fetal thalassemia. Best Pract. Res. Clin. Obstet. Gynaecol. 2017, 39, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Vrettou, C.; Kakourou, G.; Mamas, T.; Traeger-Synodinos, J. Prenatal and preimplantation diagnosis of hemoglobinopathies. Int J. Lab. Hem. 2018, 40, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Skirton, H.; Goldsmith, L.; Chitty, L.S. An easy test but a hard decision: Ethical issues concerning non-invasive prenatal testing for autosomal recessive disorders. Eur J. Hum. Genet. 2015, 23, 1004–1009. [Google Scholar] [CrossRef] [PubMed]
- Dormandy, E.; Gulliford, M.; Bryan, S.; Roberts, T.E.; Calnan, M.; Atkin, K.; Karnon, J.; Logan, J.; Kavalier, F.; Harris, H.J.; et al. Effectiveness of earlier antenatal screening for sickle cell disease and thalassaemia in primary care: Cluster randomised trial. BMJ 2010, 341, c5132. [Google Scholar] [CrossRef]
- Lal, A. Alpha thalassemia major: Prenatal and postnatal management. In UpToDate; Post, T.W., Ed.; UpToDate: Waltham, MA, USA, 2022. [Google Scholar]
- Ghi, T.; Sotiriadis, A.; Calda, P.; Da Silva Costa, F.; Raine-Fenning, N.; Alfirevic, Z.; McGillivray, G. International Society of Ultrasound in Obstetrics and Gynecology (ISUOG) ISUOG Practice Guidelines: Invasive procedures for prenatal diagnosis. Ultrasound Obs. Gynecol 2016, 48, 256–268. [Google Scholar] [CrossRef]
- Giambona, A.; Makrydimas, G.; Leto, F.; Damiani, G.; Jakil, M.C.; Picciotto, F.; Renda, D.; Fiorino, R.; Renda, M.C.; Schillaci, G.; et al. Feasibility of DNA diagnosis of haemoglobinopathies on coelocentesis: DNA Diagnosis of Haemoglobinopathies on Coelocentesis. Br. J. Haematol. 2011, 153, 268–272. [Google Scholar] [CrossRef]
- Li, X.; Zhou, Q.; Zhang, M.; Tian, X.; Zhao, Y. Sonographic Markers of Fetal α-Thalassemia Major. J. Ultrasound Med. 2015, 34, 197–206. [Google Scholar] [CrossRef]
- Cell-free DNA to Screen for Single-Gene Disorders. Available online: https://www.acog.org/clinical/clinical-guidance/practice-advisory/articles/2019/02/cell-free-dna-to-screen-for-single-gene-disorders (accessed on 14 September 2022).
- Yates, A.M. Prenatal screening and testing for hemoglobinopathy. In UpToDate; Post, T.W., Ed.; UpToDate: Waltham, MA, USA, 2019. [Google Scholar]

{kind=link}
| Conditions Where 1st Level Test Would Be Inappropriate |
|---|
|
|
|
|
|
| n | Age (Months) | ||||||
|---|---|---|---|---|---|---|---|
| 0.5 (N = 232) | 1 (N = 240) | 2 (N = 241) | 4 (N = 52) | 6 (N = 52) | 9 (N = 56) | 12 (N = 56) | |
| Hgb (mean ± SE) | 16.6 ± 0.11 | 13.9 ± 0.10 | 11.2 ± 0.06 | 12.2 ± 0.14 | 12.6 ± 0.10 | 12.7 ± 0.09 | 12.7 ± 0.09 |
| −2 SD | 13.4 | 10.7 | 9.4 | 10.3 | 11.1 | 11.4 | 11.3 |
| Hct (mean ± SE) | 53 ± 0.4 | 44 ± 0.3 | 35 ± 0.2 | 38 ± 0.4 | 36 ± 0.3 | 36 ± 0.3 | 37 ± 0.3 |
| −2 SD | 41 | 33 | 28 | 32 | 31 | 32 | 33 |
| RBC count (mean ± SE) | 4.9 ± 0.03 | 4.3 ± 0.03 | 3.7 ± 0.02 | 4.3 ± 0.06 | 4.7 ± 0.05 | 4.7 ± 0.04 | 4.7 ± 0.04 |
| −2 SD–+2 SD | 3.9–5.9 | 3.3–5.3 | 3.1–4.3 | 3.5–5.1 | 3.0–5.5 | 4.0–5.3 | 4.1–5.3 |
| MCH (mean ± SE) | 33.6 ± 0.1 | 32.5 ± 0.1 | 30.4 ± 0.1 | 28.6 ± 0.2 | 26.8 ± 0.2 | 27.3 ± 0.2 | 26.8 ± 0.2 |
| −2 SD | 30 | 29 | 27 | 25 | 24 | 25 | 24 |
| MCV (mean ± SE) | 105.3 ± 0.6 | 103.1 ± 0.3 | 94.8 ± 0.3 | 86.7 ± 0.8 | 76.3 ± 0.6 | 77.7 ± 0.5 | 77.7 ± 0.5 |
| −2 SD | 88 | 91 | 84 | 76 | 68 | 70 | 71 |
| MCHC (mean ± SE) | 314 ± 1.1 | 318 ± 1.2 | 318 ± 1.1 | 327 ± 2.7 | 350 ± 1.7 | 349 ± 1.6 | 343 ± 1.5 |
| −2 SD | 281 | 281 | 283 | 288 | 327 | 324 | 321 |
| Test | Specific Test | Confirmation Test | Other Test | Comments |
|---|---|---|---|---|
| HPLC | X | X | Separation and quantification | |
| CE | X | X | Separation and quantification | |
| Complete blood count | X | EvalueteS RBC indices: G.R (X103/pL), Hb (g/dL), mCV (fL), MCH (pg), Ht (%). | ||
| Serum iron | X | In alternatives ZnPP. | ||
| Transferrin | X | |||
| Ferritin | X | |||
| Reticulocytes | X | (%) | ||
| P50 | Arterial or venous blood test in the presence of an increased hematocrit (<45% in women and> 50% in men) | |||
| Haptoglobin | X | If haemolytic anemia is present | ||
| Methemoglobinemia | X | If cyanosis is present | ||
| Bilirubin | X | If haemolytic anemia is present |
| Increased HbA2 | Reduced HbA2 |
|---|---|
| Hyperthyroidism | Severe Iron-deficiency anemia |
| Megaloblastic anaemia | Sideroblastic anemia |
| Antiretroviral therapy for HIV | δ-Thalassaemia (b) |
| Hb unstable variants | δ -Thalassaemia chain variants (c) |
| Supernumerary alpha genes variants (i.e., ααα/αα) | α-Thalassaemia chain variants(d) |
| Glycated component of the Hb B variant if present (a) | Same type of HpFH due to defect in the Gamma gene promoter (e) |
| Liver disease/alcohol | δβ-Thalassaemia |
| Hypertrophic osteoarthropathy | α-Thalassaemia: borderline in Alf + or ALPHA while it is marked in H hemoglobinosis |
| KLF1 mutations | Hb Lepore (f) |
| HbD (a) e C (f) |
| Hb Abruzzo | Hb Kenya |
| Hb Akron | Hb Korle Bu * |
| Hb Boras | Hb Lepore Baltimore |
| Hb Bethesda * | Hb Lepore Boston |
| Hb Chandigarth | Hb Lepore Hollandia |
| Hb Deer Lodge | Hb Loves Park * |
| Hb D Iran * | Hb M Saskatoon |
| Hb Denver * | Hb Muravera |
| Hb D-Ouled Rabah | Hb Nebraska |
| Hb E | Hb Ocho Rios |
| Hb Ethiopia * | Hb Osu Christiansborg * |
| Hb Fort Worth | Hb Paddington |
| Hb G Copenhagen | Hb Rocky Mountain |
| Hb G Coushatta * | Hb San Bruno * |
| Hb G Ferrara | Hb Santa Juana * |
| Hb G Galveston | Hb SId (the aged adduct of Hb due to glutathione) |
| Hb G Honolulu * | Hb Spanish Town |
| Hb G Taipei | Hb Toulon |
| Hb Hoshida | Hb Tubingen |
| Hb Hamadan | Hb Zuri |
| HPLC, high-performance liquid chromatography. | |
| Hb Chad |
| Hb E-Saskatoon |
| Hb O-Arab |
| Hb C Harlem * |
| Congenital or acquired anemias from primitive bone marrow failure with or without displasia | Neoplasias | Conditions associated with specific therapeutic treatments | Other conditions |
| Congenital or acquired aplastic anemia | Hepatocarcinoma | Chemotherapies for leukemias | Monoclonal gammopathy of uncertain significance |
| Megaloblastic anemia from vitamin deficiency | Myeloid acute leukemias | Therapy with hydroxyurea, butyrates and erythropoiesis-stimulating agents | Pregnancy |
| Diamond-Blackfan anemia | Primitive myelofibrosis | Chronic renal insufficiency | |
| Some forms of normoblastic anemia | Juvenile chronic myelomonocytic leukemia | Hyperthyroidism | |
| Congenital sideroblastic anemias | Trisomy 13 | ||
| Acquired sideroblastic anemias | |||
| Nocturnal paroxysmal hemoglobinuria |
| Age | Subject | Number | HbA2 (%) | HbF (%) | Hb (g/dl) | MCV (fl) | MCH (pg) |
|---|---|---|---|---|---|---|---|
| At birth | Normal β Thalassaemia | 16 31 | 0.4 (0.2) 0.5 (0.2) NS | 65.1 (7.5) 73.8 (10.1) p < 0.05 | 18.1 (2.3) 18.3 (2.3) NS | 101.3 (6.9) 98.5 (8.1) NS | 35.1 (3.5) 33.8 (2.6) NS |
| 3 months | Normal β Thalassaemia | 8 12 | 1.7 (0.3) 3.2 (0.7) p < 0.01 | 18.1 (3.6) 27.0 (10.5) p < 0.05 | 11.0 (0.7) 10.0 (1.1) NS | 82.5 (3.6) 69.9 (5.8) p < 0.001 | 27.9 (2.0) 22.8 (1.8) p < 0.001 |
| 6 months | Normal β Thalassaemia | 8 10 | 2.5 (0.3) 4.8 (0.7) p < 0.001 | 3.2 (1.1) 8.2 (4.0) p < 0.001 | 11.5 (0.8) 10.5 (0.8) p < 0.05 | 74.7 (2.9) 59.2 (3.5) p < 0.001 | 24.9 (1.4) 19.2 (1.2) p < 0.001 |
| 9–10 months | Normal β Thalassaemia | 6 14 | 2.5 (0.4) 5.1 (0.5) p < 0.001 | 2.6 (1.4) 4.4 (2.1) NS | 12.5 (1.0) 11.1 (0.9) p < 0.005 | 76.8 (5.2) 58.7 (1.6) p < 0.001 | 25.9 (1.7) 19.6 (0.9) p < 0.001 |
| 1 year | Normal β Thalassaemia | 5 8 | 2.5 (0.3) 4.8 (0.4) p < 0.001 | 1.4 (0.6) 4.1 (2.1) p < 0.02 | 12.3 (1.0) 11.2 (0.9) p < 0.005 | 74.6 (5.0) 57.5 (2.4) p < 0.001 | 24.8 (2.7) 18.7 (0.9) p < 0.001 |
| Age | Number of Subjects | HbA | HbF | ||||
|---|---|---|---|---|---|---|---|
| N | G | RBC | N | G | RBC | ||
| At birth | 30 | 0.64 ± 0.15 | 0.42 ± 0.07 | 0.49 ± 0.12 | 54.0 ± 8.8 | 72.0 ± 9.2 | 66.0 ± 7.6 |
| 1 month | 10 | 0.91 ± 0.25 | 0.40 ± 0.12 | 0.72 ± 0.29 | 44.0 ± 5.7 | 68.0 ± 6.2 | 52.0 ± 5.5 |
| 2 months | 6 | 2.07 ± 0.29 | 0.96 ± 0.15 | 1.14 ± 0.32 | 24.3 ± 6.8 | 37.5 ± 6.2 | 33.0 ± 6.5 |
| 3 months | 8 | 2.13 ± 0.32 | 1.35 ± 0.19 | 1.55 ± 0.31 | 18.5 ± 5.5 | 25.6 ± 4.8 | 21.0 ± 6.1 |
| 4 months | 8 | 2.27 ± 0.26 | 1.72 ± 0.25 | 1.93 ± 0.28 | 8.0 ± 3.3 | 13.6 ± 3.6 | 10.5 ± 3.5 |
| 5 months | 6 | 2.39 ± 0.26 | 1.86 ± 0.30 | 2.18 ± 0.21 | 3.2 ± 1.1 | 5.6 ± 0.9 | 4.6 ± 0.9 |
| 6 months | 6 | 2.50 ± 0.22 | 2.10 ± 0.32 | 2.25 ± 0.30 | 2.6 ± 0.4 | 3.9 ± 1.1 | 3.3 ± 0.9 |
| 7 months | 5 | 2.50 ± 0.28 | 2.19 ± 0.31 | 2.28 ± 0.27 | 1.7 ± 0.5 | 3.3 ± 1.2 | 2.8 ± 1.1 |
| 8 months | 5 | 2.48 ± 0.32 | 2.25 ± 0.29 | 2.34 ± 0.36 | 1.2 ± 0.4 | 2.3 ± 0.5 | 1.9 ± 0.7 |
| 9–10 months | 5 | 2.53 ± 0.26 | 2.30 ± 0.28 | 2.42 ± 0.34 | 1.1 ± 0.6 | 2.2 ± 0.6 | 1.7 ± 0.4 |
| 11–12 months | 5 | 2.61 ± 0.24 | 2.50 ± 0.28 | 2.53 ± 0.29 | 1.0 ± 0.3 | 2.0 ± 0.5 | 1.4 ± 0.4 |
| 2–12 years | 20 | 2.64 ± 0.28 | 2.56 ± 0.24 | 2.60 ± 0.30 | 0.4 ± 0.2 | 0.7 ± 0.3 | 0.6 ± 0.2 |
| Method | Advantages | Limits |
|---|---|---|
| known mutations detection (Reverse dot blot hybridization, ASO, allele-specific PCR—ARMS PCR, GAP-PCR) | economic easy to perform rapid possible use of commercial kits | can recognize only known mutations may not be standardizable for some specific mutations generally non–“high-throughput” if not using commercial kits, it must be validated risk of allele drop-out |
| Sanger Sequencing | economic platform utilizable for different analyses can detect mutations in the whole gene | dedicated personnel requires specific expertise not posssible a conclusive diagnosis of large deletions |
| Pyrosequencing | rapid easy to perform | allows to perform short sequences of DNA (20–50 nucleotides) dedicated personnel specific expertise in the analysis not very widespread equipment |
| Next generation sequencing (NGS) | utilizable for different analyses can detect mutations in the whole gene sequence allows gene panel analysis | dedicated personnel specific expertise in bioinformatics analyses high costs |
| High Resolution Melt Analysis (HRMA) | rapid sensitive “high-throughput” can be used also for other analyses | technically more difficult to design the sample in some cases diagnostic confirmation by another method is required dedicated personnel requires specific expertise high costs of the instrument |
| Real Time PCR | allows to identify qualitative and quantitative variations rapid “high-throughput” | requires specific expertise high costs of the instrument |
| MLPA | simple, rapid validated commercial kits can detect any copy number variant at the locus | quality and concentration of DNA are critical instrumentation with dedicated personnel specific expertise |
| arrayCGH | can detect any copy number variant at the locus | specific kits for globin loci does not allow precise characterization of deletion/duplication breakpoints dedicated personnel specific expertise potentially high costs |
| Reverse dot blot hybridization, ASO, allele specific PCR—ARMS PCR | Known point mutations/deletions of α and β globin genes |
| GAP-PCR | Known deletions/duplications of α and β globin genes. Note: amplification of GC-rich regions may be difficult, risk of allele drop out: not recommended in PND |
| Sanger Sequencing/NGS | point mutations in the whole sequence of α and β globin genes |
| High Resolution Melt Analysis (HRMA) | point mutations in the whole sequence of α and β globin genes |
| MLPA | Deletions/duplications in the whole α and β globin gene locus |
| arrayCGH | Deletions/duplications in the whole α and β globin gene locus |
| Genotype | Expected Phenotype | Degree of Uncertainty in Predicting Phenotype | Agreement among Experts | Indication to Make PND Available | Agreement among Experts |
|---|---|---|---|---|---|
| 2 severe β0 or β+ mutations | thalassemia major | low | 100% | strong | 94% |
| Hb Lepore + severe β0 or β+ mutations | thalassemia major | low | 100% | strong | 94% |
| δβ0 + severe β0 or β+ mutations | severe thalassemia intermedia/thalassemia major | low | 100% | strong | 94% |
| HbE + severe β0 or β+ mutations | severe thalassemia intermedia/thalassemia major | average | 100% | strong | 94% |
| Hb O-Arab + severe β0 or β+ mutations | thalassemia intermedia/thalassemia major | average | 94% | strong | 94% |
| Homozygous Hb Lepore | thalassemia intermedia/thalassemia major | low | 100% | strong | 94% |
| Homozygous HbS | drepanocytic syndrome | high | 96% | clear | 90% |
| Heterozygous HbS + HbC/ Hb O-Arab/ HbD-Punjab | drepanocytic syndrome | average | 100% | clear | 94% |
| Homozygous α0 thalassemia | fetal hydrops | low | 100% | absolute | 100% |
| 2 mild β+ mutations | thalassemia intermedia | average | 84% | open | 98% |
| Homozygous δβ0 thalassemia | thalassemia intermedia | low | 100% | open | 100% |
| δβ0 + mild β+ mutations | thalassemia intermedia | high | 88% | open | 98% |
| δβ0 + Hb Lepore/HbE/Hb O-Arab | thalassemia intermedia | average | 88% | open | 100% |
| HbC + severe β0 or β+ mutations | thalassemia intermedia | average | 88% | open | 100% |
| Homozygous HbC /HbE/HbD Punjab/Hb O-Arab | thalassemia intermedia | high | 88% | low | 98% |
| Hb D-Punjab/Hb O-Arab + severe β0 or β+ mutations | thalassemia intermedia | high | 94% | clear | 88% |
| HbS/HbE | drepanocytic syndrome with intermediate course | average | 88% | open | 94% |
| HbS + severe β0 or β+ mutation | drepanocytic syndrome | low | 88% | clear | 100% |
| HbS + mild β+ mutations | drepanocytic syndrome with intermediate course | average | 100% | open | 92% |
| HbS +δβ0 or Hb Lepore | drepanocytic syndrome with intermediate course | average | 100% | open | 100% |
| HbS + HbD Punjab | drepanocytic syndrome | high | 100% | clear | 100% |
| α0 + α+ thalassemia (--/-α) | HbH disease | average | 94% | open | 100% |
| ααα o αααα + severe β0 or β+ mutations | thalassemia intermedia with variable clinical picture | average | 86% | open | 94% |
| ααα o αααα + β+ mutation | mild thalassemia intermedia | average | 82% | low | 100% |
| 2 silent β+ mutations | very mild thalassemia intermedia | low | 100% | low | 96% |
| HbC + mild β+ mutations | mild thalassemia intermedia | average | 84% | low | 92% |
| HPFH | not clinically significant | low | 100% | none | 100% |
| Homozygous α+ thalassemia | not clinically significant | low | 100% | none | 100% |
| Homozygous ααα | not clinically significant | low | 100% | none | 100% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mandrile, G.; Barella, S.; Giambona, A.; Gigante, A.; Grosso, M.; Perrotta, S.; Scianguetta, S.; Forni, G.L. First and Second Level Haemoglobinopathies Diagnosis: Best Practices of the Italian Society of Thalassemia and Haemoglobinopathies (SITE). J. Clin. Med. 2022, 11, 5426. https://doi.org/10.3390/jcm11185426
Mandrile G, Barella S, Giambona A, Gigante A, Grosso M, Perrotta S, Scianguetta S, Forni GL. First and Second Level Haemoglobinopathies Diagnosis: Best Practices of the Italian Society of Thalassemia and Haemoglobinopathies (SITE). Journal of Clinical Medicine. 2022; 11(18):5426. https://doi.org/10.3390/jcm11185426
Chicago/Turabian StyleMandrile, Giorgia, Susanna Barella, Antonino Giambona, Antonia Gigante, Michela Grosso, Silverio Perrotta, Saverio Scianguetta, and Gian Luca Forni. 2022. "First and Second Level Haemoglobinopathies Diagnosis: Best Practices of the Italian Society of Thalassemia and Haemoglobinopathies (SITE)" Journal of Clinical Medicine 11, no. 18: 5426. https://doi.org/10.3390/jcm11185426
APA StyleMandrile, G., Barella, S., Giambona, A., Gigante, A., Grosso, M., Perrotta, S., Scianguetta, S., & Forni, G. L. (2022). First and Second Level Haemoglobinopathies Diagnosis: Best Practices of the Italian Society of Thalassemia and Haemoglobinopathies (SITE). Journal of Clinical Medicine, 11(18), 5426. https://doi.org/10.3390/jcm11185426