
Host Epigenetic Alterations and Hepatitis B Virus-Associated Hepatocellular Carcinoma


Abstract
1. Introduction
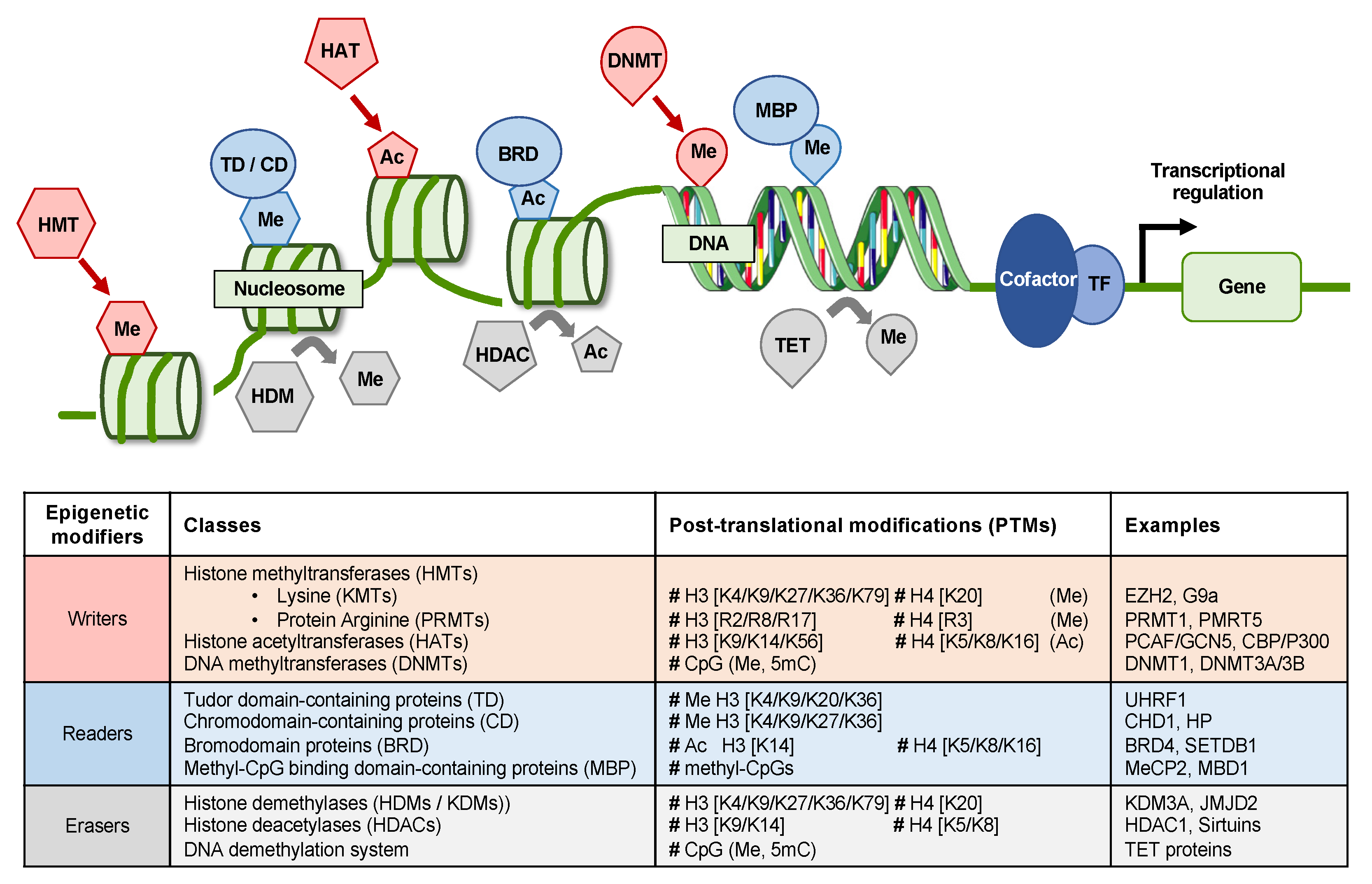
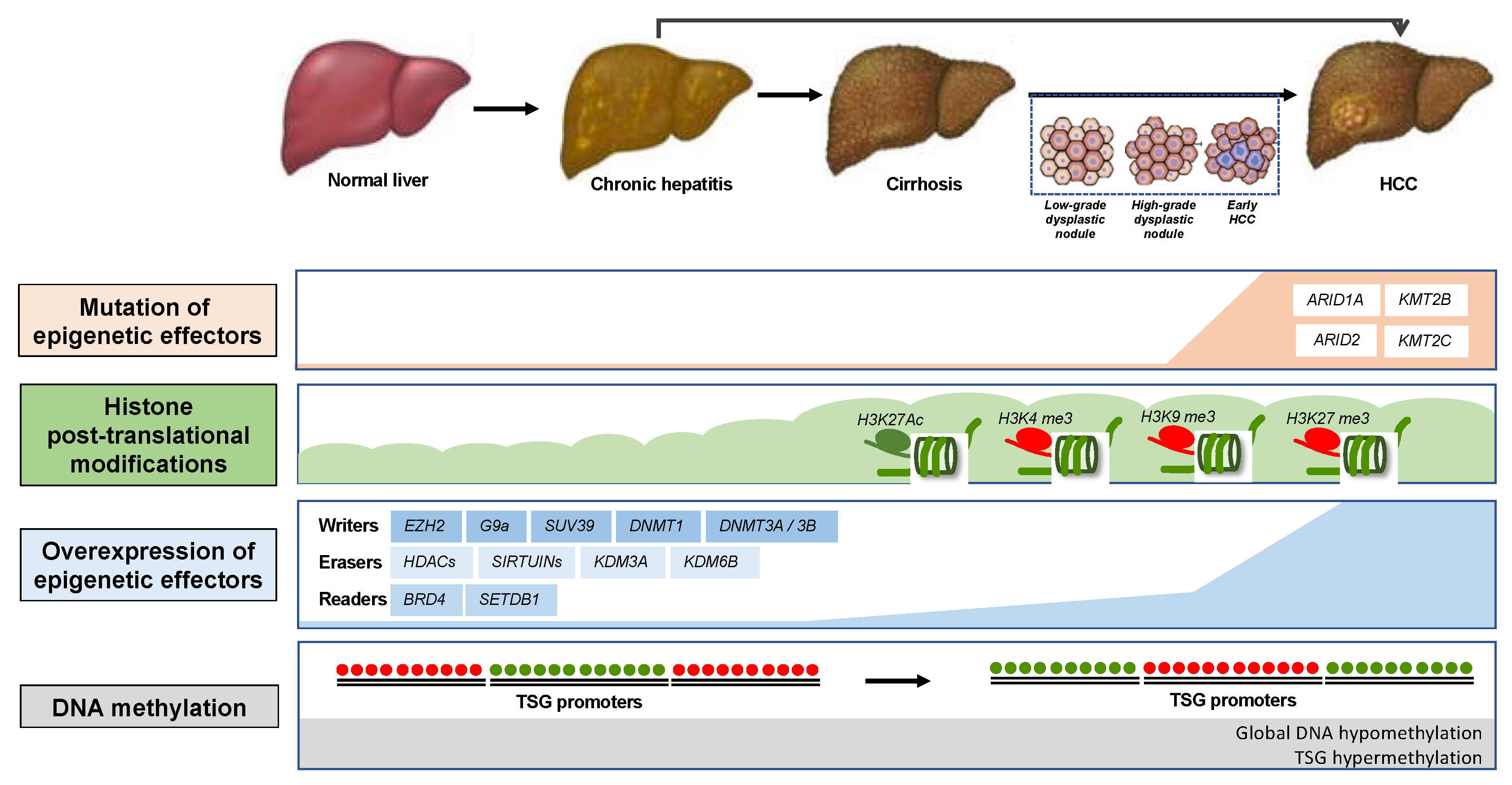
2. Epigenetic Mechanisms in HCC
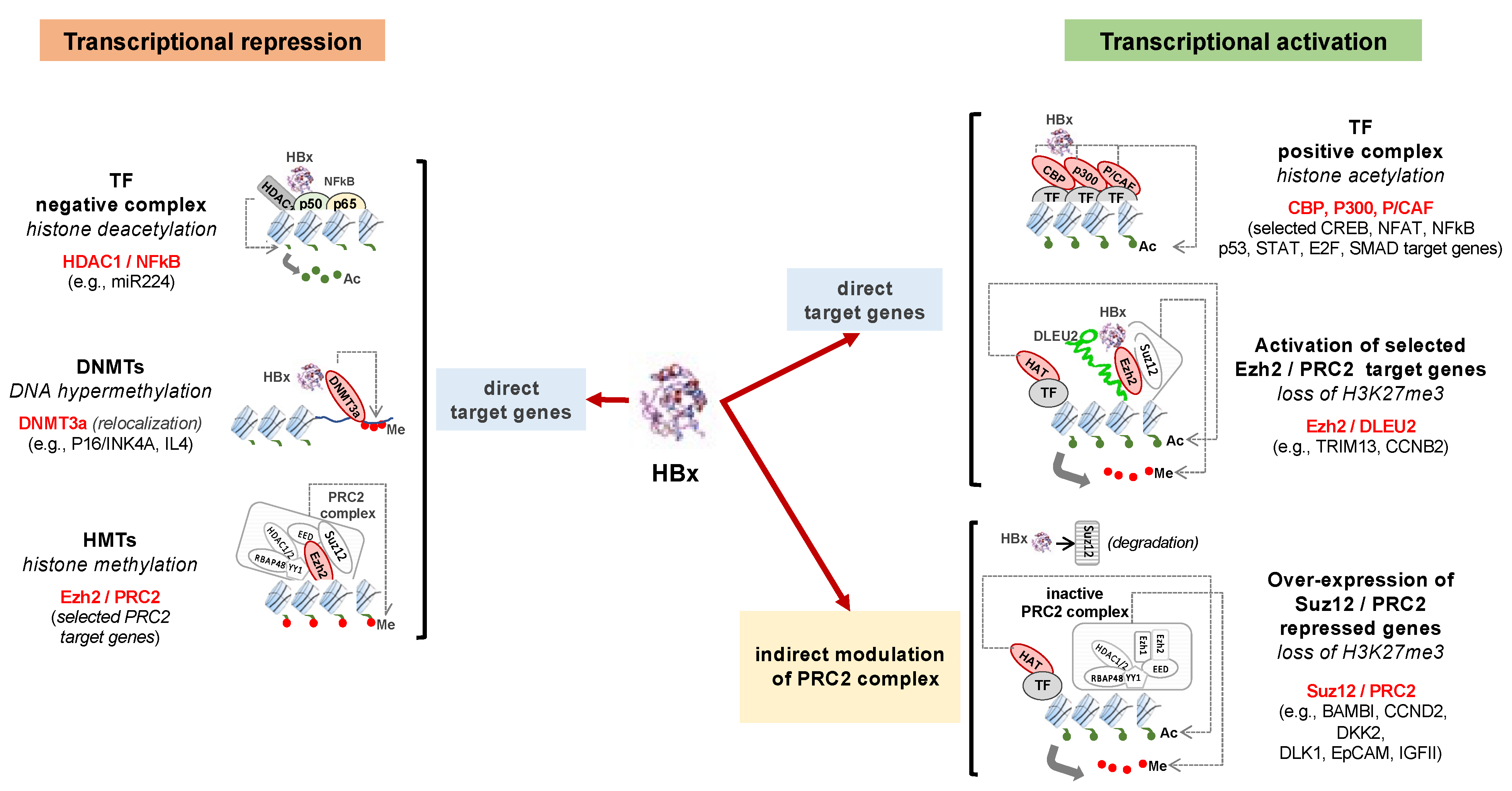
3. Deregulation of Epigenetic Mechanisms by HBV
3.1. Alteration of Host DNA Methylation
3.2. Modification of Histone Marks
3.3. Modulation of ncRNAs
4. Potential of Targeting Epigenetic Alterations in HBV-Associated HCC
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Observatory, I.-W.G.C. Globocan 2020 Database. Available online: https://gco.iarc.fr/ (accessed on 17 August 2020).
- European Association for the Study of the Liver. EASL Clinical Practice Guidelines: Management of hepatocellular carcinoma. J. Hepatol. 2018, 69, 182–236. [Google Scholar] [CrossRef]
- Levrero, M.; Zucman-Rossi, J. Mechanisms of HBV-induced hepatocellular carcinoma. J. Hepatol. 2016, 64, S84–S101. [Google Scholar] [CrossRef] [PubMed]
- Caruso, S.; O’Brien, D.R.; Cleary, S.P.; Roberts, L.R.; Zucman-Rossi, J. Genetics of HCC: Novel approaches to explore molecular diversity. Hepatology 2020. [Google Scholar] [CrossRef]
- Calderaro, J.; Ziol, M.; Paradis, V.; Zucman-Rossi, J. Molecular and histological correlations in liver cancer. J. Hepatol. 2019, 71, 616–630. [Google Scholar] [CrossRef]
- Losic, B.; Craig, A.J.; Villacorta-Martin, C.; Martins-Filho, S.N.; Akers, N.; Chen, X.; Ahsen, M.E.; von Felden, J.; Labgaa, I.; D’Avola, D.; et al. Intratumoral heterogeneity and clonal evolution in liver cancer. Nat. Commun. 2020, 11, 291. [Google Scholar] [CrossRef] [PubMed]
- Draper, A. A concise review of the changing landscape of hepatocellular carcinoma. Am. J. Manag. Care 2020, 26, S211–S219. [Google Scholar] [CrossRef]
- Faivre, S.; Rimassa, L.; Finn, R.S. Molecular therapies for HCC: Looking outside the box. J. Hepatol. 2020, 72, 342–352. [Google Scholar] [CrossRef]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef]
- Pinter, M.; Jain, R.K.; Duda, D.G. The Current Landscape of Immune Checkpoint Blockade in Hepatocellular Carcinoma: A Review. JAMA Oncol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Pinter, M.; Scheiner, B.; Peck-Radosavljevic, M. Immunotherapy for advanced hepatocellular carcinoma: A focus on special subgroups. Gut 2021, 70, 204–214. [Google Scholar] [CrossRef]
- Sung, W.K.; Zheng, H.; Li, S.; Chen, R.; Liu, X.; Li, Y.; Lee, N.P.; Lee, W.H.; Ariyaratne, P.N.; Tennakoon, C.; et al. Genome-wide survey of recurrent HBV integration in hepatocellular carcinoma. Nat. Genet. 2012, 44, 765–769. [Google Scholar] [CrossRef]
- Peneau, C.; Imbeaud, S.; La Bella, T.; Hirsch, T.Z.; Caruso, S.; Calderaro, J.; Paradis, V.; Blanc, J.F.; Letouze, E.; Nault, J.C.; et al. Hepatitis B virus integrations promote local and distant oncogenic driver alterations in hepatocellular carcinoma. Gut 2021. [Google Scholar] [CrossRef] [PubMed]
- Pollicino, T.; Belloni, L.; Raffa, G.; Pediconi, N.; Squadrito, G.; Raimondo, G.; Levrero, M. Hepatitis B virus replication is regulated by the acetylation status of hepatitis B virus cccDNA-bound H3 and H4 histones. Gastroenterology 2006, 130, 823–837. [Google Scholar] [CrossRef] [PubMed]
- Belloni, L.; Pollicino, T.; De Nicola, F.; Guerrieri, F.; Raffa, G.; Fanciulli, M.; Raimondo, G.; Levrero, M. Nuclear HBx binds the HBV minichromosome and modifies the epigenetic regulation of cccDNA function. Proc. Natl. Acad. Sci. USA 2009, 106, 19975–19979. [Google Scholar] [CrossRef]
- Guerrieri, F.; Belloni, L.; Pediconi, N.; Levrero, M. Molecular mechanisms of HBV-associated hepatocarcinogenesis. Semin. Liver Dis. 2013, 33, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Riviere, L.; Gerossier, L.; Ducroux, A.; Dion, S.; Deng, Q.; Michel, M.L.; Buendia, M.A.; Hantz, O.; Neuveut, C. HBx relieves chromatin-mediated transcriptional repression of hepatitis B viral cccDNA involving SETDB1 histone methyltransferase. J. Hepatol. 2015, 63, 1093–1102. [Google Scholar] [CrossRef]
- Tropberger, P.; Mercier, A.; Robinson, M.; Zhong, W.; Ganem, D.E.; Holdorf, M. Mapping of histone modifications in episomal HBV cccDNA uncovers an unusual chromatin organization amenable to epigenetic manipulation. Proc. Natl. Acad. Sci. USA 2015, 112, E5715–E5724. [Google Scholar] [CrossRef]
- Hong, X.; Kim, E.S.; Guo, H. Epigenetic regulation of hepatitis B virus covalently closed circular DNA: Implications for epigenetic therapy against chronic hepatitis B. Hepatology 2017, 66, 2066–2077. [Google Scholar] [CrossRef] [PubMed]
- Hama, N.; Totoki, Y.; Miura, F.; Tatsuno, K.; Saito-Adachi, M.; Nakamura, H.; Arai, Y.; Hosoda, F.; Urushidate, T.; Ohashi, S.; et al. Epigenetic landscape influences the liver cancer genome architecture. Nat. Commun. 2018, 9, 1643. [Google Scholar] [CrossRef]
- Guerrieri, F.; Belloni, L.; D’Andrea, D.; Pediconi, N.; Le Pera, L.; Testoni, B.; Scisciani, C.; Floriot, O.; Zoulim, F.; Tramontano, A.; et al. Genome-wide identification of direct HBx genomic targets. BMC Genom. 2017, 18, 184. [Google Scholar] [CrossRef]
- Fujimoto, A.; Totoki, Y.; Abe, T.; Boroevich, K.A.; Hosoda, F.; Nguyen, H.H.; Aoki, M.; Hosono, N.; Kubo, M.; Miya, F.; et al. Whole-genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nat. Genet. 2012, 44, 760–764. [Google Scholar] [CrossRef]
- Schulze, K.; Imbeaud, S.; Letouze, E.; Alexandrov, L.B.; Calderaro, J.; Rebouissou, S.; Couchy, G.; Meiller, C.; Shinde, J.; Soysouvanh, F.; et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat. Genet. 2015, 47, 505–511. [Google Scholar] [CrossRef]
- Bayo, J.; Fiore, E.J.; Dominguez, L.M.; Real, A.; Malvicini, M.; Rizzo, M.; Atorrasagasti, C.; Garcia, M.G.; Argemi, J.; Martinez, E.D.; et al. A comprehensive study of epigenetic alterations in hepatocellular carcinoma identifies potential therapeutic targets. J. Hepatol. 2019, 71, 78–90. [Google Scholar] [CrossRef]
- Hu, B.; Lin, J.Z.; Yang, X.B.; Sang, X.T. The roles of mutated SWI/SNF complexes in the initiation and development of hepatocellular carcinoma and its regulatory effect on the immune system: A review. Cell Prolif. 2020, 53, e12791. [Google Scholar] [CrossRef]
- Manna, D.; Sarkar, D. Non-Coding RNAs: Regulating Disease Progression and Therapy Resistance in Hepatocellular Carcinoma. Cancers 2020, 12, 1243. [Google Scholar] [CrossRef]
- Toh, T.B.; Lim, J.J.; Chow, E.K. Epigenetics of hepatocellular carcinoma. Clin. Transl. Med. 2019, 8, 13. [Google Scholar] [CrossRef]
- Fernández-Barrena, M.G.; Arechederra, M.; Colyn, L.; Berasain, C.; Avila, M.A. Epigenetics in hepatocellular carcinoma development and therapy: The tip of a big iceberg. JHEP Rep. 2020, 100167. [Google Scholar] [CrossRef]
- Diab, A.; Foca, A.; Zoulim, F.; Durantel, D.; Andrisani, O. The diverse functions of the hepatitis B core/capsid protein (HBc) in the viral life cycle: Implications for the development of HBc-targeting antivirals. Antivir. Res. 2018, 149, 211–220. [Google Scholar] [CrossRef]
- Fernandez, M.; Quiroga, J.A.; Carreno, V. Hepatitis B virus downregulates the human interferon-inducible MxA promoter through direct interaction of precore/core proteins. J. Gen. Virol. 2003, 84, 2073–2082. [Google Scholar] [CrossRef]
- Du, J.; Liang, X.; Liu, Y.; Qu, Z.; Gao, L.; Han, L.; Liu, S.; Cui, M.; Shi, Y.; Zhang, Z.; et al. Hepatitis B virus core protein inhibits TRAIL-induced apoptosis of hepatocytes by blocking DR5 expression. Cell Death Differ. 2009, 16, 219–229. [Google Scholar] [CrossRef]
- Xiang, A.; Ren, F.; Lei, X.; Zhang, J.; Guo, R.; Lu, Z.; Guo, Y. The hepatitis B virus (HBV) core protein enhances the transcription activation of CRE via the CRE/CREB/CBP pathway. Antivir. Res. 2015, 120, 7–15. [Google Scholar] [CrossRef]
- Salerno, D.; Chiodo, L.; Alfano, V.; Floriot, O.; Cottone, G.; Paturel, A.; Pallocca, M.; Plissonnier, M.L.; Jeddari, S.; Belloni, L.; et al. Hepatitis B protein HBx binds the DLEU2 lncRNA to sustain cccDNA and host cancer-related gene transcription. Gut 2020. [Google Scholar] [CrossRef] [PubMed]
- Gruffaz, M.; Testoni, B.; Luangsay, S.; Fusil, F.; Malika, A.; Mancip, J.; Petit, M.; Javanbakht, H.; Cosset, F.; Zoulim, F. The nuclear function of Hepatitis B capsid (HBc) protein is to inhibit IFN response very early after infection of hepatocytes. Hepatology 2013, 58, 276A. [Google Scholar]
- Gruffaz, M.; Testoni, B.; Luangsay, S.; Malika, A.; Petit, M.; Ma, H.; Klumpp, K.; Javanbakht, H.; Durantel, D.; Zoulim, F. Hepatitis B core (HBc) protein is a key and very early negative regulator of the interferon response. J. Hepatol. 2013, 58, S155–S156. [Google Scholar] [CrossRef]
- Belloni, L.; Li, L.; Palumbo, G.; Chirapu, S.; Calvo, L.; Finn, M.; Lopatin, U.; Zlotnick, A.; Levrero, M. HAPs hepatitis B virus (HBV) capsid inhibitors block core protein interaction with the viral minichromosome and host cell genes and affect cccDNA transcription. Hepatology 2013, 58, 277A. [Google Scholar]
- Guo, Y.; Kang, W.; Lei, X.; Li, Y.; Xiang, A.; Liu, Y.; Zhao, J.; Zhang, J.; Yan, Z. Hepatitis B viral core protein disrupts human host gene expression by binding to promoter regions. BMC Genom. 2012, 13, 563. [Google Scholar] [CrossRef]
- Calvisi, D.F.; Ladu, S.; Gorden, A.; Farina, M.; Lee, J.S.; Conner, E.A.; Schroeder, I.; Factor, V.M.; Thorgeirsson, S.S. Mechanistic and prognostic significance of aberrant methylation in the molecular pathogenesis of human hepatocellular carcinoma. J. Clin. Investig. 2007, 117, 2713–2722. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, A.; Portela, A.; Sayols, S.; Battiston, C.; Hoshida, Y.; Mendez-Gonzalez, J.; Imbeaud, S.; Letouze, E.; Hernandez-Gea, V.; Cornella, H.; et al. DNA methylation-based prognosis and epidrivers in hepatocellular carcinoma. Hepatology 2015, 61, 1945–1956. [Google Scholar] [CrossRef]
- Hernandez-Vargas, H.; Lambert, M.P.; Le Calvez-Kelm, F.; Gouysse, G.; McKay-Chopin, S.; Tavtigian, S.V.; Scoazec, J.Y.; Herceg, Z. Hepatocellular carcinoma displays distinct DNA methylation signatures with potential as clinical predictors. PLoS ONE 2010, 5, e9749. [Google Scholar] [CrossRef]
- Lambert, M.P.; Paliwal, A.; Vaissiere, T.; Chemin, I.; Zoulim, F.; Tommasino, M.; Hainaut, P.; Sylla, B.; Scoazec, J.Y.; Tost, J.; et al. Aberrant DNA methylation distinguishes hepatocellular carcinoma associated with HBV and HCV infection and alcohol intake. J. Hepatol. 2011, 54, 705–715. [Google Scholar] [CrossRef]
- Sun, S.; Li, Y.; Han, S.; Jia, H.; Li, X.; Li, X. A comprehensive genome-wide profiling comparison between HBV and HCV infected hepatocellular carcinoma. BMC Med. Genom. 2019, 12, 147. [Google Scholar] [CrossRef] [PubMed]
- Kuss-Duerkop, S.K.; Westrich, J.A.; Pyeon, D. DNA Tumor Virus Regulation of Host DNA Methylation and Its Implications for Immune Evasion and Oncogenesis. Viruses 2018, 10, 82. [Google Scholar] [CrossRef]
- Park, I.Y.; Sohn, B.H.; Yu, E.; Suh, D.J.; Chung, Y.H.; Lee, J.H.; Surzycki, S.J.; Lee, Y.I. Aberrant epigenetic modifications in hepatocarcinogenesis induced by hepatitis B virus X protein. Gastroenterology 2007, 132, 1476–1494. [Google Scholar] [CrossRef]
- Lee, S.M.; Lee, Y.G.; Bae, J.B.; Choi, J.K.; Tayama, C.; Hata, K.; Yun, Y.; Seong, J.K.; Kim, Y.J. HBx induces hypomethylation of distal intragenic CpG islands required for active expression of developmental regulators. Proc. Natl. Acad. Sci. USA 2014, 111, 9555–9560. [Google Scholar] [CrossRef]
- Okamoto, Y.; Shinjo, K.; Shimizu, Y.; Sano, T.; Yamao, K.; Gao, W.; Fujii, M.; Osada, H.; Sekido, Y.; Murakami, S.; et al. Hepatitis virus infection affects DNA methylation in mice with humanized livers. Gastroenterology 2014, 146, 562–572. [Google Scholar] [CrossRef]
- Lambert, M.P.; Ancey, P.B.; Esposti, D.D.; Cros, M.P.; Sklias, A.; Scoazec, J.Y.; Durantel, D.; Hernandez-Vargas, H.; Herceg, Z. Aberrant DNA methylation of imprinted loci in hepatocellular carcinoma and after in vitro exposure to common risk factors. Clin. Epigenetics 2015, 7, 15. [Google Scholar] [CrossRef]
- Ancey, P.B.; Testoni, B.; Gruffaz, M.; Cros, M.P.; Durand, G.; Le Calvez-Kelm, F.; Durantel, D.; Herceg, Z.; Hernandez-Vargas, H. Genomic responses to hepatitis B virus (HBV) infection in primary human hepatocytes. Oncotarget 2015, 6, 44877–44891. [Google Scholar] [CrossRef]
- Liu, J.; Jiang, J.; Mo, J.; Liu, D.; Cao, D.; Wang, H.; He, Y.; Wang, H. Global DNA 5-Hydroxymethylcytosine and 5-Formylcytosine Contents Are Decreased in the Early Stage of Hepatocellular Carcinoma. Hepatology 2019, 69, 196–208. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.O.; Kwun, H.J.; Jung, J.K.; Choi, K.H.; Min, D.S.; Jang, K.L. Hepatitis B virus X protein represses E-cadherin expression via activation of DNA methyltransferase 1. Oncogene 2005, 24, 6617–6625. [Google Scholar] [CrossRef]
- Jung, J.K.; Arora, P.; Pagano, J.S.; Jang, K.L. Expression of DNA methyltransferase 1 is activated by hepatitis B virus X protein via a regulatory circuit involving the p16INK4a-cyclin D1-CDK 4/6-pRb-E2F1 pathway. Cancer Res. 2007, 67, 5771–5778. [Google Scholar] [CrossRef]
- Zheng, D.L.; Zhang, L.; Cheng, N.; Xu, X.; Deng, Q.; Teng, X.M.; Wang, K.S.; Zhang, X.; Huang, J.; Han, Z.G. Epigenetic modification induced by hepatitis B virus X protein via interaction with de novo DNA methyltransferase DNMT3A. J. Hepatol. 2009, 50, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Wu, G.; Bu, F.; Lu, B.; Liang, A.; Cao, L.; Tong, X.; Lu, X.; Wu, M.; Guo, Y. Epigenetic silence of ankyrin-repeat-containing, SH3-domain-containing, and proline-rich-region- containing protein 1 (ASPP1) and ASPP2 genes promotes tumor growth in hepatitis B virus-positive hepatocellular carcinoma. Hepatology 2010, 51, 142–153. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.H.; Na, H.; Na, T.Y.; Shin, Y.K.; Seong, J.K.; Lee, M.O. Epigenetic control of metastasis-associated protein 1 gene expression by hepatitis B virus X protein during hepatocarcinogenesis. Oncogenesis 2012, 1, e25. [Google Scholar] [CrossRef]
- Xie, Q.; Chen, L.; Shan, X.; Shan, X.; Tang, J.; Zhou, F.; Chen, Q.; Quan, H.; Nie, D.; Zhang, W.; et al. Epigenetic silencing of SFRP1 and SFRP5 by hepatitis B virus X protein enhances hepatoma cell tumorigenicity through Wnt signaling pathway. Int. J. Cancer 2014, 135, 635–646. [Google Scholar] [CrossRef]
- Fu, X.; Song, X.; Li, Y.; Tan, D.; Liu, G. Hepatitis B virus X protein upregulates DNA methyltransferase 3A/3B and enhances SOCS-1CpG island methylation. Mol. Med. Rep. 2016, 13, 301–308. [Google Scholar] [CrossRef]
- Ying, J.; Li, H.; Seng, T.J.; Langford, C.; Srivastava, G.; Tsao, S.W.; Putti, T.; Murray, P.; Chan, A.T.; Tao, Q. Functional epigenetics identifies a protocadherin PCDH10 as a candidate tumor suppressor for nasopharyngeal, esophageal and multiple other carcinomas with frequent methylation. Oncogene 2006, 25, 1070–1080. [Google Scholar] [CrossRef]
- Qiu, X.; Zhang, L.; Lu, S.; Song, Y.; Lao, Y.; Hu, J.; Fan, H. Upregulation of DNMT1 mediated by HBx suppresses RASSF1A expression independent of DNA methylation. Oncol. Rep. 2014, 31, 202–208. [Google Scholar] [CrossRef]
- Fang, S.; Huang, S.F.; Cao, J.; Wen, Y.A.; Zhang, L.P.; Ren, G.S. Silencing of PCDH10 in hepatocellular carcinoma via de novo DNA methylation independent of HBV infection or HBX expression. Clin. Exp. Med. 2013, 13, 127–134. [Google Scholar] [CrossRef]
- Arzumanyan, A.; Friedman, T.; Kotei, E.; Ng, I.O.; Lian, Z.; Feitelson, M.A. Epigenetic repression of E-cadherin expression by hepatitis B virus x antigen in liver cancer. Oncogene 2012, 31, 563–572. [Google Scholar] [CrossRef]
- Rasmussen, K.D.; Helin, K. Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev. 2016, 30, 733–750. [Google Scholar] [CrossRef]
- Pfeifer, G.P.; Szabo, P.E.; Song, J. Protein Interactions at Oxidized 5-Methylcytosine Bases. J. Mol. Biol. 2019, 432, 1718–1730. [Google Scholar] [CrossRef]
- Yuan, J.H.; Yang, F.; Chen, B.F.; Lu, Z.; Huo, X.S.; Zhou, W.P.; Wang, F.; Sun, S.H. The histone deacetylase 4/SP1/microrna-200a regulatory network contributes to aberrant histone acetylation in hepatocellular carcinoma. Hepatology 2011, 54, 2025–2035. [Google Scholar] [CrossRef] [PubMed]
- Freese, K.; Seitz, T.; Dietrich, P.; Lee, S.M.L.; Thasler, W.E.; Bosserhoff, A.; Hellerbrand, C. Histone Deacetylase Expressions in Hepatocellular Carcinoma and Functional Effects of Histone Deacetylase Inhibitors on Liver Cancer Cells In Vitro. Cancers 2019, 11, 1587. [Google Scholar] [CrossRef]
- Shon, J.K.; Shon, B.H.; Park, I.Y.; Lee, S.U.; Fa, L.; Chang, K.Y.; Shin, J.H.; Lee, Y.I. Hepatitis B virus-X protein recruits histone deacetylase 1 to repress insulin-like growth factor binding protein 3 transcription. Virus Res. 2009, 139, 14–21. [Google Scholar] [CrossRef]
- Srisuttee, R.; Koh, S.S.; Kim, S.J.; Malilas, W.; Boonying, W.; Cho, I.R.; Jhun, B.H.; Ito, M.; Horio, Y.; Seto, E.; et al. Hepatitis B virus X (HBX) protein upregulates beta-catenin in a human hepatic cell line by sequestering SIRT1 deacetylase. Oncol. Rep. 2012, 28, 276–282. [Google Scholar] [CrossRef]
- Cougot, D.; Wu, Y.; Cairo, S.; Caramel, J.; Renard, C.A.; Levy, L.; Buendia, M.A.; Neuveut, C. The hepatitis B virus X protein functionally interacts with CREB-binding protein/p300 in the regulation of CREB-mediated transcription. J. Biol. Chem. 2007, 282, 4277–4287. [Google Scholar] [CrossRef]
- Liu, X.Y.; Tang, S.H.; Wu, S.L.; Luo, Y.H.; Cao, M.R.; Zhou, H.K.; Jiang, X.W.; Shu, J.C.; Bie, C.Q.; Huang, S.M.; et al. Epigenetic modulation of insulin-like growth factor-II overexpression by hepatitis B virus X protein in hepatocellular carcinoma. Am. J. Cancer Res. 2015, 5, 956–978. [Google Scholar]
- Qian, J.; Yao, D.; Dong, Z.; Wu, W.; Qiu, L.; Yao, N.; Li, S.; Bian, Y.; Wang, Z.; Shi, G. Characteristics of hepatic igf-ii expression and monitored levels of circulating igf-ii mRNA in metastasis of hepatocellular carcinoma. Am. J. Clin. Pathol. 2010, 134, 799–806. [Google Scholar] [CrossRef] [PubMed]
- Black, J.C.; Van Rechem, C.; Whetstine, J.R. Histone lysine methylation dynamics: Establishment, regulation, and biological impact. Mol. Cell 2012, 48, 491–507. [Google Scholar] [CrossRef]
- Sasaki, M.; Ikeda, H.; Itatsu, K.; Yamaguchi, J.; Sawada, S.; Minato, H.; Ohta, T.; Nakanuma, Y. The overexpression of polycomb group proteins Bmi1 and EZH2 is associated with the progression and aggressive biological behavior of hepatocellular carcinoma. Lab. Investig. 2008, 88, 873–882. [Google Scholar] [CrossRef]
- Cai, M.Y.; Tong, Z.T.; Zheng, F.; Liao, Y.J.; Wang, Y.; Rao, H.L.; Chen, Y.C.; Wu, Q.L.; Liu, Y.H.; Guan, X.Y.; et al. EZH2 protein: A promising immunomarker for the detection of hepatocellular carcinomas in liver needle biopsies. Gut 2011, 60, 967–976. [Google Scholar] [CrossRef] [PubMed]
- Au, S.L.; Ng, I.O.; Wong, C.M. Epigenetic dysregulation in hepatocellular carcinoma: Focus on polycomb group proteins. Front. Med. 2013, 7, 231–241. [Google Scholar] [CrossRef]
- Fan, D.N.; Tsang, F.H.; Tam, A.H.; Au, S.L.; Wong, C.C.; Wei, L.; Lee, J.M.; He, X.; Ng, I.O.; Wong, C.M. Histone lysine methyltransferase, suppressor of variegation 3–9 homolog 1, promotes hepatocellular carcinoma progression and is negatively regulated by microRNA-125b. Hepatology 2013, 57, 637–647. [Google Scholar] [CrossRef]
- Wong, C.M.; Wei, L.; Law, C.T.; Ho, D.W.; Tsang, F.H.; Au, S.L.; Sze, K.M.; Lee, J.M.; Wong, C.C.; Ng, I.O. Up-regulation of histone methyltransferase SETDB1 by multiple mechanisms in hepatocellular carcinoma promotes cancer metastasis. Hepatology 2016, 63, 474–487. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Chiu, D.K.; Tsang, F.H.; Law, C.T.; Cheng, C.L.; Au, S.L.; Lee, J.M.; Wong, C.C.; Ng, I.O.; Wong, C.M. Histone methyltransferase G9a promotes liver cancer development by epigenetic silencing of tumor suppressor gene RARRES3. J. Hepatol. 2017, 67, 758–769. [Google Scholar] [CrossRef]
- Wang, D.Y.; An, S.H.; Liu, L.; Bai, S.S.; Wu, K.X.; Zhu, R.; Wang, Z.J. Hepatitis B virus X protein influences enrichment profiles of H3K9me3 on promoter regions in human hepatoma cell lines. Oncotarget 2016, 7, 84883–84892. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, Y.; Tsuge, M.; Tsushima, K.; Suehiro, Y.; Fujino, H.; Ono, A.; Yamauchi, M.; Makokha, G.N.; Nakahara, T.; Murakami, E.; et al. Signal activation of hepatitis B virus-related hepatocarcinogenesis by upregulation of SUV39h1. J. Infect. Dis. 2020, 222, 2061–2070. [Google Scholar] [CrossRef]
- Kim, G.; Kim, J.Y.; Lim, S.C.; Lee, K.Y.; Kim, O.; Choi, H.S. SUV39H1/DNMT3A-dependent methylation of the RB1 promoter stimulates PIN1 expression and melanoma development. FASEB J. 2018, 32, 5647–5660. [Google Scholar] [CrossRef]
- Zhang, H.; Diab, A.; Fan, H.; Mani, S.K.; Hullinger, R.; Merle, P.; Andrisani, O. PLK1 and HOTAIR Accelerate Proteasomal Degradation of SUZ12 and ZNF198 during Hepatitis B Virus-Induced Liver Carcinogenesis. Cancer Res. 2015, 75, 2363–2374. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Xing, Z.; Mani, S.K.; Bancel, B.; Durantel, D.; Zoulim, F.; Tran, E.J.; Merle, P.; Andrisani, O. RNA helicase DEAD box protein 5 regulates Polycomb repressive complex 2/Hox transcript antisense intergenic RNA function in hepatitis B virus infection and hepatocarcinogenesis. Hepatology 2016, 64, 1033–1048. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Jia, Z.; Tian, Y.; Yang, P.; Sun, H.; Wang, C.; Ding, Y.; Zhang, M.; Zhang, Y.; Yang, D.; et al. HBx Protein Contributes to Liver Carcinogenesis by H3K4me3 Modification Through Stabilizing WD Repeat Domain 5 Protein. Hepatology 2020, 71, 1678–1695. [Google Scholar] [CrossRef]
- Trievel, R.C.; Shilatifard, A. WDR5, a complexed protein. Nat. Struct. Mol. Biol. 2009, 16, 678–680. [Google Scholar] [CrossRef]
- Yang, L.; He, J.; Chen, L.; Wang, G. Hepatitis B virus X protein upregulates expression of SMYD3 and C-MYC in HepG2 cells. Med. Oncol. 2009, 26, 445–451. [Google Scholar] [CrossRef]
- Blanc, R.S.; Richard, S. Arginine Methylation: The Coming of Age. Mol. Cell 2017, 65, 8–24. [Google Scholar] [CrossRef]
- Guccione, E.; Richard, S. The regulation, functions and clinical relevance of arginine methylation. Nat. Rev. Mol. Cell Biol. 2019, 20, 642–657. [Google Scholar] [CrossRef]
- Ryu, J.W.; Kim, S.K.; Son, M.Y.; Jeon, S.J.; Oh, J.H.; Lim, J.H.; Cho, S.; Jung, C.R.; Hamamoto, R.; Kim, D.S.; et al. Novel prognostic marker PRMT1 regulates cell growth via downregulation of CDKN1A in HCC. Oncotarget 2017, 8, 115444–115455. [Google Scholar] [CrossRef]
- Zhang, X.P.; Jiang, Y.B.; Zhong, C.Q.; Ma, N.; Zhang, E.B.; Zhang, F.; Li, J.J.; Deng, Y.Z.; Wang, K.; Xie, D.; et al. PRMT1 Promoted HCC Growth and Metastasis In Vitro and In Vivo via Activating the STAT3 Signalling Pathway. Cell Physiol. Biochem. 2018, 47, 1643–1654. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Yan, C.; Xie, P.; Cao, Y.; Shao, J.; Ge, J. PRMT2 accelerates tumorigenesis of hepatocellular carcinoma by activating Bcl2 via histone H3R8 methylation. Exp. Cell Res. 2020, 394, 112152. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Dong, S.; Li, Z.; Lu, L.; Zhang, S.; Chen, X.; Cen, X.; Wu, Y. Targeting protein arginine methyltransferase 5 inhibits human hepatocellular carcinoma growth via the downregulation of beta-catenin. J. Transl. Med. 2015, 13, 349. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, D.; Kanda, M.; Sugimoto, H.; Shibata, M.; Tanaka, H.; Takami, H.; Iwata, N.; Hayashi, M.; Tanaka, C.; Kobayashi, D.; et al. The protein arginine methyltransferase 5 promotes malignant phenotype of hepatocellular carcinoma cells and is associated with adverse patient outcomes after curative hepatectomy. Int. J. Oncol. 2017, 50, 381–386. [Google Scholar] [CrossRef]
- Jeon, J.Y.; Lee, J.S.; Park, E.R.; Shen, Y.N.; Kim, M.Y.; Shin, H.J.; Joo, H.Y.; Cho, E.H.; Moon, S.M.; Shin, U.S.; et al. Protein arginine methyltransferase 5 is implicated in the aggressiveness of human hepatocellular carcinoma and controls the invasive activity of cancer cells. Oncol. Rep. 2018, 40, 536–544. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, J.; Liu, X.; Li, S.; Wang, Q.; Di, C.; Hu, Z.; Yu, T.; Ding, J.; Li, J.; et al. The LINC01138 drives malignancies via activating arginine methyltransferase 5 in hepatocellular carcinoma. Nat. Commun. 2018, 9, 1572. [Google Scholar] [CrossRef]
- Jiang, H.; Zhou, Z.; Jin, S.; Xu, K.; Zhang, H.; Xu, J.; Sun, Q.; Wang, J.; Xu, J. PRMT9 promotes hepatocellular carcinoma invasion and metastasis via activating PI3K/Akt/GSK-3beta/Snail signaling. Cancer Sci. 2018, 109, 1414–1427. [Google Scholar] [CrossRef] [PubMed]
- Chan, L.H.; Zhou, L.; Ng, K.Y.; Wong, T.L.; Lee, T.K.; Sharma, R.; Loong, J.H.; Ching, Y.P.; Yuan, Y.F.; Xie, D.; et al. PRMT6 Regulates RAS/RAF Binding and MEK/ERK-Mediated Cancer Stemness Activities in Hepatocellular Carcinoma through CRAF Methylation. Cell Rep. 2018, 25, 690–701. [Google Scholar] [CrossRef]
- Wong, T.L.; Ng, K.Y.; Tan, K.V.; Chan, L.H.; Zhou, L.; Che, N.; Hoo, R.L.C.; Lee, T.K.; Richard, S.; Lo, C.M.; et al. CRAF Methylation by PRMT6 Regulates Aerobic Glycolysis-Driven Hepatocarcinogenesis via ERK-Dependent PKM2 Nuclear Relocalization and Activation. Hepatology 2020, 71, 1279–1296. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Chen, J.; Wu, M.; Zhang, X.; Zhang, M.; Yue, L.; Li, Y.; Liu, J.; Li, B.; Shen, F.; et al. PRMT5 restricts hepatitis B virus replication through epigenetic repression of covalently closed circular DNA transcription and interference with pregenomic RNA encapsidation. Hepatology 2017, 66, 398–415. [Google Scholar] [CrossRef] [PubMed]
- Benhenda, S.; Ducroux, A.; Riviere, L.; Sobhian, B.; Ward, M.D.; Dion, S.; Hantz, O.; Protzer, U.; Michel, M.L.; Benkirane, M.; et al. Methyltransferase PRMT1 is a binding partner of HBx and a negative regulator of hepatitis B virus transcription. J. Virol. 2013, 87, 4360–4371. [Google Scholar] [CrossRef]
- Kapranov, P.; Cheng, J.; Dike, S.; Nix, D.A.; Duttagupta, R.; Willingham, A.T.; Stadler, P.F.; Hertel, J.; Hackermuller, J.; Hofacker, I.L.; et al. RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science 2007, 316, 1484–1488. [Google Scholar] [CrossRef]
- Ura, S.; Honda, M.; Yamashita, T.; Ueda, T.; Takatori, H.; Nishino, R.; Sunakozaka, H.; Sakai, Y.; Horimoto, K.; Kaneko, S. Differential microRNA expression between hepatitis B and hepatitis C leading disease progression to hepatocellular carcinoma. Hepatology 2009, 49, 1098–1112. [Google Scholar] [CrossRef]
- Diaz, G.; Melis, M.; Tice, A.; Kleiner, D.E.; Mishra, L.; Zamboni, F.; Farci, P. Identification of microRNAs specifically expressed in hepatitis C virus-associated hepatocellular carcinoma. Int. J. Cancer 2013, 133, 816–824. [Google Scholar] [CrossRef] [PubMed]
- Hou, W.; Bonkovsky, H.L. Non-coding RNAs in hepatitis C-induced hepatocellular carcinoma: Dysregulation and implications for early detection, diagnosis and therapy. World J. Gastroenterol. 2013, 19, 7836–7845. [Google Scholar] [CrossRef]
- Kumar, A. MicroRNA in HCV infection and liver cancer. Biochim. Biophys. Acta 2011, 1809, 694–699. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Kim, J.H.; Lee, S.W. The role of microRNAs in hepatitis C virus replication and related liver diseases. J. Microbiol. 2014, 52, 445–451. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.M.; Tsang, F.H.; Ng, I.O. Non-coding RNAs in hepatocellular carcinoma: Molecular functions and pathological implications. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 137–151. [Google Scholar] [CrossRef]
- Bandiera, S.; Pfeffer, S.; Baumert, T.F.; Zeisel, M.B. miR-122--a key factor and therapeutic target in liver disease. J. Hepatol. 2015, 62, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Plissonnier, M.L.; Herzog, K.; Levrero, M.; Zeisel, M.B. Non-Coding RNAs and Hepatitis C Virus-Induced Hepatocellular Carcinoma. Viruses 2018, 10, 591. [Google Scholar] [CrossRef]
- Xu, J.; An, P.; Winkler, C.A.; Yu, Y. Dysregulated microRNAs in Hepatitis B Virus-Related Hepatocellular Carcinoma: Potential as Biomarkers and Therapeutic Targets. Front. Oncol. 2020, 10, 1271. [Google Scholar] [CrossRef]
- Tricoli, L.; Niture, S.; Chimeh, U.; Kumar, D. Role of microRNAs in the development of hepatocellular carcinoma and acquired drug resistance. Front. Biosci. (Landmark Ed.) 2019, 24, 545–554. [Google Scholar]
- Lamontagne, J.; Steel, L.F.; Bouchard, M.J. Hepatitis B virus and microRNAs: Complex interactions affecting hepatitis B virus replication and hepatitis B virus-associated diseases. World J. Gastroenterol. 2015, 21, 7375–7399. [Google Scholar] [CrossRef]
- Chen, Y.; Shen, A.; Rider, P.J.; Yu, Y.; Wu, K.; Mu, Y.; Hao, Q.; Liu, Y.; Gong, H.; Zhu, Y.; et al. A liver-specific microRNA binds to a highly conserved RNA sequence of hepatitis B virus and negatively regulates viral gene expression and replication. FASEB J. 2011, 25, 4511–4521. [Google Scholar] [CrossRef]
- Damania, P.; Sen, B.; Dar, S.B.; Kumar, S.; Kumari, A.; Gupta, E.; Sarin, S.K.; Venugopal, S.K. Hepatitis B virus induces cell proliferation via HBx-induced microRNA-21 in hepatocellular carcinoma by targeting programmed cell death protein4 (PDCD4) and phosphatase and tensin homologue (PTEN). PLoS ONE 2014, 9, e91745. [Google Scholar] [CrossRef]
- Meng, F.; Henson, R.; Wehbe-Janek, H.; Ghoshal, K.; Jacob, S.T.; Patel, T. MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology 2007, 133, 647–658. [Google Scholar] [CrossRef] [PubMed]
- Li, C.H.; Xu, F.; Chow, S.; Feng, L.; Yin, D.; Ng, T.B.; Chen, Y. Hepatitis B virus X protein promotes hepatocellular carcinoma transformation through interleukin-6 activation of microRNA-21 expression. Eur. J. Cancer 2014, 50, 2560–2569. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.J.; Song, W.; Zhang, S.D.; Shen, X.H.; Qiu, X.M.; Wu, H.Z.; Gong, P.H.; Lu, S.; Zhao, Z.J.; He, M.L.; et al. HBx-upregulated lncRNA UCA1 promotes cell growth and tumorigenesis by recruiting EZH2 and repressing p27Kip1/CDK2 signaling. Sci. Rep. 2016, 6, 23521. [Google Scholar] [CrossRef]
- Pfister, S.X.; Ashworth, A. Marked for death: Targeting epigenetic changes in cancer. Nat. Rev. Drug Discov. 2017, 16, 241–263. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, A.; Arimondo, P.B.; Rots, M.G.; Jeronimo, C.; Berdasco, M. The timeline of epigenetic drug discovery: From reality to dreams. Clin. Epigenet. 2019, 11, 174. [Google Scholar] [CrossRef]
- Lu, Y.; Chan, Y.T.; Tan, H.Y.; Li, S.; Wang, N.; Feng, Y. Epigenetic regulation in human cancer: The potential role of epi-drug in cancer therapy. Mol. Cancer 2020, 19, 79. [Google Scholar] [CrossRef] [PubMed]
- Morel, D.; Jeffery, D.; Aspeslagh, S.; Almouzni, G.; Postel-Vinay, S. Combining epigenetic drugs with other therapies for solid tumours - past lessons and future promise. Nat. Rev. Clin. Oncol. 2020, 17, 91–107. [Google Scholar] [CrossRef]
- Soukupova, J.; Bertran, E.; Penuelas-Haro, I.; Urdiroz-Urricelqui, U.; Borgman, M.; Kohlhof, H.; Fabregat, I. Resminostat induces changes in epithelial plasticity of hepatocellular carcinoma cells and sensitizes them to sorafenib-induced apoptosis. Oncotarget 2017, 8, 110367–110379. [Google Scholar] [CrossRef]
- Llopiz, D.; Ruiz, M.; Villanueva, L.; Iglesias, T.; Silva, L.; Egea, J.; Lasarte, J.J.; Pivette, P.; Trochon-Joseph, V.; Vasseur, B.; et al. Enhanced anti-tumor efficacy of checkpoint inhibitors in combination with the histone deacetylase inhibitor Belinostat in a murine hepatocellular carcinoma model. Cancer Immunol. Immunother. 2019, 68, 379–393. [Google Scholar] [CrossRef]
- Mei, Q.; Chen, M.; Lu, X.; Li, X.; Duan, F.; Wang, M.; Luo, G.; Han, W. An open-label, single-arm, phase I/II study of lower-dose decitabine based therapy in patients with advanced hepatocellular carcinoma. Oncotarget 2015, 6, 16698–16711. [Google Scholar] [CrossRef]
- Yeo, W.; Chung, H.C.; Chan, S.L.; Wang, L.Z.; Lim, R.; Picus, J.; Boyer, M.; Mo, F.K.; Koh, J.; Rha, S.Y.; et al. Epigenetic therapy using belinostat for patients with unresectable hepatocellular carcinoma: A multicenter phase I/II study with biomarker and pharmacokinetic analysis of tumors from patients in the Mayo Phase II Consortium and the Cancer Therapeutics Research Group. J. Clin. Oncol. 2012, 30, 3361–3367. [Google Scholar] [CrossRef]
- Bitzer, M.; Horger, M.; Giannini, E.G.; Ganten, T.M.; Worns, M.A.; Siveke, J.T.; Dollinger, M.M.; Gerken, G.; Scheulen, M.E.; Wege, H.; et al. Resminostat plus sorafenib as second-line therapy of advanced hepatocellular carcinoma—The SHELTER study. J. Hepatol. 2016, 65, 280–288. [Google Scholar] [CrossRef]
- Rechtman, M.M.; Har-Noy, O.; Bar-Yishay, I.; Fishman, S.; Adamovich, Y.; Shaul, Y.; Halpern, Z.; Shlomai, A. Curcumin inhibits hepatitis B virus via down-regulation of the metabolic coactivator PGC-1alpha. FEBS Lett. 2010, 584, 2485–2490. [Google Scholar] [CrossRef] [PubMed]
- Araki, K.; Miyazaki, J.; Tsurimoto, T.; Inomoto, T.; Iwanaga, T.; Matsubara, K.; Yamamura, K. Demethylation by 5-azacytidine results in the expression of hepatitis B virus surface antigen in transgenic mice. Jpn. J. Cancer Res. 1989, 80, 295–298. [Google Scholar] [CrossRef]
- Miyoshi, E.; Fujii, J.; Hayashi, N.; Ueda, K.; Towata, T.; Fusamoto, H.; Kamada, T.; Taniguchi, N. Enhancement of hepatitis-B surface-antigen expression by 5-azacytidine in a hepatitis-B-virus-transfected cell line. Int. J. Cancer 1992, 52, 137–140. [Google Scholar] [CrossRef]
- Ritchie, D.; Piekarz, R.L.; Blombery, P.; Karai, L.J.; Pittaluga, S.; Jaffe, E.S.; Raffeld, M.; Janik, J.E.; Prince, H.M.; Bates, S.E. Reactivation of DNA viruses in association with histone deacetylase inhibitor therapy: A case series report. Haematologica 2009, 94, 1618–1622. [Google Scholar] [CrossRef]
- Wang, Y.C.; Yang, X.; Xing, L.H.; Kong, W.Z. Effects of SAHA on proliferation and apoptosis of hepatocellular carcinoma cells and hepatitis B virus replication. World J. Gastroenterol. 2013, 19, 5159–5164. [Google Scholar] [CrossRef]
- Zhong, S.; Tang, M.W.; Yeo, W.; Liu, C.; Lo, Y.M.; Johnson, P.J. Silencing of GSTP1 gene by CpG island DNA hypermethylation in HBV-associated hepatocellular carcinomas. Clin. Cancer Res. 2002, 8, 1087–1092. [Google Scholar]
- Lin, H.C.; Chen, Y.F.; Hsu, W.H.; Yang, C.W.; Kao, C.H.; Tsai, T.F. Resveratrol helps recovery from fatty liver and protects against hepatocellular carcinoma induced by hepatitis B virus X protein in a mouse model. Cancer Prev. Res. 2012, 5, 952–962. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Lim, J.; Kim, J.R.; Cho, S. Inhibitory effects of resveratrol on hepatitis B virus X protein-induced hepatocellular carcinoma. J. Vet. Sci. 2017, 18, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Srisuttee, R.; Koh, S.S.; Malilas, W.; Moon, J.; Cho, I.R.; Jhun, B.H.; Horio, Y.; Chung, Y.H. SIRT1 sensitizes hepatocellular carcinoma cells expressing hepatitis B virus X protein to oxidative stress-induced apoptosis. Biochem. Biophys. Res. Commun. 2012, 429, 45–50. [Google Scholar] [CrossRef]
- Hsieh, Y.H.; Su, I.J.; Yen, C.J.; Tsai, T.F.; Tsai, H.W.; Tsai, H.N.; Huang, Y.J.; Chen, Y.Y.; Ai, Y.L.; Kao, L.Y.; et al. Histone deacetylase inhibitor suberoylanilide hydroxamic acid suppresses the pro-oncogenic effects induced by hepatitis B virus pre-S2 mutant oncoprotein and represents a potential chemopreventive agent in high-risk chronic HBV patients. Carcinogenesis 2013, 34, 475–485. [Google Scholar] [CrossRef] [PubMed]
- Hamamoto, H.; Maemura, K.; Matsuo, K.; Taniguchi, K.; Tanaka, Y.; Futaki, S.; Takeshita, A.; Asai, A.; Hayashi, M.; Hirose, Y.; et al. Delta-like 3 is silenced by HBx via histone acetylation in HBV-associated HCCs. Sci. Rep. 2018, 8, 4842. [Google Scholar] [CrossRef] [PubMed]
- Wahlestedt, C. Targeting long non-coding RNA to therapeutically upregulate gene expression. Nat. Rev. Drug. Discov. 2013, 12, 433–446. [Google Scholar] [CrossRef]
- Janssen, H.L.; Kauppinen, S.; Hodges, M.R. HCV infection and miravirsen. N. Engl. J. Med. 2013, 369, 878. [Google Scholar] [CrossRef]
- van der Ree, M.H.; de Vree, J.M.; Stelma, F.; Willemse, S.; van der Valk, M.; Rietdijk, S.; Molenkamp, R.; Schinkel, J.; van Nuenen, A.C.; Beuers, U.; et al. Safety, tolerability, and antiviral effect of RG-101 in patients with chronic hepatitis C: A phase 1B, double-blind, randomised controlled trial. Lancet 2017, 389, 709–717. [Google Scholar] [CrossRef]
- Dandri, M. Epigenetic modulation in chronic hepatitis B virus infection. Semin. Immunopathol. 2020, 42, 173–185. [Google Scholar] [CrossRef]
- Hensel, K.O.; Rendon, J.C.; Navas, M.C.; Rots, M.G.; Postberg, J. Virus-host interplay in hepatitis B virus infection and epigenetic treatment strategies. FEBS J. 2017, 284, 3550–3572. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Compound | Epidrug Target 1 | Stage of Development | Clinical Trial |
|---|---|---|---|
| Belinostat | HDAC | Phase 1/2 | NCT00321594 |
| Decitabine + chemo-/immunotherapy | DNMT | Phase 1/2 | NCT01799083 |
| Guadecitabine + durvalumab | DNMT | Phase 1 | NCT03257761 |
| Guadecitabine + sorafenib + oxaliplatin | DNMT | Phase 2 | NCT01752933 |
| Resminostat + sorafenib | HDAC | Phase 1/2 | NCT00943449 |
| Compound | Target 1 | Model System | Reference |
|---|---|---|---|
| 5-aza-2′-deoxycytidine (decitabine) | DNMTs | Cell lines | [130] |
| Nicotinamide | Sirt1 | HBx transgenic mice | [66] |
| Resveratrol | Sirt1 | Cell line, Huh7-HBx xenograft mice, HBx transgenic mice | [66,131,132,133] |
| Suberoylanilide hydroxamic acid (SAHA) | HDACs | Cell lines, pre-S 2 mutant LHBS transgenic mice | [129,134] |
| Trichostatin A (TSA) | HDACs | Cell lines | [135] |
| WDR5-0103 | WDR5 | Cell lines, MHCC97H and Huh7-HBx xenograft mice | [82] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeisel, M.B.; Guerrieri, F.; Levrero, M. Host Epigenetic Alterations and Hepatitis B Virus-Associated Hepatocellular Carcinoma. J. Clin. Med. 2021, 10, 1715. https://doi.org/10.3390/jcm10081715
Zeisel MB, Guerrieri F, Levrero M. Host Epigenetic Alterations and Hepatitis B Virus-Associated Hepatocellular Carcinoma. Journal of Clinical Medicine. 2021; 10(8):1715. https://doi.org/10.3390/jcm10081715
Chicago/Turabian StyleZeisel, Mirjam B., Francesca Guerrieri, and Massimo Levrero. 2021. "Host Epigenetic Alterations and Hepatitis B Virus-Associated Hepatocellular Carcinoma" Journal of Clinical Medicine 10, no. 8: 1715. https://doi.org/10.3390/jcm10081715
APA StyleZeisel, M. B., Guerrieri, F., & Levrero, M. (2021). Host Epigenetic Alterations and Hepatitis B Virus-Associated Hepatocellular Carcinoma. Journal of Clinical Medicine, 10(8), 1715. https://doi.org/10.3390/jcm10081715