
The Role of Human Herpesvirus 6 Infection in Alzheimer’s Disease Pathogenicity—A Theoretical Mosaic


Abstract
:1. Introduction
2. Methodology
3. Evidence of HHV-6 Infection and Alzheimer’s Disease

{kind=link}
{kind=link}
| Study Cohort, n (Controls) | Study Design | Main Results | Reference |
|---|---|---|---|
| Research reporting the possible role of HHV-6 in AD pathogenesis | |||
| 98 AD (no controls) | Materials: whole blood samples Method: genotyping kits for KIR and HLA alleles Research question: KIR/HLA genetic background in AD | KIR2DS2/KIR2DL2/C1 correlated with patients with lower MMSE score Indirect marker for increased susceptibility to HHV-6A infection | Rizzo et al., 2019 [16] |
| 643 AD (no controls) | Materials: brains from AD patients Method: functional genomic analysis, a multiscale network of LOAD-associated virome Research question: pathogenic HHV-6 regulation of molecular, clinical, and neuropathological networks | Increased HHV-6A from subjects with AD compared with controls | Readhead et al., 2018 [17] |
| 158 AD (228 controls) | Materials: Peripheral blood leukocyte samples Method: Single nucleotide polymorphism detection, genotyping Research question: Specific gene mutations associated with factors regulating antiviral response in AD patients | HHV-6 DNA is (statistically significant) more frequently encountered in AD groups vs. controls Overexpression of IL-28B TT carriers in AD patients Med23 and IRF7 GG genotypes correlated with HHV-6 risk for AD | Licastro et al., 2015 [18] |
| 93 AD (164 ND) | Materials: Peripheral blood leukocyte samples Method: qPCR, genotyping Research question: HHV-6 presence in peripheral blood of AD patients | Significantly increased positivity of HHV-6 in peripheral blood leukocyte samples and brain tissue in AD patients | Carbone et al., 2014 [19] |
| 27 AD (13 controls) | Materials: CSF and serum samples Method: ELISA, PCR Research question: Assessment of the immune response to HHV-6 in AD patients via the detection of intrathecal antibodies | Detectable intrathecal antibody synthesis to HHV-6 in AD patients (in low percentage) versus negative controls | Wozniak et al., 2005 [20] |
| 50 AD (35 controls) | Materials: Frozen postmortem brains Method: PCR Research question: HHV-6 detection in AD brain specimens | HHV-6 is present in the brain of a far higher proportion of AD patients than of age-matched controls | Lin et al., 2002 [15] |
| Research refuting HHV-6 involvement in AD pathogenesis | |||
| 575 definite AD (341 ND) | Materials: 3 independent AD cohorts Method: RNA-seq, PCR Research question: Screening for pathogens (including 118 human viruses) in AD patients | Little specificity of HHV-6 to AD brains over controls by both RNA-Seq and droplet digital PCR methods (no differences in viral detection between the two groups) | Allnutt et al., 2020 [21] |
| 602 AD (no controls) | Materials: Brain samples Method: KrakenUniq (highly sensitive method) Research question: Detection of extremely low HHV-6 read counts in AD brains | Identification via KrakenUniq of HHV-6A reads in only 2 out of the top 15 samples sorted by reported HHV-6A abundance | Chorlton et al., 2020 [22] |
| 50 AD (52 ND) | Materials: Blood samples Method: PCR, multiplex immunoassay Research question: Analysis of IgG reactivity toward several viruses in AD patients | HHV-6 IgG reactivity was significantly lower in AD compared to controls | Westman et al., 2017 [23] |
| 59 AD, 60 aMCI (61 controls) | Materials: Whole blood and serum samples Method: ELISA, MRI, and genotyping Research question: The analysis of HHV-6-specific humoral immunity in AD patients | HHV-6 seroprevalence, antibody titers, and avidity were similar in all three groups | Agostini et al., 2016 [24] |
| 34 AD (40 controls) | Materials: Brain specimens Method: PCR Research question: Detection in brain specimens for HHV-6 DNA | No significant difference for HHV-6 DNA in AD groups compared to the control group HHV-6 is no additional risk factor for AD | Hemling et al., 2003 [25] |
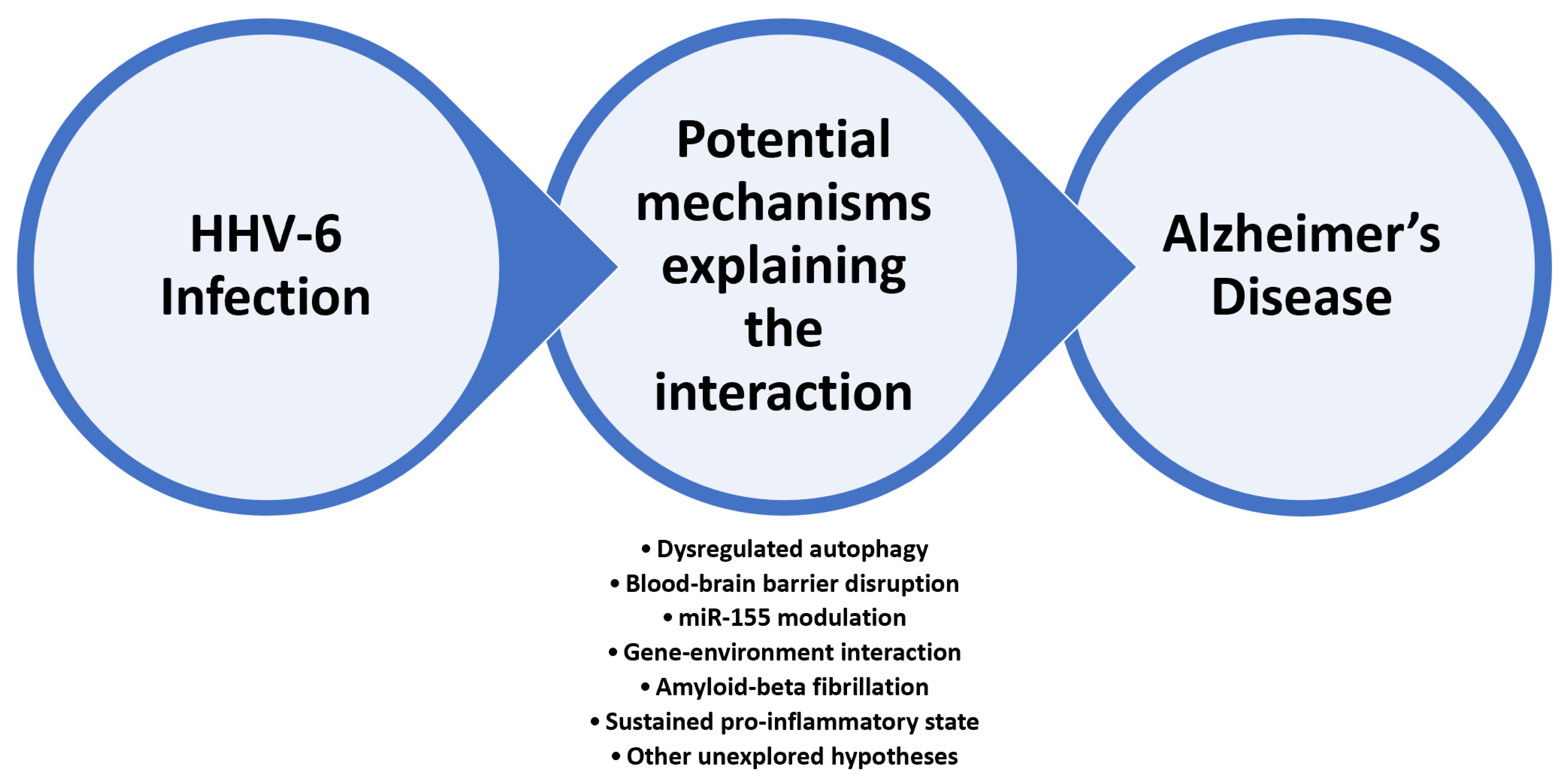
4. Research Directions Investigating HHV-6 Involvement in Alzheimer’s Disease—Selected Examples
4.1. HHV-6 as the Main Deregulator of Autophagy Mechanism at the CNS Level
4.2. miRNAs as a Valuable Link between HHV-6 Infection and Alzheimer’s Disease
4.3. HHV-6 and Amyloid Beta Fibrillation—Reframing the Amyloid Hypothesis
5. Related Mechanisms and Future Research Directions
5.1. From HHV-6 Childhood Infection to AD in Older Individuals—The Neuroinflammatory Hypothesis
5.2. HHV-6 CNS versus Peripheral Infection
5.3. From Neuroprotection to Neuroinflammation—The Double-Edged Blade
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Salahuddin, S.Z.; Ablashi, D.V.; Markham, P.D.; Josephs, S.F.; Sturzenegger, S.; Kaplan, M.; Halligan, G.; Biberfeld, P.; Wong-Staal, F.; Kramarsky, B.; et al. Isolation of a new virus, HBLV, in patients with lymphoproliferative disorders. Science 1986, 234, 596–601. [Google Scholar] [CrossRef] [PubMed]
- Pormohammad, A.; Azimi, T.; Falah, F.; Faghihloo, E. Relationship of human herpes virus 6 and multiple sclerosis: A systematic review and meta-analysis. J. Cell Physiol. 2018, 233, 2850–2862. [Google Scholar] [CrossRef] [PubMed]
- Bartolini, L.; Theodore, W.H.; Jacobson, S.; Gaillard, W.D. Infection with HHV-6 and its role in epilepsy. Epilepsy Res. 2019, 153, 34–39. [Google Scholar] [CrossRef]
- Santpere, G.; Telford, M.; Andrés-Benito, P.; Navarro, A.; Ferrer, I. The Presence of Human Herpesvirus 6 in the Brain in Health and Disease. Biomolecules. 2020, 10, 1520. [Google Scholar] [CrossRef] [PubMed]
- Emery, V.C.; Clark, D.A. HHV-6A, 6B, and 7: Persistence in the population, epidemiology and transmission. Vincent C Emery and Duncan A. Clark. In Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis; Arvin, A., Campadelli-Fiume, G., Mocarski, E., Moore, P.S., Roizman, B., Whitley, R., Yamanishi, K., Eds.; Cambridge University Press: Cambridge, UK, 2007; Chapter 49. [Google Scholar]
- Stone, R.C.; Micali, G.A.; Schwartz, R.A. Roseola infantum and its causal human herpesviruses. Int. J. Dermatol. 2014, 53, 397–403. [Google Scholar] [CrossRef]
- Eliassen, E.; Hemond, C.C.; Santoro, J.D. HHV-6-Associated Neurological Disease in Children: Epidemiologic, Clinical, Diagnostic, and Treatment Considerations. Pediatr. Neurol. 2020, 105, 10–20. [Google Scholar] [CrossRef]
- Soria Lopez, J.A.; González, H.M.; Léger, G.C. Alzheimer’s disease. Handb. Clin. Neurol. 2019, 167, 231–255. [Google Scholar] [CrossRef]
- Folch, J.; Petrov, D.; Ettcheto, M.; Abad, S.; Sánchez-López, E.; García, M.L.; Olloquequi, J.; Beas-Zarate, C.; Auladell, C.; Camins, A. Current Research Therapeutic Strategies for Alzheimer’s Disease Treatment. Neural. Plast. 2016, 2016, 8501693. [Google Scholar] [CrossRef] [Green Version]
- Cline, E.N.; Bicca, M.A.; Viola, K.L.; Klein, W.L. The Amyloid-β Oligomer Hypothesis: Beginning of the Third Decade. J. Alzheimers Dis. 2018, 64 (Suppl. S1), S567–S610. [Google Scholar] [CrossRef] [Green Version]
- Arnsten, A.F.T.; Datta, D.; Del Tredici, K.; Braak, H. Hypothesis: Tau pathology is an initiating factor in sporadic Alzheimer’s disease. Alzheimers Dement. 2021, 17, 115–124. [Google Scholar] [CrossRef]
- Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Tönnies, E.; Trushina, E. Oxidative Stress, Synaptic Dysfunction, and Alzheimer’s Disease. J. Alzheimers Dis. 2017, 57, 1105–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wouk, J.; Rechenchoski, D.Z.; Rodrigues, B.; Ribelato, E.V.; Faccin-Galhardi, L.C. Viral infections and their relationship to neurological disorders. Arch. Virol. 2021, 166, 733–753. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.R.; Wozniak, M.A.; Cooper, R.J.; Wilcock, G.K. Itzhaki RF. Herpesviruses in brain and Alzheimer’s disease. J. Pathol. 2002, 197, 395–402. [Google Scholar] [CrossRef]
- Rizzo, R.; Bortolotti, D.; Gentili, V.; Rotola, A.; Bolzani, S.; Caselli, E.; Tola, M.R.; Di Luca, D. KIR2DS2/KIR2DL2/HLA-C1 Haplotype Is Associated with Alzheimer’s Disease: Implication for the Role of Herpesvirus Infections. J. Alzheimers Dis. 2019, 67, 1379–1389. [Google Scholar] [CrossRef]
- Readhead, B.; Haure-Mirande, J.V.; Funk, C.C.; Richards, M.A.; Shannon, P.; Haroutunian, V.; Sano, M.; Liang, W.S.; Beckmann, N.D.; Price, N.; et al. Multiscale Analysis of Independent Alzheimer’s Cohorts Finds Disruption of Molecular, Genetic, and Clinical Networks by Human Herpesvirus. Neuron 2018, 99, 64–82.e7. [Google Scholar] [CrossRef] [Green Version]
- Licastro, F.; Raschi, E.; Carbone, I.; Porcellini, E. Variants in Antiviral Genes are Risk Factors for Cognitive Decline and Dementia. J. Alzheimers Dis. 2015, 46, 655–663. [Google Scholar] [CrossRef]
- Carbone, I.; Lazzarotto, T.; Ianni, M.; Porcellini, E.; Forti, P.; Masliah, E.; Gabrielli, L.; Licastro, F. Herpes virus in Alzheimer’s disease: Relation to progression of the disease. Neurobiol. Aging. 2014, 35, 122–912. [Google Scholar] [CrossRef]
- Wozniak, M.A.; Shipley, S.J.; Combrinck, M.; Wilcock, G.K.; Itzhaki, R.F. Productive herpes simplex virus in brain of elderly normal subjects and Alzheimer’s disease patients. J. Med. Virol. 2004, 75, 300–306. [Google Scholar] [CrossRef]
- Allnutt, M.A.; Johnson, K.; Bennett, D.A.; Connor, S.M.; Troncoso, J.C.; Pletnikova, O.; Albert, M.S.; Resnick, S.M.; Scholz, S.W.; De Jager, P.L.; et al. Human Herpesvirus 6 Detection in Alzheimer’s Disease Cases and Controls across Multiple Cohorts. Neuron 2020, 105, 1027–1035.e2. [Google Scholar] [CrossRef]
- Chorlton, S.D. Reanalysis of Alzheimer’s brain sequencing data reveals absence of purported HHV6A and HHV7. J. Bioinform Comput Biol. 2020, 18, 2050012. [Google Scholar] [CrossRef] [PubMed]
- Westman, G.; Blomberg, J.; Yun, Z.; Lannfelt, L.; Ingelsson, M.; Eriksson, B.M. Decreased HHV-6 IgG in Alzheimer’s Disease. Front. Neurol. 2017, 8, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agostini, S.; Mancuso, R.; Baglio, F.; Cabinio, M.; Hernis, A.; Guerini, F.R.; Calabrese, E.; Nemni, R.; Clerici, M. Lack of evidence for a role of HHV-6 in the pathogenesis of Alzheimer’s disease. J. Alzheimers Dis. 2016, 49, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Hemling, N.; Röyttä, M.; Rinne, J.; Pöllänen, P.; Broberg, E.; Tapio, V.; Vahlberg, T.; Hukkanen, V. Herpesviruses in brains in Alzheimer’s and Parkinson’s diseases. Ann. Neurol. 2003, 54, 267–271. [Google Scholar] [CrossRef] [PubMed]
- Bigley, T.M.; Xiong, M.; Ali, M.; Chen, Y.; Wang, C.; Serrano, J.R.; Eteleeb, A.; Harari, O.; Yang, L.; Patel, S.J.; et al. Murine roseolovirus does not accelerate amyloid-β pathology and human roseoloviruses are not over-represented in Alzheimer disease brains. Mol. Neurodegener. 2022, 17, 10. [Google Scholar] [CrossRef]
- Jeong, H.H.; Liu, Z. Are HHV-6A and HHV-7 Really More Abundant in Alzheimer’s Disease? Neuron 2019, 104, 1034–1035. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Q.; Ni, Y.; Le, W. Autophagy and Alzheimer’s Disease. Adv. Exp. Med. Biol. 2020, 1207, 3–19. [Google Scholar] [CrossRef]
- Menzies, F.M.; Fleming, A.; Caricasole, A.; Bento, C.F.; Andrews, S.P.; Ashkenazi, A.; Füllgrabe, J.; Jackson, A.; Jimenez Sanchez, M.; Karabiyik, C.; et al. Autophagy and Neurodegeneration: Pathogenic Mechanisms and Therapeutic Opportunities. Neuron 2017, 93, 1015–1034. [Google Scholar] [CrossRef] [Green Version]
- Ashrafian, H.; Zadeh, E.H.; Khan, R.H. Review on Alzheimer’s disease: Inhibition of amyloid beta and tau tangle formation. Int J. Biol. Macromol. 2021, 167, 382–394. [Google Scholar] [CrossRef]
- Xin, S.H.; Tan, L.; Cao, X.; Yu, J.T.; Tan, L. Clearance of Amyloid Beta and Tau in Alzheimer’s Disease: From Mechanisms to Therapy. Neurotox. Res. 2018, 34, 733–748. [Google Scholar] [CrossRef]
- Liu, J.; Li, L. Targeting Autophagy for the Treatment of Alzheimer’s Disease: Challenges and Opportunities. Front. Mol. Neurosci. 2019, 12, 203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, M.A.; Rahman, M.S.; Rahman, M.D.H.; Mamun-Or-Rashid, A.; Uddin, J.; Rahman; Hwang, H.; Pang, M.-G.; Rhim, H. Modulatory Effects of Autophagy on APP Processing as a Potential Treatment Target for Alzheimer’s Disease. Biomedicines 2020, 9, 5. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Wolfe, D.M.; Darji, S.; McBrayer, M.K.; Colacurcio, D.J.; Kumar, A.; Stavrides, P.; Mohan, P.S.; Nixon, R.A. β2-adrenergic Agonists Rescue Lysosome Acidification and Function in PSEN1 Deficiency by Reversing Defective ER-to-lysosome Delivery of ClC-7. J. Mol. Biol. 2020, 432, 2633–2650. [Google Scholar] [CrossRef]
- Romeo, M.A.; Masuelli, L.; Gaeta, A.; Nazzari, C.; Granato, M.; Gilardini Montani, M.S.; Faggioni, A.; Cirone, M. Impact of HHV-6A and HHV-6B lytic infection on autophagy and endoplasmic reticulum stress. J. Gen. Virol. 2019, 100, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Romeo, M.A.; Gilardini Montani, M.S.; Benedetti, R.; Giambelli, L.; D’Aprile, R.; Gaeta, A.; Faggioni, A.; Cirone, M. The cross-talk between STAT1/STAT3 and ROS up-regulates PD-L1 and promotes the release of pro-inflammatory/immune suppressive cytokines in primary monocytes infected by HHV-6B. Virus Res. 2021, 292, 198231. [Google Scholar] [CrossRef]
- Juźwik, C.A.; Drake, S.; Zhang, Y.; Paradis-Isler, N.; Sylvester, A.; Amar-Zifkin, A.; Douglas, C.; Morquette, B.; Moore, C.S.; Fournier, A.E. microRNA dysregulation in neurodegenerative diseases: A systematic review. Prog Neurobiol. 2019, 182, 101664. [Google Scholar] [CrossRef]
- Tribolet, L.; Kerr, E.; Cowled, C.; Bean, A.G.D.; Stewart, C.R.; Dearnley, M.; Farr, R.J. MicroRNA Biomarkers for Infectious Diseases: From Basic Research to Biosensing. Front. Microbiol. 2020, 11, 1197. [Google Scholar] [CrossRef]
- Adlakha, Y.K.; Neeru, S. Brain microRNAs and insights into biological functions and therapeutic potential of brain enriched miRNA-128. Mol. Cancer 2014, 13, 33. [Google Scholar] [CrossRef] [Green Version]
- McNeill, E.M.; Warinner, C.; Alkins, S.; Taylor, A.; Heggeness, H.; DeLuca, T.F.; Fulga, T.A.; Wall, D.P.; Griffith, L.C.; Van Vactor, D. The conserved microRNA miR-34 regulates synaptogenesis via coordination of distinct mechanisms in presynaptic and postsynaptic cells. Nat. Commun. 2020, 11, 1092. [Google Scholar] [CrossRef] [Green Version]
- Rastegar-Moghaddam, S.H.; Ebrahimzadeh-Bideskan, A.; Shahba, S.; Malvandi, A.M.; Mohammadipour, A. Roles of the miR-155 in Neuroinflammation and Neurological Disorders: A Potent Biological and Therapeutic Target. Cell Mol. Neurobiol. 2022. [Google Scholar] [CrossRef]
- Zhao, Y.; Jaber, V.; Alexandrov, P.N.; Vergallo, A.; Lista, S.; Hampel, H.; Lukiw, W.J. microRNA-Based Biomarkers in Alzheimer’s Disease (AD). Front. Neurosci. 2020, 14, 585432. [Google Scholar] [CrossRef] [PubMed]
- Simion, V.; Nadim, W.D.; Benedetti, H.; Pichon, C.; Morisset-Lopez, S.; Baril, P. Pharmacomodulation of microRNA Expression in Neurocognitive Diseases: Obstacles and Future Opportunities. Curr. Neuropharmacol. 2017, 15, 276–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guedes, J.R.; Santana, I.; Cunha, C.; Duro, D.; Almeida, M.; Cardoso, A.; de Lima, M.P. MicroRNA deregulation and chemotaxis and phagocytosis impairment in Alzheimer’s disease. Alzheimers Dement. 2015, 3, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Caselli, E.; D’Accolti, M.; Soffritti, I.; Zatelli, M.C.; Rossi, R.; Degli Uberti, E.; Di Luca, D. HHV-6A in vitro infection of thyrocytes and T cells alters the expression of miRNA associated to autoimmune thyroiditis. Virol. J. 2017, 14, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sierksma, A.; Lu, A.; Salta, E.; Eynden, E.V.; Callaerts-Vegh, Z.; D’Hooge, R.; Blum, D.; Buée, L.; Fiers, M.; De Strooper, B. Deregulation of neuronal miRNAs induced by amyloid-β or TAU pathology. Mol. Neurodegener. 2018, 13, 54. [Google Scholar] [CrossRef] [Green Version]
- Patrick, E.; Rajagopal, S.; Wong, H.A.; McCabe, C.; Xu, J.; Tang, A.; Imboywa, S.H.; Schneider, J.A.; Pochet, N.; Krichevsky, A.M.; et al. Dissecting the role of non-coding RNAs in the accumulation of amyloid and tau neuropathologies in Alzheimer’s disease. Mol. Neurodegener. 2017, 12, 51. [Google Scholar] [CrossRef]
- Sun, X.H.; Song, M.F.; Song, H.D.; Wang, Y.W.; Luo, M.J.; Yin, L.M. miR155 mediates inflammatory injury of hippocampal neuronal cells via the activation of microglia. Mol. Med. Rep. 2019, 19, 2627–2635. [Google Scholar] [CrossRef]
- Nguyen, A.T.; Wang, K.; Hu, G.; Wang, X.; Miao, Z.; Azevedo, J.A.; Suh, E.; Van Deerlin, V.M.; Choi, D.; Roeder, K.; et al. APOE and TREM2 regulate amyloid-responsive microglia in Alzheimer’s disease. Acta Neuropathol. 2020, 140, 477–493. [Google Scholar] [CrossRef]
- Zheng, H.; Cheng, B.; Li, Y.; Li, X.; Chen, X.; Zhang, Y.W. TREM2 in Alzheimer’s Disease: Microglial Survival and Energy Metabolism. Front. Aging Neurosci. 2018, 10, 395. [Google Scholar] [CrossRef] [Green Version]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017, 47, 566–581.e9. [Google Scholar] [CrossRef] [Green Version]
- Vigorito, E.; Perks, K.L.; Abreu-Goodger, C.; Bunting, S.; Xiang, Z.; Kohlhaas, S.; Das, P.P.; Miska, E.; Rodriguez, A.; Bradley, A.; et al. microRNA-155 regulates the generation of immunoglobulin class-switched plasma cells. Immunity 2007, 27, 847–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pashangzadeh, S.; Motallebnezhad, M.; Vafashoar, F.; Khalvandi, A.; Mojtabavi, N. Implications the Role of miR-155 in the Pathogenesis of Autoimmune Diseases. Front. Immunol. 2021, 12, 669382. [Google Scholar] [CrossRef] [PubMed]
- Dunand-Sauthier, I.; Santiago-Raber, M.L.; Capponi, L.; Vejnar, C.; Schaad, O.; Irla, M.; Estévez, M.Q.S.; Descombes, P.; Zdobnov, E.M.; Acha-Orbea, H.; et al. Silencing of c-Fos expression by microRNA-155 is critical for dendritic cell maturation and function. Blood 2011, 117, 4490–4500. [Google Scholar] [CrossRef]
- Pasca, S.; Jurj, A.; Petrushev, B.; Tomuleasa, C.; Matei, D. MicroRNA-155 Implication in M1 Polarization and the Impact in Inflammatory Diseases. Front. Immunol. 2020, 11, 625. [Google Scholar] [CrossRef] [PubMed]
- Readhead, B.; Haure-Mirande, J.V.; Mastroeni, D.; Audrain, M.; Fanutza, T.; Kim, S.H.; Blitzer, R.D.; Gandy, S.; Dudley, J.T.; Ehrlich, M.E.; et al. miR155 regulation of behavior, neuropathology, and cortical transcriptomics in Alzheimer’s disease. Acta Neuropathol. 2020, 140, 295–315. [Google Scholar] [CrossRef]
- Eimer, W.A.; Vijaya Kumar, D.K.; Navalpur Shanmugam, N.K.; Rodriguez, A.S.; Mitchell, T.; Washicosky, K.J.; György, B.; Breakefield, X.O.; Tanzi, R.E.; Moir, R.D. Alzheimer’s Disease-Associated β-Amyloid Is Rapidly Seeded by Herpesviridae to Protect against Brain Infection [published correction appears in Neuron. Neuron 2018, 99, 56–63.e3. [Google Scholar] [CrossRef] [Green Version]
- Coates, M.M.; Kintu, A.; Gupta, N.; Wroe, E.B.; Adler, A.J.; Kwan, G.F.; Park, P.H.; Rajbhandari, R.; Byrne, A.L.; Casey, D.C.; et al. Burden of non-communicable diseases from infectious causes in 2017: A modelling study. Lancet Glob. Health 2020, 8, e1489–e1498. [Google Scholar] [CrossRef]
- Lotz, S.K.; Blackhurst, B.M.; Reagin, K.L.; Funk, K.E. Microbial Infections Are a Risk Factor for Neurodegenerative Diseases. Front. Cell Neurosci. 2021, 15, 691136. [Google Scholar] [CrossRef]
- Li, L.; Mao, S.; Wang, J.; Ding, X.; Zen, J.Y. Viral infection and neurological disorders—potential role of extracellular nucleotides in neuroinflammation. ExRNA 2019, 1, 26. [Google Scholar] [CrossRef] [Green Version]
- Bradburn, S.; Murgatroyd, C.; Ray, N. Neuroinflammation in mild cognitive impairment and Alzheimer’s disease: A meta-analysis. Ageing Res. Rev. 2019, 50, 1–8. [Google Scholar] [CrossRef]
- Fan, L.; Mao, C.; Hu, X.; Zhang, S.; Yang, Z.; Hu, Z.; Sun, H.; Fan, Y.; Dong, Y.; Yang, J.; et al. New Insights Into the Pathogenesis of Alzheimer’s Disease. Front. Neurol. 2020, 10, 1312. [Google Scholar] [CrossRef] [PubMed]
- Urban, S.L.; Jensen, I.J.; Shan, Q.; Pewe, L.L.; Xue, H.H.; Badovinac, V.P.; Harty, J.T. Peripherally induced brain tissue-resident memory CD8+ T cells mediate protection against CNS infection. Nat. Immunol. 2020, 21, 938–949. [Google Scholar] [CrossRef] [PubMed]
- Rutsch, A.; Kantsjö, J.B.; Ronchi, F. The Gut-Brain Axis: How Microbiota and Host Inflammasome Influence Brain Physiology and Pathology. Front. Immunol. 2020, 11, 604179. [Google Scholar] [CrossRef] [PubMed]
- Skuja, S.; Svirskis, S.; Murovska, M. Human Herpesvirus-6 and -7 in the Brain Microenvironment of Persons with Neurological Pathology and Healthy People. Int. J. Mol. Sci. 2021, 22, 2364. [Google Scholar] [CrossRef] [PubMed]
- Bortolotti, D.; Gentili, V.; Rotola, A.; Caselli, E.; Rizzo, R. HHV-6A infection induces amyloid-beta expression and activation of microglial cells. Alzheimers Res. Ther. 2019, 11, 104. [Google Scholar] [CrossRef] [Green Version]
- Woodburn, S.C.; Bollinger, J.L.; Wohleb, E.S. The semantics of microglia activation: Neuroinflammation, homeostasis, and stress. J. Neuroinflamm. 2021, 18, 258. [Google Scholar] [CrossRef]
- Telford, M.; Navarro, A.; Santpere, G. Whole genome diversity of inherited chromosomally integrated HHV-6 derived from healthy individuals of diverse geographic origin. Sci. Rep. 2018, 8, 3472. [Google Scholar] [CrossRef] [Green Version]
- Pantry, S.N.; Medveczky, P.G. Latency, Integration, and Reactivation of Human Herpesvirus-6. Viruses 2017, 9, 194. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Ding, L.; Zhu, Q.; Shu, M.; Cai, Q. Common Infections May Lead to Alzheimer’s Disease. Virol. Sin. 2018, 33, 456–458. [Google Scholar] [CrossRef]
- Shao, Q.; Lin, Z.; Wu, X.; Tang, J.; Lu, S.; Feng, D.; Cheng, C.; Qing, L.; Yao, K.; Chen, Y. Transcriptome sequencing of neurologic diseases associated genes in HHV-6A infected human astrocyte. Oncotarget 2016, 7, 48070–48080. [Google Scholar] [CrossRef] [Green Version]
- Costa Sa, A.C.; Madsen, H.; Brown, J.R. Shared Molecular Signatures Across Neurodegenerative Diseases and Herpes Virus Infections Highlights Potential Mechanisms for Maladaptive Innate Immune Responses. Sci. Rep. 2019, 9, 8795. [Google Scholar] [CrossRef] [PubMed]
- Mor, A.; Tankiewicz-Kwedlo, A.; Krupa, A.; Pawlak, D. Role of Kynurenine Pathway in Oxidative Stress during Neurodegenerative Disorders. Cells. 2021, 10, 1603. [Google Scholar] [CrossRef] [PubMed]
- Harberts, E.; Yao, K.; Wohler, J.E.; Maric, D.; Ohayon, J.; Henkin, R.; Jacobson, S. Human herpesvirus-6 entry into the central nervous system through the olfactory pathway. Proc. Natl. Acad. Sci. USA 2011, 108, 13734–13739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Profaci, C.P.; Munji, R.N.; Pulido, R.S.; Daneman, R. The blood-brain barrier in health and disease: Important unanswered questions. J. Exp. Med. 2020, 217, e20190062. [Google Scholar] [CrossRef]
- Liebner, S.; Dijkhuizen, R.M.; Reiss, Y.; Plate, K.H.; Agalliu, D.; Constantin, G. Functional morphology of the blood-brain barrier in health and disease. Acta Neuropathol. 2018, 135, 311–336. [Google Scholar] [CrossRef] [Green Version]
- Schreiner, T.G.; Romanescu, C.; Popescu, B.O. The Blood–Brain Barrier—A Key Player in Multiple Sclerosis Disease Mechanisms. Biomolecules 2022, 12, 538. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Li, G. Immune response and blood-brain barrier dysfunction during viral neuroinvasion. Innate Immun. 2021, 27, 109–117. [Google Scholar] [CrossRef]
- Tansey, M.G.; Wallings, R.L.; Houser, M.C.; Herrick, M.K.; Keating, C.E.; Joers, V. Inflammation and immune dysfunction in Parkinson disease. Nat. Rev. Immunol. 2022, 122, 1–17. [Google Scholar] [CrossRef]
- Bettcher, B.M.; Tansey, M.G.; Dorothée, G.; Heneka, M.T. Peripheral and central immune system crosstalk in Alzheimer disease—A research prospectus [published correction appears.]. Nat. Rev. Neurol. 2021, 17, 689–701. [Google Scholar] [CrossRef]
- Konsman, J.P. Cytokines in the Brain and Neuroinflammation: We Didn’t Starve the Fire! Pharmaceuticals 2022, 15, 140. [Google Scholar] [CrossRef]
- Bettcher, B.M.; Johnson, S.C.; Fitch, R.; Casaletto, K.B.; Heffernan, K.S.; Asthana, S.; Zetterberg, H.; Blennow, K.; Carlsson, C.M.; Neuhaus, J.; et al. Cerebrospinal Fluid and Plasma Levels of Inflammation Differentially Relate to CNS Markers of Alzheimer’s Disease Pathology and Neuronal Damage. J. Alzheimers Dis. 2018, 62, 385–397. [Google Scholar] [CrossRef] [PubMed]
- Faridar, A.; Thome, A.D.; Zhao, W.; Thonhoff, J.R.; Beers, D.R.; Pascual, B.; Masdeu, J.C.; Appel, S.H. Restoring regulatory T-cell dysfunction in Alzheimer’s disease through ex vivo expansion. Brain. Commun. 2020, 2, fcaa112. [Google Scholar] [CrossRef] [PubMed]
- Gate, D.; Saligrama, N.; Leventhal, O.; Yang, A.C.; Unger, M.S.; Middeldorp, J.; Chen, K.; Lehallier, B.; Channappa, D.; De Los Santos, M.B.; et al. Clonally expanded CD8 T cells patrol the cerebrospinal fluid in Alzheimer’s disease. Nature 2020, 577, 399–404. [Google Scholar] [CrossRef] [PubMed]
- Merlini, M.; Kirabali, T.; Kulic, L.; Nitsch, R.M.; Ferretti, M.T. Extravascular CD3+ T Cells in Brains of Alzheimer Disease Patients Correlate with Tau but Not with Amyloid Pathology: An Immunohistochemical Study. Neurodegener Dis. 2018, 18, 49–56. [Google Scholar] [CrossRef]
- Murray, E.R.; Kemp, M.; Nguyen, T.T. The Microbiota-Gut-Brain Axis in Alzheimer’s Disease: A Review of Taxonomic Alterations and Potential Avenues for Interventions. Arch. Clin. Neuropsychol. 2022, 37, 595–607. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Lee, J.E. miR-155 is involved in Alzheimer’s disease by regulating T lymphocyte function. Front. Aging Neurosci. 2015, 7, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Chen, Q.; Nai, Y.; Cao, C. Suppression of miR-155 attenuates neuropathic pain by inducing an M1 to M2 switch in microglia. Folia Neuropathol. 2020, 58, 70–82. [Google Scholar] [CrossRef]
- Komaroff, A.L.; Pellett, P.E.; Jacobson, S. Human Herpesviruses 6A and 6B in Brain Diseases: Association versus Causation. Clin. Microbiol Rev. 2020, 34, e00143-20. [Google Scholar] [CrossRef]

| Search | Keywords |
|---|---|
| #1 | “HHV-6” OR “HHV-6A” OR “HHV-6B” OR “Human herpesvirus |
| #2 | “Alzheimer’s disease” OR “Alzheimer’s dementia” OR “dementia” |
| #3 | #1 AND #2 |
| Final results | |
| Identified records | PubMed (n = 44) Google Scholar (n = 639) Science Direct (n = 228) |
| Excluded records (duplicates, not eligible) | n = 891 |
| Included records in the review | n = 20 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Romanescu, C.; Schreiner, T.G.; Mukovozov, I. The Role of Human Herpesvirus 6 Infection in Alzheimer’s Disease Pathogenicity—A Theoretical Mosaic. J. Clin. Med. 2022, 11, 3061. https://doi.org/10.3390/jcm11113061
Romanescu C, Schreiner TG, Mukovozov I. The Role of Human Herpesvirus 6 Infection in Alzheimer’s Disease Pathogenicity—A Theoretical Mosaic. Journal of Clinical Medicine. 2022; 11(11):3061. https://doi.org/10.3390/jcm11113061
Chicago/Turabian StyleRomanescu, Constantin, Thomas Gabriel Schreiner, and Ilya Mukovozov. 2022. "The Role of Human Herpesvirus 6 Infection in Alzheimer’s Disease Pathogenicity—A Theoretical Mosaic" Journal of Clinical Medicine 11, no. 11: 3061. https://doi.org/10.3390/jcm11113061
APA StyleRomanescu, C., Schreiner, T. G., & Mukovozov, I. (2022). The Role of Human Herpesvirus 6 Infection in Alzheimer’s Disease Pathogenicity—A Theoretical Mosaic. Journal of Clinical Medicine, 11(11), 3061. https://doi.org/10.3390/jcm11113061