
De Novo L509P Mutation of the TGFBI Gene Associated with Slit-Lamp Findings of Lattice Corneal Dystrophy Type IIIA

 ,
,  ,
, {kind=link}
{kind=link}
Abstract
:1. Background
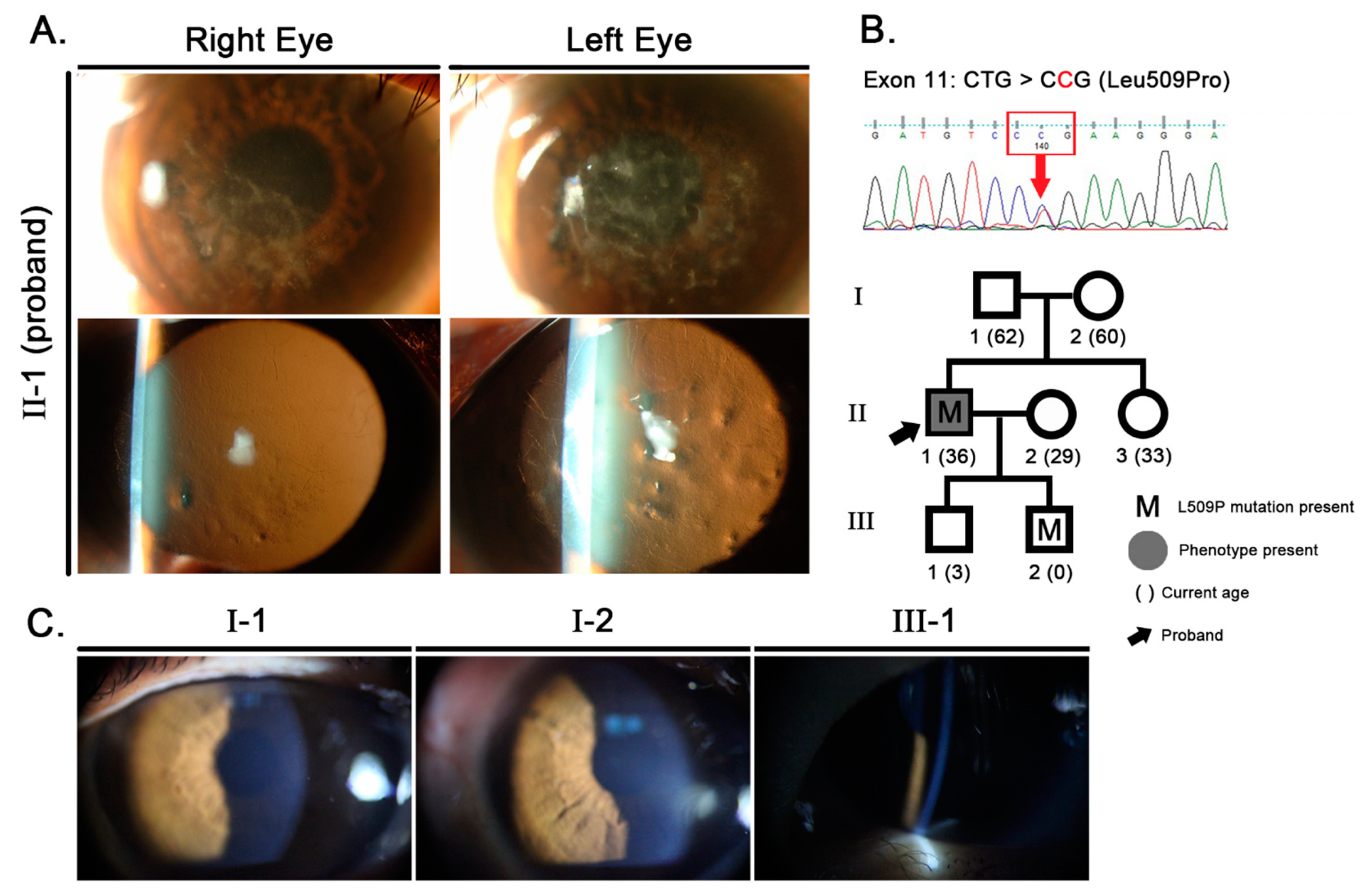
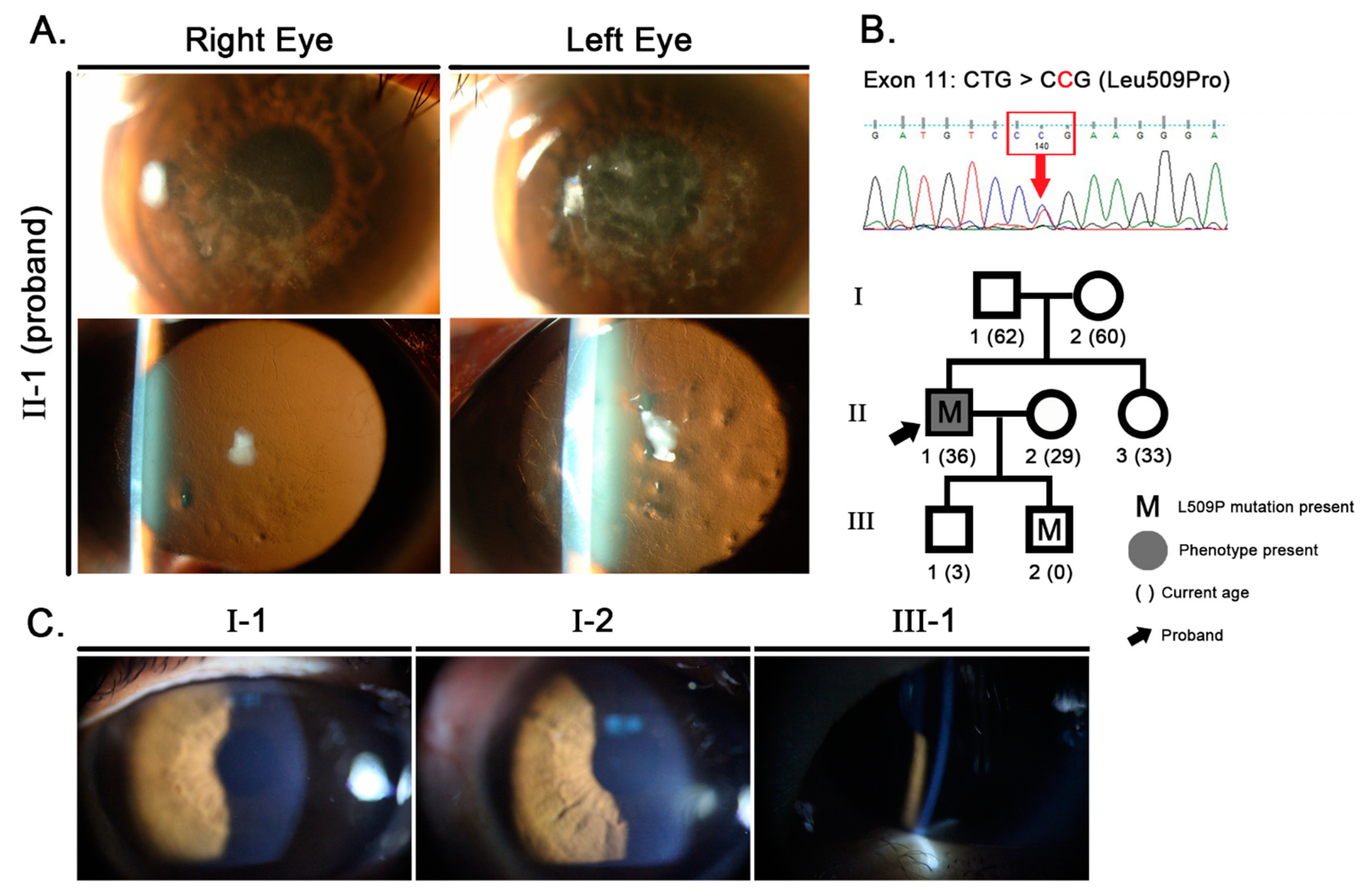
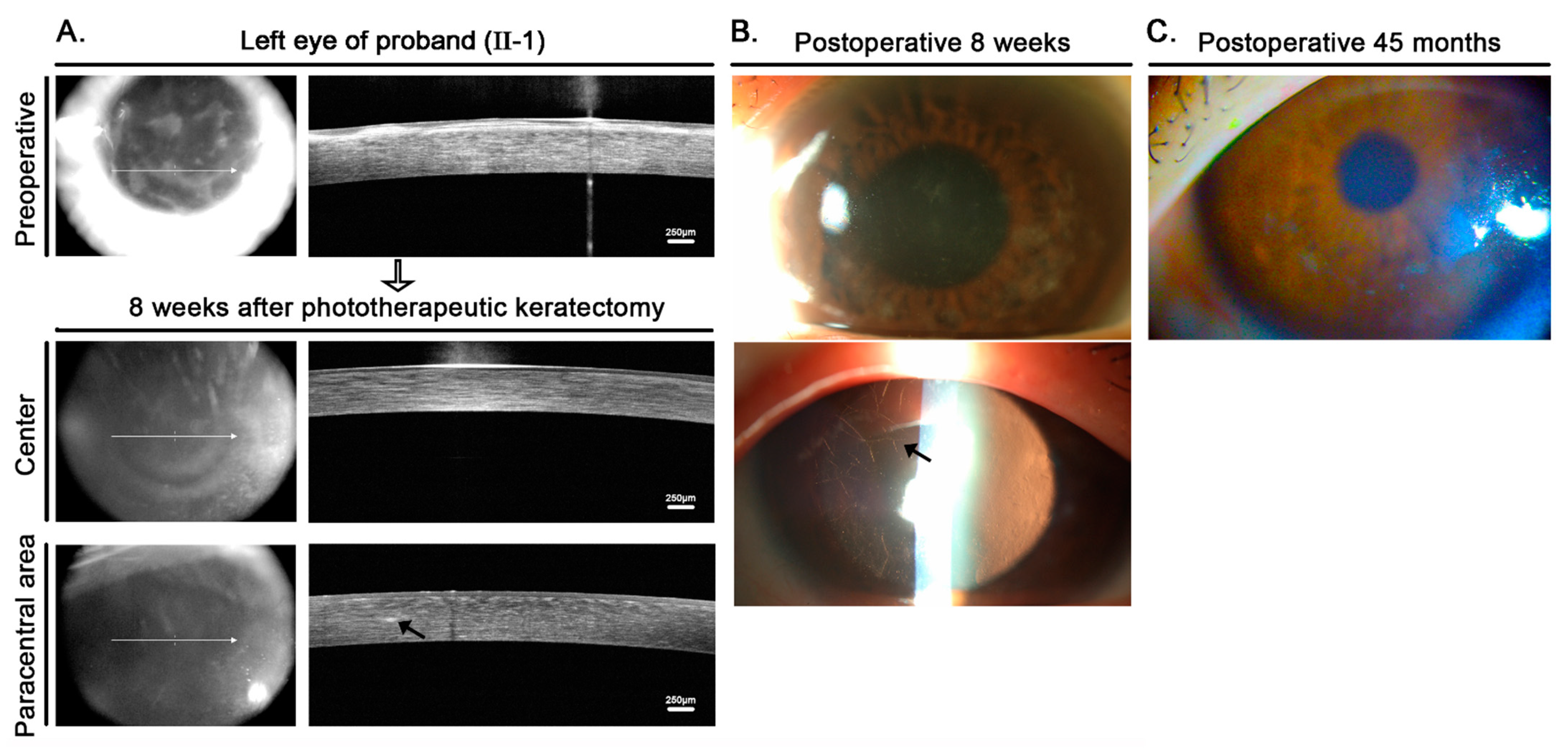
2. Case Presentation
2.1. Clinical Analysis
2.2. Molecular Genetic Analysis
2.3. Confirmation of Paternity
3. Discussion and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
Appendix A. Materials and Methods
Appendix A.1. Patients and Clinical Evaluation
Appendix A.2. Genomic DNA Preparation and Mutation Analysis
References
- Hoersch, S.; Andrade-Navarro, M.A. Periostin shows increased evolutionary plasticity in its alternatively spliced region. BMC Evol. Biol. 2010, 10, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, X.; Cai, L.; Li, Y.; Zhu, J.; Jin, P.; Chen, L.; Ma, F. Identification and characterization of transforming growth factor beta induced gene (TGFBIG) from Branchiostoma belcheri: Insights into evolution of TGFBI family. Genomics 2014, 103, 147–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, J.S.; Moller, H.U.; Aldave, A.J.; Seitz, B.; Bredrup, C.; Kivela, T.; Munier, F.L.; Rapuano, C.J.; Nischal, K.K.; Kim, E.K.; et al. IC3D Classification of Corneal Dystrophies-Edition 2. Cornea 2015, 34, 117–159. [Google Scholar] [CrossRef] [Green Version]
- Han, K.E.; Choi, S.I.; Kim, T.I.; Maeng, Y.S.; Stulting, R.D.; Ji, Y.W.; Kim, E.K. Pathogenesis and Treatments of TGFBI Corneal Dystrophies. Prog. Retin. Eye Res. 2015, 50, 67–88. [Google Scholar] [CrossRef]
- Han, K.E.; Choi, S.I.; Chung, W.S.; Jung, S.H.; Katsanis, N.; Kim, T.I.; Kim, E.K. Extremely varied phenotypes in granular corneal dystrophy type 2 heterozygotes. Mol. Vis. 2012, 18, 1755–1762. [Google Scholar]
- Lakshminarayanan, R.; Chaurasia, S.S.; Murugan, E.; Venkatraman, A.; Chai, S.M.; Vithana, E.N.; Beuerman, R.W.; Mehta, J.S. Biochemical properties and aggregation propensity of transforming growth factor-induced protein (TGFBIp) and the amyloid forming mutants. Ocul. Surf. 2015, 13, 9–25. [Google Scholar] [CrossRef]
- Gruenauer-Kloevekorn, C.; Clausen, I.; Weidle, E.; Wolter-Roessler, M.; Tost, F.; Volcker, H.E.; Schulze, D.P.; Heinritz, W.; Reinhard, T.; Froster, U.; et al. TGFBI (BIGH3) gene mutations in German families: Two novel mutations associated with unique clinical and histopathological findings. Br. J. Ophthalmol. 2009, 93, 932–937. [Google Scholar] [CrossRef]
- Niel-Butschi, F.; Kantelip, B.; Iwaszkiewicz, J.; Zoete, V.; Boimard, M.; Delpech, M.; Bourges, J.L.; Renard, G.; D’Hermies, F.; Pisella, P.J.; et al. Genotype-phenotype correlations of TGFBI p.Leu509Pro, p.Leu509Arg, p.Val613Gly, and the allelic association of p.Met502Val-p.Arg555Gln mutations. Mol. Vis. 2011, 17, 1192–1202. [Google Scholar]
- Lisch, W.; Seitz, B. Lattice corneal dystrophy type 1: An epithelial or stromal entity? Cornea 2014, 33, 1109–1112. [Google Scholar] [CrossRef]
- Crow, J.F. The origins, patterns and implications of human spontaneous mutation. Nat. Rev. Genet. 2000, 1, 40–47. [Google Scholar] [CrossRef]
- Haldane, J.B. The rate of spontaneous mutation of a human gene. 1935. J. Genet. 2004, 83, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Drake, J.W.; Charlesworth, B.; Charlesworth, D.; Crow, J.F. Rates of spontaneous mutation. Genetics 1998, 148, 1667–1686. [Google Scholar] [CrossRef] [PubMed]
- Drost, J.B.; Lee, W.R. Biological basis of germline mutation: Comparisons of spontaneous germline mutation rates among drosophila, mouse, and human. Environ. Mol. Mutagen. 1995, 25 (Suppl. 26), 48–64. [Google Scholar] [CrossRef]
- Drayna, D. Founder mutations. Sci. Am. 2005, 293, 78–85. [Google Scholar] [CrossRef]
- Tanhehco, T.Y.; Eifrig, D.E.; Schwab, I.R., Jr.; Rapuano, C.J.; Klintworth, G.K. Two cases of Reis-Bucklers corneal dystrophy (granular corneal dystrophy type III) caused by spontaneous mutations in the TGFBI gene. Arch. Ophthalmol. 2006, 124, 589–593. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.C.; Nakamura, H.; Subramanyam, S.; Stock, L.E.; Gillette, T.E.; Yoshikawa, S.; Ma, X.; Yee, R.W. Spontaneous and inheritable R555Q mutation in the TGFBI/BIGH3 gene in two unrelated families exhibiting Bowman’s layer corneal dystrophy. Ophthalmology 2007, 114, e39–e46. [Google Scholar] [CrossRef]
- Hou, Y.C.; Wang, I.J.; Hsiao, C.H.; Chen, W.L.; Hu, F.R. Phenotype-genotype correlations in patients with TGFBI-linked corneal dystrophies in Taiwan. Mol. Vis. 2012, 18, 362–371. [Google Scholar]
- Korvatska, E.; Munier, F.L.; Djemai, A.; Wang, M.X.; Frueh, B.; Chiou, A.G.; Uffer, S.; Ballestrazzi, E.; Braunstein, R.E.; Forster, R.K.; et al. Mutation hot spots in 5q31-linked corneal dystrophies. Am. J. Hum. Genet. 1998, 62, 320–324. [Google Scholar] [CrossRef] [Green Version]
- Maki, H. Origins of spontaneous mutations: Specificity and directionality of base-substitution, frameshift, and sequence-substitution mutageneses. Annu. Rev. Genet. 2002, 36, 279–303. [Google Scholar] [CrossRef]
- Griffiths, J.F.; Griffiths, A.J.; Wessler, S.R.; Lewontin, R.C.; Gelbart, W.M.; Suzuki, D.T.; Miller, J.H. An Introduction to Genetic Analysis, 10th ed.; W. H. Freeman & Company: New York, NY, USA, 2012. [Google Scholar]
- Das, S.; Langenbucher, A.; Seitz, B. Excimer laser phototherapeutic keratectomy for granular and lattice corneal dystrophy: A comparative study. J. Refract. Surg. 2005, 21, 727–731. [Google Scholar] [CrossRef]
- Jung, S.H.; Han, K.E.; Stulting, R.D.; Sgrignoli, B.; Kim, T.I.; Kim, E.K. Phototherapeutic keratectomy in diffuse stromal haze in granular corneal dystrophy type 2. Cornea 2013, 32, 296–300. [Google Scholar] [CrossRef]
- Chen, M.; Xie, L. Features of recurrence after excimer laser phototherapeutic keratectomy for anterior corneal pathologies in North China. Ophthalmology 2013, 120, 1179–1185. [Google Scholar] [CrossRef]
- Jun, I.; Jung, J.W.; Choi, Y.J.; Kim, T.-i.; Seo, K.Y.; Kim, E.K. Long-term Clinical Outcomes of Phototherapeutic Keratectomy in Corneas With Granular Corneal Dystrophy Type 2 Exacerbated After LASIK. J. Refract. Surg. 2018, 34, 132–139. [Google Scholar] [CrossRef] [Green Version]
- Jung, J.W.; Kim, S.A.; Kang, E.M.; Kim, T.-i.; Cho, H.S.; Kim, E.K. Lattice corneal dystrophy type IIIA with hyaline component from a novel A620P mutation and distinct surgical treatments. Cornea 2014, 33, 1324–1331. [Google Scholar] [CrossRef]
- Kocak-Altintas, A.G.; Kocak-Midillioglu, I.; Akarsu, A.N.; Duman, S. BIGH3 gene analysis in the differential diagnosis of corneal dystrophies. Cornea 2001, 20, 64–68. [Google Scholar] [CrossRef]
- Klintworth, G.K. Advances in the molecular genetics of corneal dystrophies. Am. J. Ophthalmol. 1999, 128, 747–754. [Google Scholar] [CrossRef]
- Jun, I.; Lee, J.S.; Lee, J.H.; Lee, C.S.; Choi, S.I.; Gee, H.Y.; Lee, M.G.; Kim, E.K. Adult-Onset Vitelliform Macular Dystrophy caused by BEST1 p.Ile38Ser Mutation is a Mild Form of Best Vitelliform Macular Dystrophy. Sci. Rep. 2017, 7, 9146. [Google Scholar] [CrossRef]
- Thomson, D.M.; Brown, N.N.; Clague, A.E. Routine use of hair root or buccal swab specimens for PCR analysis: Advantages over using blood. Clin. Chim. Acta. 1992, 207, 169–174. [Google Scholar] [CrossRef]
- Joki-Erkkila, M.; Tuomisto, S.; Seppanen, M.; Huhtala, H.; Ahola, A.; Karhunen, P.J. Urine specimen collection following consensual intercourse—A forensic evidence collection method for Y-DNA and spermatozoa. J. Forensic Legal. Med. 2016, 37, 50–54. [Google Scholar] [CrossRef]
- Park, J.H.; Hong, S.B.; Kim, J.Y.; Chong, Y.; Han, S.; Jeon, C.H.; Ahn, H.J. Genetic variation of 23 autosomal STR loci in Korean population. Forensic Sci. Int. Genet. 2013, 7, e76–e77. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ji, Y.W.; Ahn, H.; Shin, K.-J.; Kim, T.-i.; Seo, K.Y.; Stulting, R.D.; Kim, E.K. De Novo L509P Mutation of the TGFBI Gene Associated with Slit-Lamp Findings of Lattice Corneal Dystrophy Type IIIA. J. Clin. Med. 2022, 11, 3055. https://doi.org/10.3390/jcm11113055
Ji YW, Ahn H, Shin K-J, Kim T-i, Seo KY, Stulting RD, Kim EK. De Novo L509P Mutation of the TGFBI Gene Associated with Slit-Lamp Findings of Lattice Corneal Dystrophy Type IIIA. Journal of Clinical Medicine. 2022; 11(11):3055. https://doi.org/10.3390/jcm11113055
Chicago/Turabian StyleJi, Yong Woo, Hyunmin Ahn, Kyoung-Jin Shin, Tae-im Kim, Kyoung Yul Seo, R. Doyle Stulting, and Eung Kweon Kim. 2022. "De Novo L509P Mutation of the TGFBI Gene Associated with Slit-Lamp Findings of Lattice Corneal Dystrophy Type IIIA" Journal of Clinical Medicine 11, no. 11: 3055. https://doi.org/10.3390/jcm11113055
APA StyleJi, Y. W., Ahn, H., Shin, K.-J., Kim, T.-i., Seo, K. Y., Stulting, R. D., & Kim, E. K. (2022). De Novo L509P Mutation of the TGFBI Gene Associated with Slit-Lamp Findings of Lattice Corneal Dystrophy Type IIIA. Journal of Clinical Medicine, 11(11), 3055. https://doi.org/10.3390/jcm11113055