
Altered Electrical, Biomolecular, and Immunologic Phenotypes in a Novel Patient-Derived Stem Cell Model of Desmoglein-2 Mutant ARVC

 , , ,
, , ,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. hiPSC Line Generation
2.2. hiPSC-CM Differentiation and Culture
2.3. Biomolecular Characterization Using Immunostaining, Real-Time Quantitative PCR, Western Blot, and Cytokine Arrays
2.4. RNA Sequencing and Analysis
2.5. Optical Mapping of Calcium and Transmembrane Potential
2.6. siRNA Transfection
2.7. Statistics
3. Results
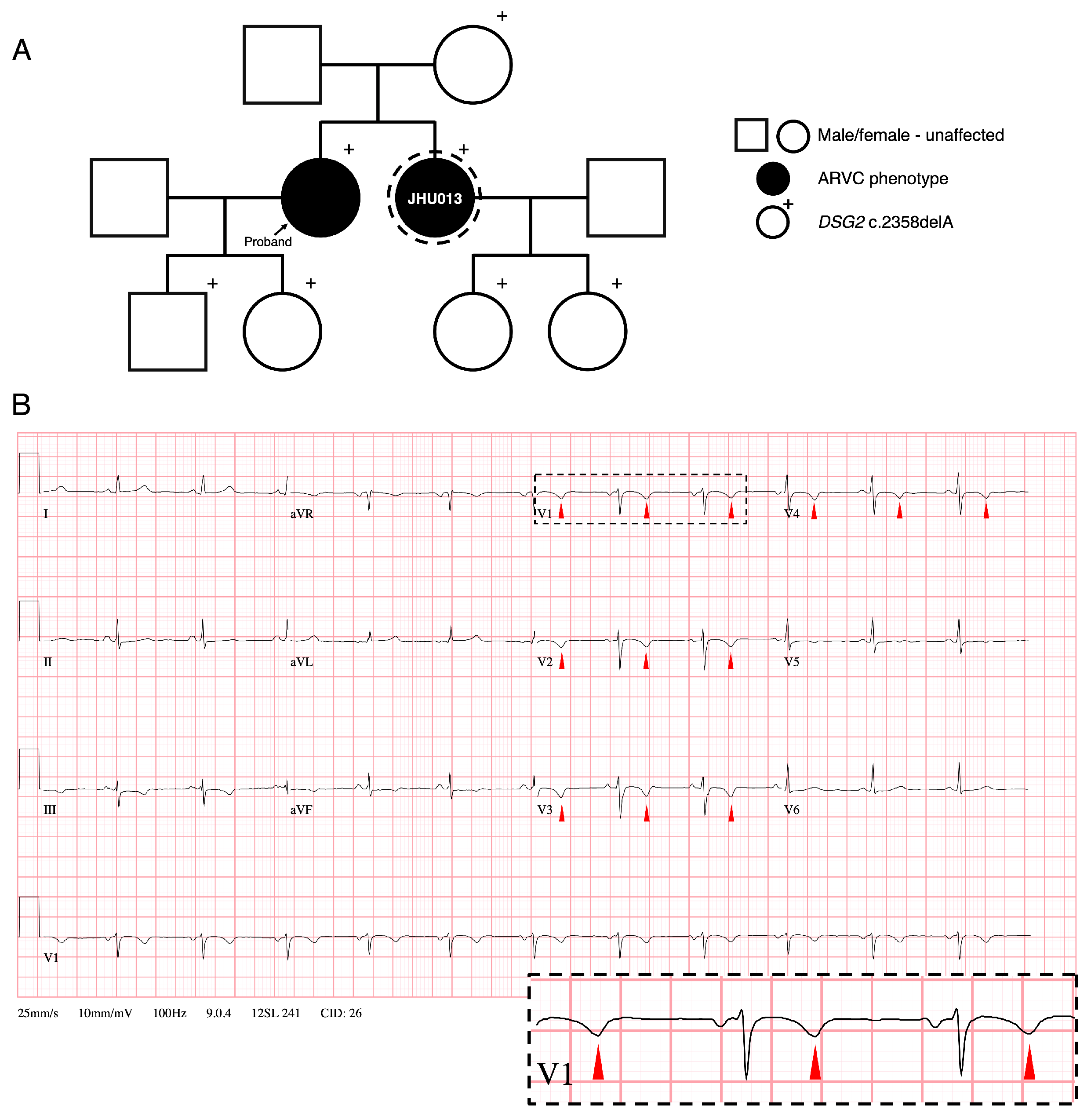
3.1. Family History and Clinical Phenotypes of ARVC Patient Donor
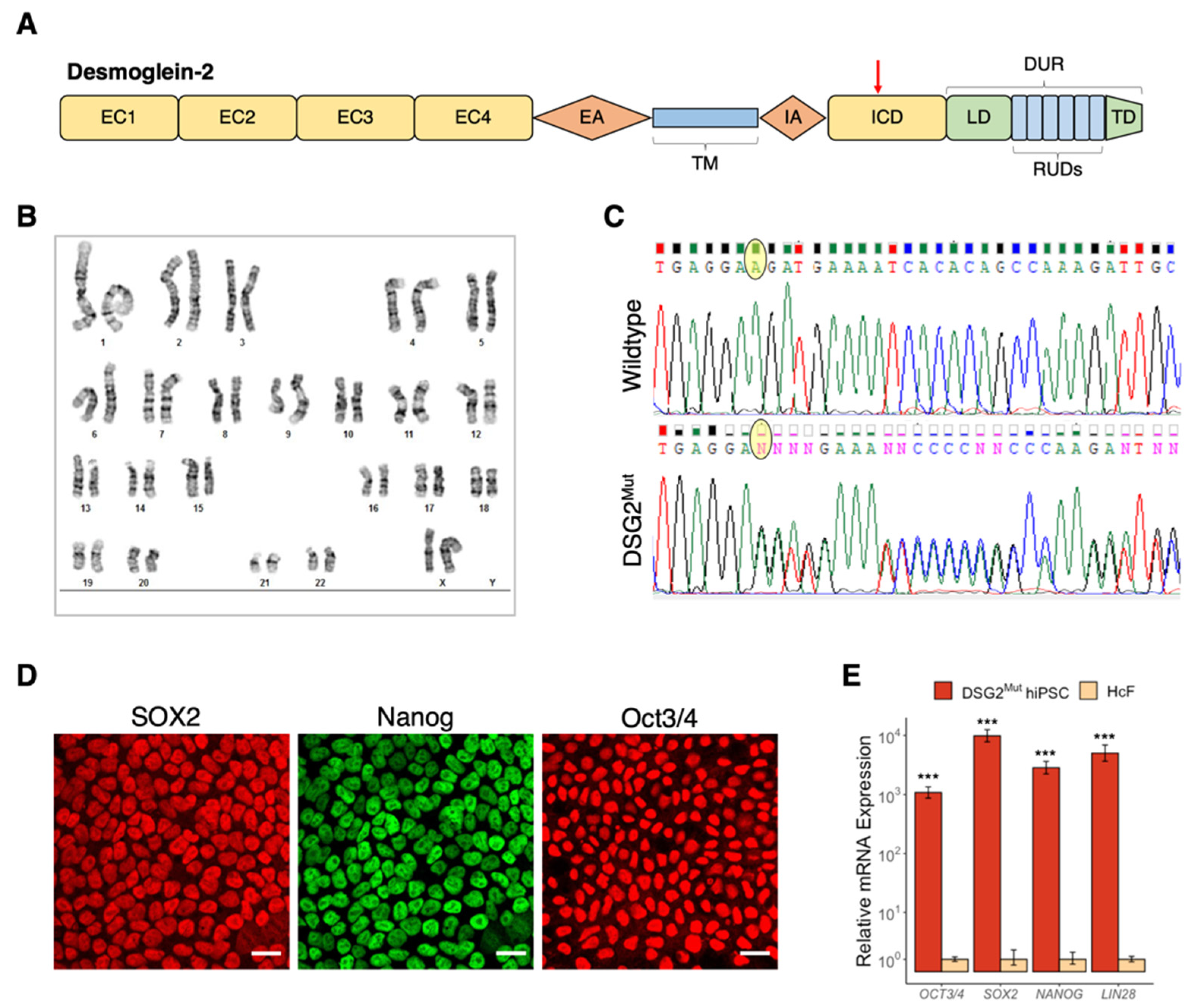
3.2. Establishment of Patient-Specific Human Induced Pluripotent Stem Cell Line with Novel Dsg2 Pathogenic Variant
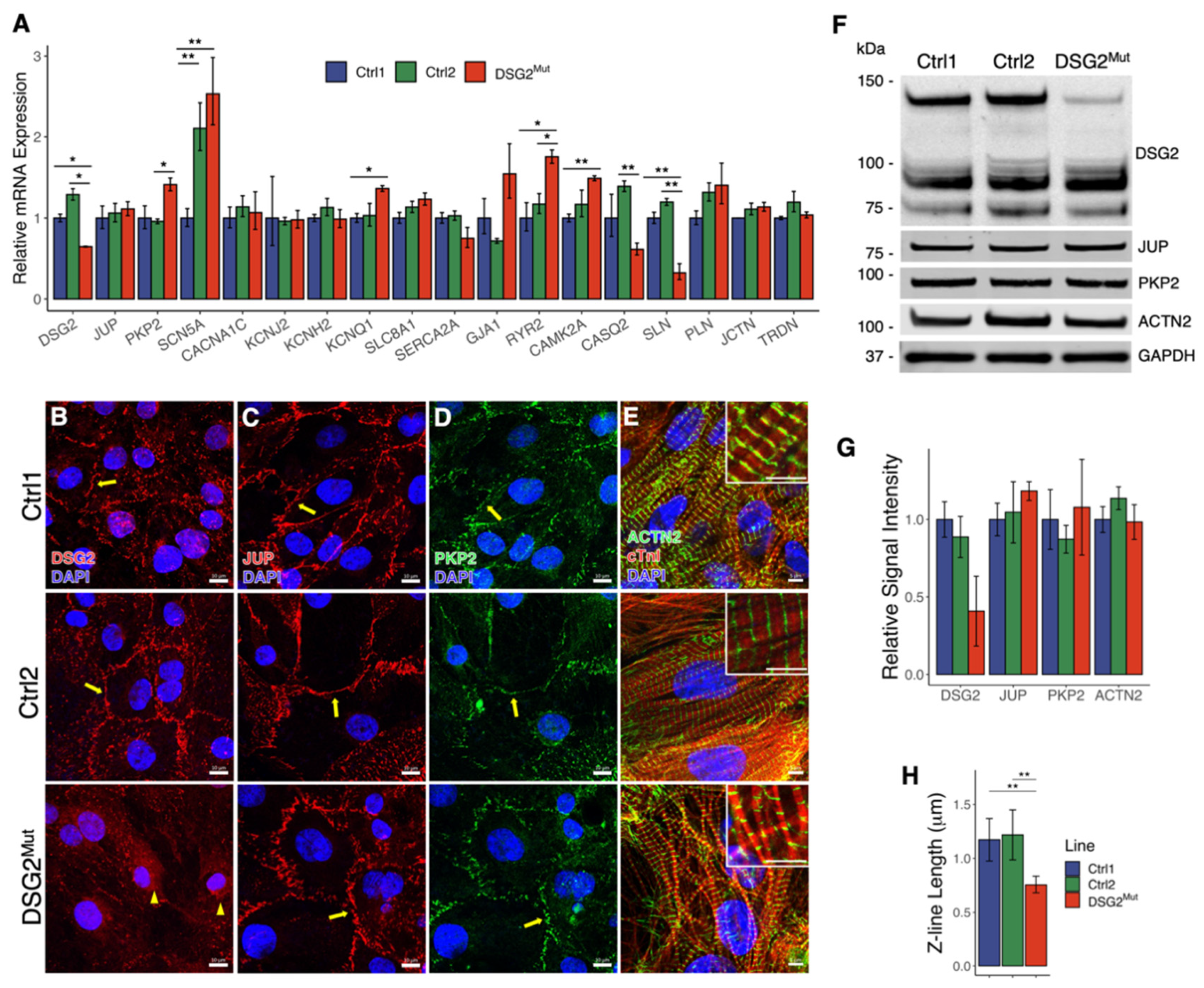
3.3. Biomolecular Characterization of DSG2Mut hiPSC-CM Model
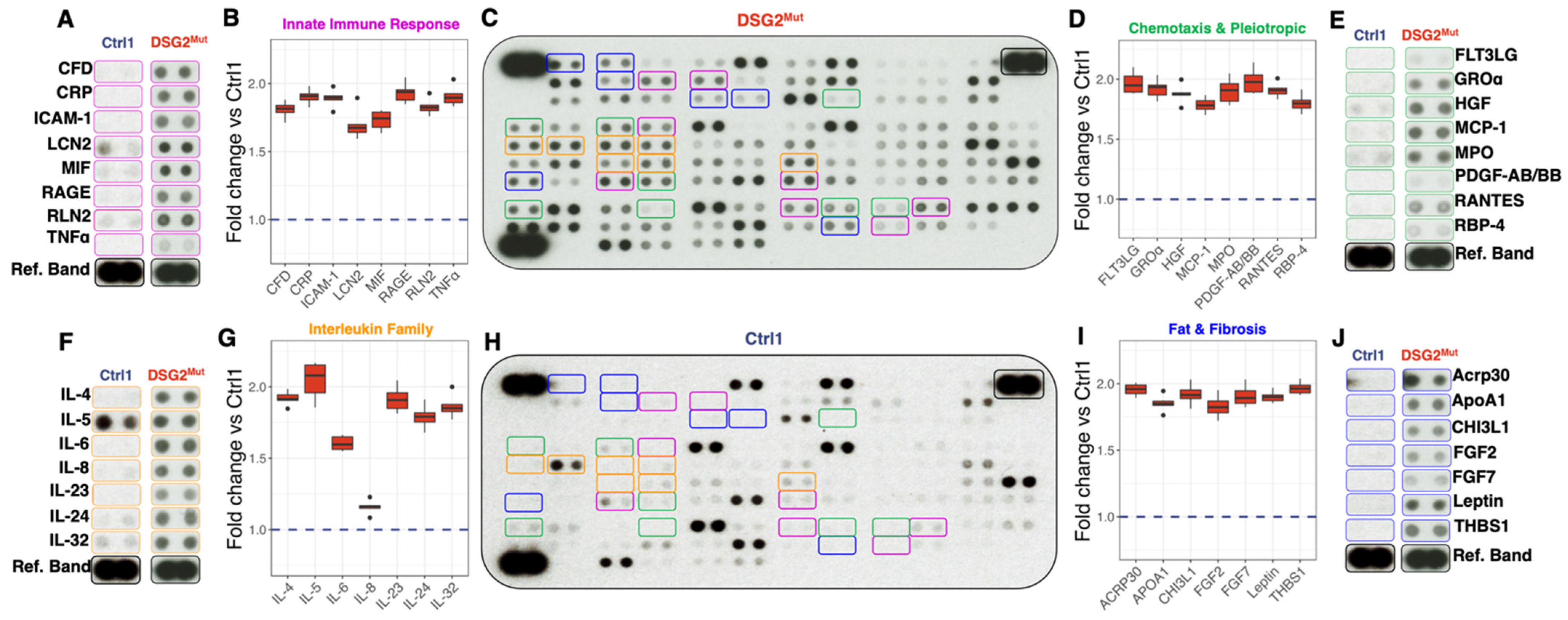
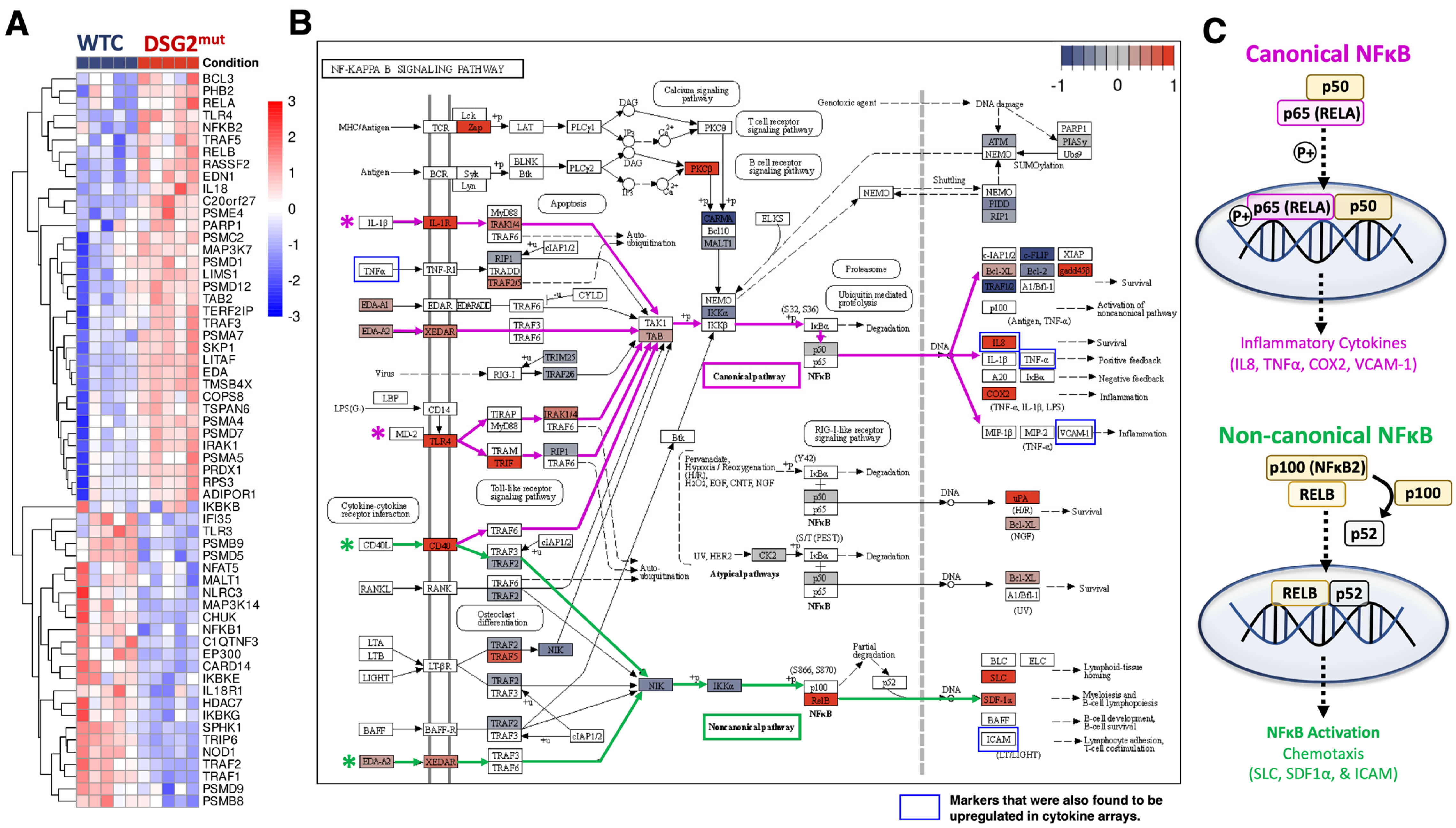
3.4. Cytokine and Chemokine Expression in DSG2Mut hiPSC-CMs
3.5. Immune Signaling in DSG2Mut hiPSC-CMs Is Regulated by Canonical and Non-Canonical NFĸB Pathways
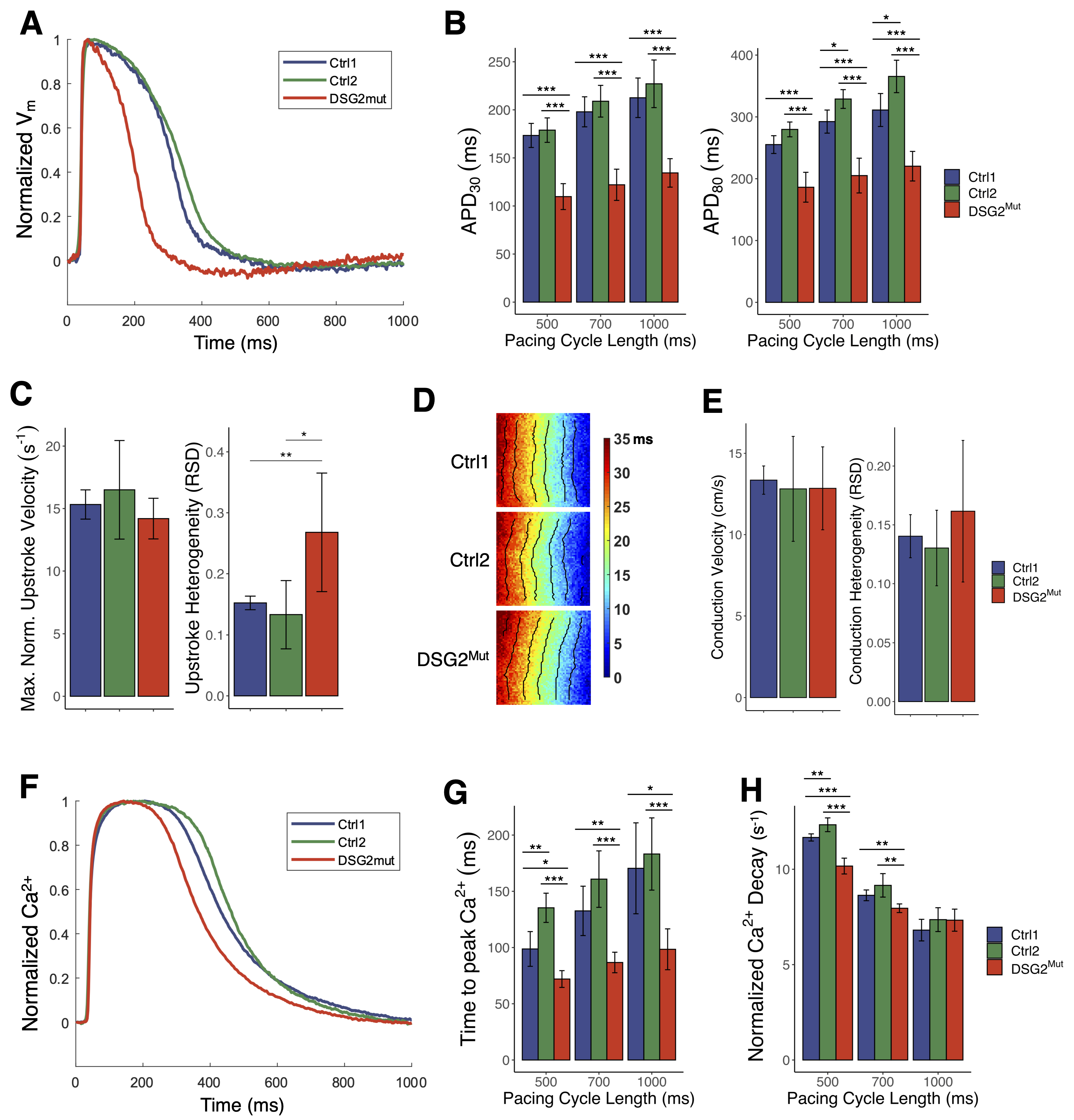
3.6. Shortened Action Potential Durations, Heterogeneous Excitation, and Altered Calcium Handling in DSG2Mut CMs
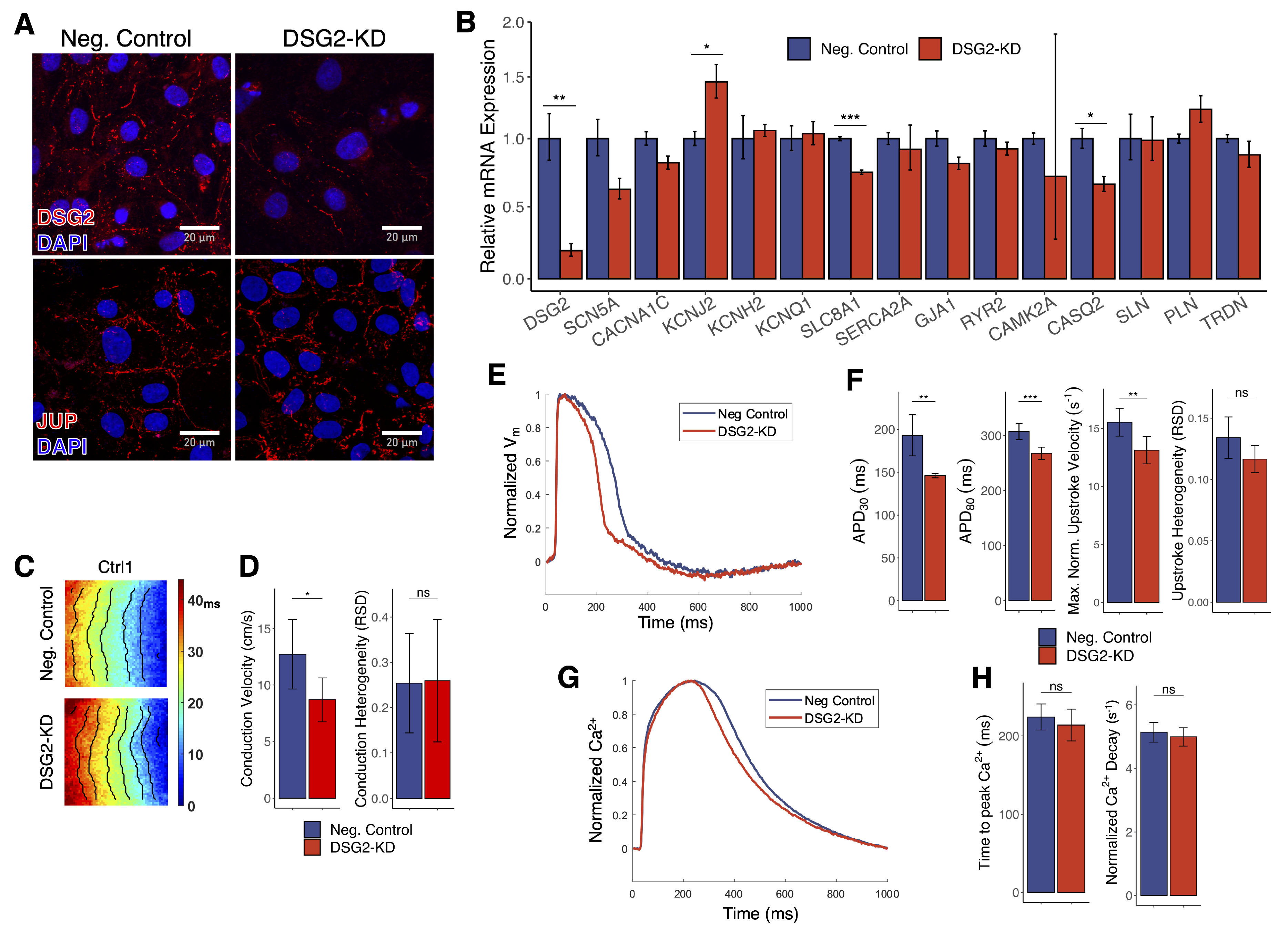
3.7. DSG2 Knockdown Recapitulates Some, but Not All, Effects of DSG2 c.2358 Variant in Control hiPSC-CMs
4. Discussion
Study Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Saguner, A.M.; Brunckhorst, C.; Duru, F. Arrhythmogenic ventricular cardiomyopathy: A paradigm shift from right to biventricular disease. World J. Cardiol. 2014, 6, 154. [Google Scholar] [CrossRef]
- Basso, C.; Corrado, D.; Thiene, G. Cardiovascular causes of sudden death in young individuals including athletes. Cardiol. Rev. 1999, 7, 127–135. [Google Scholar] [CrossRef]
- Tabib, A.; Loire, R.; Chalabreysse, L.; Meyronnet, D.; Miras, A.; Malicier, D.; Thivolet, F.; Chevalier, P.; Bouvagnet, P. Circum-stances of death and gross and microscopic observations in a series of 200 cases of sudden death associated with arrhythmo-genic right ventricular cardiomyopathy and/or dysplasia. Circulation 2003, 108, 3000–3005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basso, C.; Corrado, D.; Marcus, F.I.; Nava, A.; Thiene, G. Arrhythmogenic right ventricular cardiomyopathy. Lancet 2009, 373, 1289–1300. [Google Scholar] [CrossRef]
- Peters, S.; Trümmel, M.; Meyners, W. Prevalence of right ventricular dysplasia-cardiomyopathy in a non-referral hospital. Int. J. Cardiol. 2004, 97, 499–501. [Google Scholar] [CrossRef]
- Groeneweg, J.A.; Bhonsale, A.; James, C.; Riele, A.S.T.; Dooijes, D.; Tichnell, C.; Murray, B.; Wiesfeld, A.C.P.; Sawant, A.; Kassamali, B.; et al. Clinical presentation, long-term follow-up, and outcomes of 1001 arrhythmogenic right ventricular dysplasia/cardiomyopathy patients and family members. Circ. Cardiovasc. Genet. 2015, 8, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Vatta, M.; Marcus, F.; Towbin, J.A. Arrhythmogenic right ventricular cardiomyopathy: A ’final common pathway’ that defines clinical phenotype. Eur. Heart J. 2006, 28, 529–530. [Google Scholar] [CrossRef] [Green Version]
- Towbin, J.A.; McKenna, W.J.; Abrams, D.; Ackerman, M.J.; Calkins, H.; Darrieux, F.C.; Daubert, J.P.; de Chillou, C.; DePasquale, E.C.; Desai, M.Y.; et al. 2019 HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy. Heart Rhythm. 2019, 16, e301–e372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, C.L.; Sutanto, H.; Dalageorgou, C.; McKenna, W.J.; Syrris, P.; Futema, M. Frequency of genetic variants associated with arrhythmogenic right ventricular cardiomyopathy in the genome aggregation database. Eur. J. Hum. Genet. 2018, 26, 1312–1318. [Google Scholar] [CrossRef]
- van Lint, F.H.; Murray, B.; Tichnell, C.; Zwart, R.; Amat, N.; Deprez, R.H.L.; Dittmann, S.; Stallmeyer, B.; Calkins, H.; van der Smagt, J.J.; et al. Arrhythmogenic right ventricular cardiomyopathy-associated desmosomal variants are rarely de novo. Circ. Genom. Precis. Med. 2019, 12, e002467. [Google Scholar] [CrossRef] [Green Version]
- Rizzo, S.; Lodder, E.M.; Verkerk, A.; Wolswinkel, R.; Beekman, L.; Pilichou, K.; Basso, C.; Remme, C.A.; Thiene, G.; Bezzina, C.R. Intercalated disc abnormalities, reduced Na+ current density, and conduction slowing in desmoglein-2 mutant mice prior to cardiomyopathic changes. Cardiovasc. Res. 2012, 95, 409–418. [Google Scholar] [CrossRef] [Green Version]
- Gomes, J.; Finlay, M.; Ahmed, A.K.; Ciaccio, E.J.; Asimaki, A.; Saffitz, J.E.; Quarta, G.; Nobles, M.; Syrris, P.; Chaubey, S.; et al. Electrophysiological abnormalities precede overt structural changes in arrhythmogenic right ventricular cardiomyopathy due to mutations in desmoplakin-A combined murine and human study. Eur. Heart J. 2012, 33, 1942–1953. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, S.R.; Gard, J.J.; Protonotarios, N.; Tsatsopoulou, A.; Spiliopoulou, C.; Anastasakis, A.; Squarcioni, C.P.; McKenna, W.J.; Thiene, G.; Basso, C.; et al. Remodeling of myocyte gap junctions in arrhythmogenic right ventricular cardiomyopathy due to a deletion in plakoglobin (Naxos disease). Heart Rhythm. 2004, 1, 3–11. [Google Scholar] [CrossRef]
- Chelko, S.P.; Keceli, G.; Carpi, A.; Doti, N.; Agrimi, J.; Asimaki, A.; Beti, C.B.; Miyamoto, M.; Amat-Codina, N.; Bedja, D.; et al. Exercise triggers CAPN1-mediated AIF truncation, inducing myocyte cell death in arrhythmogenic cardiomyopathy. Sci. Transl. Med. 2021, 13, eabf0891. [Google Scholar] [CrossRef] [PubMed]
- Chelko, S.; Asimaki, A.; Lowenthal, J.; Bueno-Beti, C.; Bedja, D.; Scalco, A.; Amat-Alarcon, N.; Andersen, P.; Judge, D.P.; Tung, L.; et al. Therapeutic modulation of the immune response in arrhythmogenic cardiomyopathy. Circulation 2019, 140, 1491–1505. [Google Scholar] [CrossRef] [PubMed]
- Cerrone, M.; Montnach, J.; Carolina, V.; Zhao, Y.-T.; Zhang, M.; Agullo-Pascual, E.; Leo-Macias, A.; Alvarado, F.J.; Dolgalev, I.; Karathanos, T.V.; et al. Plakophilin-2 is required for transcription of genes that control calcium cycling and cardiac rhythm. Nat. Commun. 2017, 8, 1–16. [Google Scholar] [CrossRef]
- Kim, J.-C.; Pérez-Hernández, M.; Alvarado, F.J.; Maurya, S.; Montnach, J.; Yin, Y.; Zhang, M.; Lin, X.; Vasquez, C.; Heguy, A.; et al. Disruption of Ca 2+ i Homeostasis and Connexin 43 Hemichannel Function in the Right Ventricle Precedes Overt Arrhythmogenic Cardiomyopathy in Plakophilin-2–Deficient Mice. Circulation 2019, 140, 1015–1030. [Google Scholar] [CrossRef] [PubMed]
- Vischer, A.S.; Connolly, D.J.; Coats, C.J.; Fuentes, V.L.; McKenna, W.J.; Castelletti, S.; Pantazis, A.A. Arrhythmogenic right ventricular cardiomyopathy in Boxer dogs: The diagnosis as a link to the human disease. Acta Myol. 2017, 36, 135–150. [Google Scholar] [PubMed]
- Eason, B.D.; Leach, S.B.; Kuroki, K. Arrhythmogenic Right Ventricular Cardiomyopathy in a Weimaraner. Can. Vet. J. 2015, 56, 1035–1039. [Google Scholar]
- Boukens, B.J.; Rivaud, M.R.; Rentschler, S.; Coronel, R. Misinterpretation of the mouse ECG: “Musing the waves of mus musculus ”. J. Physiol. 2014, 592, 4613–4626. [Google Scholar] [CrossRef]
- Ross, S.B.; Fraser, S.; Semsarian, C. Induced pluripotent stem cells in the inherited cardiomyopathies: From disease mechanisms to novel therapies. Trends Cardiovasc. Med. 2016, 26, 663–672. [Google Scholar] [CrossRef]
- Blazeski, A.; Lowenthal, J.; Wang, Y.; Teuben, R.; Zhu, R.; Gerecht, S.; Tomaselli, G.; Tung, L. Engineered Heart Slice Model of Arrhythmogenic Cardiomyopathy Using Plakophilin-2 Mutant Myocytes. Tissue Eng. Part. A 2019, 25, 725–735. [Google Scholar] [CrossRef]
- Kim, C.; Wong, J.; Wen, J.; Wang, S.; Wang, C.; Spiering, S.; Kan, N.G.; Forcales, S.; Puri, P.L.; Leone, T.C.; et al. Studying arrhythmogenic right ventricular dysplasia with patient-specific iPSCs. Nat. Cell Biol. 2013, 494, 105–110. [Google Scholar] [CrossRef] [Green Version]
- Khudiakov, A.; Zaytseva, A.; Perepelina, K.; Smolina, N.; Pervunina, T.; Vasichkina, E.; Karpushev, A.; Tomilin, A.; Malashicheva, A.; Kostareva, A. Sodium current abnormalities and deregulation of Wnt/β-catenin signaling in iPSC-derived cardiomyocytes generated from patient with arrhythmogenic cardiomyopathy harboring compound genetic variants in plakophilin 2 gene. Biochim. et Biophys. Acta BBA Mol. Basis Dis. 2020, 1866, 165915. [Google Scholar] [CrossRef] [PubMed]
- El-Battrawy, I.; Zhao, Z.; Lan, H.; Cyganek, L.; Tombers, C.; Li, X.; Buljubasic, F.; Lang, S.; Tiburcy, M.; Zimmermann, W.-H.; et al. Electrical dysfunctions in human-induced pluripotent stem cell-derived cardiomyocytes from a patient with an arrhythmogenic right ventricular cardiomyopathy. Europace 2018, 20, f46–f56. [Google Scholar] [CrossRef] [PubMed]
- Marcus, F.I.; McKenna, W.J.; Sherrill, D.; Basso, C.; Bauce, B.; Bluemke, D.A.; Calkins, H.; Corrado, D.; Cox, M.G.P.J.; Daubert, J.P.; et al. Diagnosis of Arrhytmogenic Right Ventricular Cardiomyopathy/Dysplasia (ARVC/D): Proposed Modification of the Task Force Criteria. Circulation 2010, 121, 1533–1541. [Google Scholar] [CrossRef] [PubMed]
- Poon, E.; Luo, X.-L.; Webb, S.; Yan, B.; Zhao, R.; Wu, S.C.M.; Yang, Y.; Zhang, P.; Bai, H.; Shao, J.; et al. The cell surface marker CD36 selectively identifies matured, mitochondria-rich hPSC-cardiomyocytes. Cell Res. 2020, 30, 626–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ong, C.S.; Fukunishi, T.; Zhang, H.; Huang, C.Y.; Nashed, A.; Blazeski, A.; DiSilvestre, D.; Vricella, L.; Conte, J.; Tung, L.; et al. Biomaterial-Free Three-Dimensional Bioprinting of Cardiac Tissue using Human Induced Pluripotent Stem Cell Derived Cardiomyocytes. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyaoka, Y.; Chan, A.H.; Judge, L.M.; Yoo, J.; Huang, M.; Nguyen, T.D.; Lizarraga, P.P.; So, P.-L.; Conklin, B.R. Isolation of single-base genome-edited human iPS cells without antibiotic selection. Nat. Methods 2014, 11, 291–293. [Google Scholar] [CrossRef] [Green Version]
- Burridge, P.W.; Matsa, E.; Shukla, P.; Lin, Z.C.; Churko, J.M.; Ebert, A.D.; Lan, F.; Diecke, S.; Huber, B.; Mordwinkin, N.M.; et al. Chemically Defined and Small Molecule-Based Generation of Human Cardiomyocytes. Nat. Methods 2014, 11, 855–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.Y.; Maia-Joca, R.P.M.; Ong, C.S.; Wilson, I.; DiSilvestre, D.; Tomaselli, G.F.; Reich, D.H. Enhancement of human iPSC-derived cardiomyocyte maturation by chemical conditioning in a 3D environment. J. Mol. Cell. Cardiol. 2020, 138, 1–11. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. The Subread aligner: Fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 2013, 41, e108. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, W.; Brouwer, C. Pathview: An R/Bioconductor package for pathway-based data integration and visualization. Bioinformatics 2013, 29, 1830–1831. [Google Scholar] [CrossRef] [Green Version]
- Sylvain Le Pepe EasyqpcR: EasyqpcR for Easy Analysis of Real-Time PCR Data. Available online: http://irtomit.labo.univ-poitiers.fr/ (accessed on 7 January 2021).
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef] [PubMed]
- Kelly, M.A.; Caleshu, C.; Morales, A.; Buchan, J.; Wolf, Z.; Harrison, S.M.; Cook, S.; Dillon, M.W.; Garcia, J.; Haverfield, E. Adaptation and validation of the ACMG/AMP variant classification framework for MYH7-associated inherited cardiomyopathies: Recommendations by ClinGen’s Inherited Cardiomyopathy Expert Panel. Genet. Med. 2018, 20, 351–359. [Google Scholar] [CrossRef] [Green Version]
- Chelko, S.; Asimaki, A.; Andersen, P.; Bedja, D.; Amat-Alarcon, N.; DeMazumder, D.; Jasti, R.; Macrae, C.A.; Leber, R.; Kleber, A.G.; et al. Central role for GSK3β in the pathogenesis of arrhythmogenic cardiomyopathy. JCI Insight 2016, 1, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asimaki, A.; Kapoor, S.; Plovie, E.; Arndt, A.K.; Adams, E.; Liu, Z.; James, C.; Judge, D.; Calkins, H.; Churko, J.; et al. Identification of a New Modulator of the Intercalated Disc in a Zebrafish Model of Arrhythmogenic Cardiomyopathy. Sci. Transl. Med. 2014, 6, 240ra74. [Google Scholar] [CrossRef] [Green Version]
- Toffolo, R.L.; David Ianuzzo, C. Myofibrillar Adaptations during Cardiac Hypertrophy; Kluwer Academic Publishers: New York, NY, USA, 1994; Volume 131. [Google Scholar]
- Scheel, P.J.; Murray, B.; Tichnell, C.; James, C.A.; Tandri, H.; Calkins, H.; Chelko, S.P.; Gilotra, N.A. Arrhythmogenic Right Ventricular Cardiomyopathy Presenting as Clinical Myocarditis in Women. Am. J. Cardiol. 2021, 145, 128–134. [Google Scholar] [CrossRef]
- Bangert, A.; Andrassy, M.; Müller, A.-M.; Bockstahler, M.; Fischer, A.; Volz, C.H.; Leib, C.; Göser, S.; Korkmaz-Icöz, S.; Zittrich, S.; et al. Critical role of RAGE and HMGB1 in inflammatory heart disease. Proc. Natl. Acad. Sci. USA 2016, 113, E155–E164. [Google Scholar] [CrossRef] [Green Version]
- Zhong, Q.; Zhou, K.; Liang, Q.; Lin, S.; Wang, Y.; Xiong, X.; Meng, Z.; Zhao, T.; Zhu, W.; Yang, Y.; et al. Interleukin-23 Secreted by Activated Macrophages Drives γδT Cell Production of Interleukin-17 to Aggravate Secondary Injury After Intracerebral Hemorrhage. J. Am. Heart Assoc. 2016, 5. [Google Scholar] [CrossRef]
- Ha, H.; Han, D.; Choi, Y. TRAF-Mediated TNFR-Family Signaling. Curr. Protoc. Immunol. 2009, 87, 11–19. [Google Scholar] [CrossRef]
- Xie, P. TRAF Molecules in Cell Signaling and in Human Diseases. J. Mol. Signal. 2013, 8, 1–31. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.-H.; Sun, S.-C. Tumor Necrosis Factor Receptor-Associated Factor Regulation of Nuclear Factor κB and Mitogen-Activated Protein Kinase Pathways. Front. Immunol. 2018, 9, 1849. [Google Scholar] [CrossRef] [PubMed]
- James, C.A.; Jongbloed, J.D.; Hershberger, R.E.; Morales, A.; Judge, D.P.; Syrris, P.; Pilichou, K.; Domingo, A.M.; Murray, B.; Cadrin-Tourigny, J.; et al. International Evidence Based Reappraisal of Genes Associated With Arrhythmogenic Right Ventricular Cardiomyopathy Using the Clinical Genome Resource Framework. Circ. Genom. Precis. Med. 2021, 14, e003273. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Nekrasova, O.E.; Patel, D.M.; Klessner, J.L.; Godsel, L.M.; Koetsier, J.L.; Amargo, E.V.; Desai, B.V.; Green, K.J. The C-terminal unique region of desmoglein 2 inhibits its internalization via tail–tail interactions. J. Cell Biol. 2012, 199, 699–711. [Google Scholar] [CrossRef] [Green Version]
- Gehmlich, K.; Asimaki, A.; Cahill, T.J.; Ehler, E.; Syrris, P.; Zachara, E.; Re, F.; Avella, A.; Monserrat, L.; Saffitz, J.E.; et al. Novel missense mutations in exon 15 of desmoglein-2: Role of the intracellular cadherin segment in arrhythmogenic right ventricular cardiomyopathy? Heart Rhythm. 2010, 7, 1446–1453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asimaki, A.; Tandri, H.; Huang, H.; Halushka, M.K.; Gautam, S.; Basso, C.; Thiene, G.; Tsatsopoulou, A.; Protonotarios, N.; McKenna, W.J.; et al. A New Diagnostic Test for Arrhythmogenic Right Ventricular Cardiomyopathy From the Departments of Pathology. New Engl. J. Med. 2009, 360, 1075–1084. [Google Scholar] [CrossRef] [PubMed]
- Munkholm, J.; Christensen, A.H.; Svendsen, J.H.; Andersen, C.B. Usefulness of Immunostaining for Plakoglobin as a Diagnostic Marker of Arrhythmogenic Right Ventricular Cardiomyopathy. Am. J. Cardiol. 2012, 109, 272–275. [Google Scholar] [CrossRef]
- Ermakov, S.; Ursell, P.C.; Johnson, C.J.; Meadows, A.; Zhao, S.; Marcus, G.M.; Scheinman, M. Plakoglobin Immunolocalization as a Diagnostic Test for Arrhythmogenic Right Ventricular Cardiomyopathy. Pacing Clin. Electrophysiol. 2014, 37, 1708–1716. [Google Scholar] [CrossRef]
- D’Amati, G.; Leone, O.; di Gioia, C.R.T.; Magelli, C.; Arpesella, G.; Grillo, P.; Marino, B.; Fiore, F.; Gallo, P. Arrhythmogenic right ventricular cardiomyopathy: Clinicopathologic correlation based on a revised definition of pathologic patterns. Hum. Pathol. 2001, 32, 1078–1086. [Google Scholar] [CrossRef]
- Hedberg, C.; Melberg, A.; Kuhl, A.; Jenne, D.; Oldfors, A. Autosomal dominant myofibrillar myopathy with arrhythmogenic right ventricular cardiomyopathy 7 is caused by a DES mutation. Eur. J. Hum. Genet. 2012, 20, 984–985. [Google Scholar] [CrossRef]
- Murray, B. Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy (ARVD/C): A Review of Molecular and Clinical Literature. J. Genet. Couns. 2012, 21, 494–504. [Google Scholar] [CrossRef]
- Gandjbakhch, E.; Redheuil, A.; Pousset, F.; Charron, P.; Frank, R. Clinical Diagnosis, Imaging, and Genetics of Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia. J. Am. Coll. Cardiol. 2018, 72, 784–804. [Google Scholar] [CrossRef] [PubMed]
- Shiba, M.; Higo, S.; Kondo, T.; Li, J.; Liu, L.; Ikeda, Y.; Kohama, Y.; Kameda, S.; Tabata, T.; Inoue, H.; et al. Phenotypic recapitulation and correction of desmoglein-2-deficient cardiomyopathy using human-induced pluripotent stem cell-derived cardiomyocytes. Hum. Mol. Genet. 2021. [Google Scholar] [CrossRef] [PubMed]
- Klabunde, R.E. Cardiac electrophysiology: Normal and ischemic ionic currents and the ECG. Adv. Physiol. Educ. 2017, 41, 29–37. [Google Scholar] [CrossRef] [Green Version]
- Willis, B.C.; Ponce-Balbuena, D.; Jalife, J. Protein assemblies of sodium and inward rectifier potassium channels control cardiac excitability and arrhythmogenesis. Am. J. Physiol. Circ. Physiol. 2015, 308, H1463–H1473. [Google Scholar] [CrossRef] [Green Version]
- Sampson, L.J.; Leyland, M.L.; Dart, C. Direct Interaction between the Actin-binding Protein Filamin-A and the Inwardly Rectifying Potassium Channel, Kir2.1. J. Biol. Chem. 2003, 278, 41988–41997. [Google Scholar] [CrossRef] [Green Version]
- Sutanto, H.; Lyon, A.; Lumens, J.; Schotten, U.; Dobrev, D.; Heijman, J. Cardiomyocyte calcium handling in health and disease: Insights from in vitro and in silico studies. Prog. Biophys. Mol. Biol. 2020, 157, 54–75. [Google Scholar] [CrossRef] [PubMed]
- Bhupathy, P.; Babu, G.J.; Periasamy, M. Sarcolipin and phospholamban as regulators of cardiac sarcoplasmic reticulum Ca2+ ATPase. J. Mol. Cell. Cardiol. 2007, 42, 903–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asahi, M.; Otsu, K.; Nakayama, H.; Hikoso, S.; Takeda, T.; Gramolini, A.O.; Trivieri, M.G.; Oudit, G.Y.; Morita, T.; Kusakari, Y.; et al. Cardiac-Specific Overexpression of Sarcolipin Inhibits Sarco(Endo)Plasmic Reticulum Ca 2 ATPase (SERCA2a) Activity and Impairs Cardiac Function in Mice. Proc. Natl. Acad. Sci. USA 2004, 110, 9199–9204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Matthews, G.D.K.; Lei, M.; Huang, C.L.H. Abnormal Ca2+ Homeostasis, Atrial Arrhythmogenesis, and Sinus Node Dysfunction in Murine Hearts Modeling RyR2 Modification. Front. Physiol. 2013, 4, 150. [Google Scholar] [CrossRef] [Green Version]
- Bartos, D.C.; Morotti, S.; Ginsburg, K.S.; Grandi, E.; Bers, N.M. Quantitative analysis of the Ca2+-dependent regulation of delayed rectifier K+currentIKsin rabbit ventricular myocytes. J. Physiol. 2017, 595, 2253–2268. [Google Scholar] [CrossRef] [Green Version]
- Kant, S.; Holthöfer, B.; Magin, T.M.; Krusche, C.A.; Leube, R.E. Desmoglein 2–Dependent Arrhythmogenic Cardiomyopathy Is Caused by a Loss of Adhesive Function. Circ. Cardiovasc. Genet. 2015, 8, 553–563. [Google Scholar] [CrossRef] [Green Version]
- Karbassi, E.; Fenix, A.; Marchiano, S.; Muraoka, N.; Nakamura, K.; Yang, X.; Murry, C.E. Cardiomyocyte maturation: Advances in knowledge and implications for regenerative medicine. Nat. Rev. Cardiol. 2020, 17, 341–359. [Google Scholar] [CrossRef]
- van de Sande, D.V.; Kopljar, I.; Maaike, A.; Teisman, A.; Gallacher, D.J.; Bart, L.; Snyders, D.J.; Leybaert, L.; Lu, H.R.; Labro, A.J. The resting membrane potential of hSC-CM in a syncytium is more hyperpolarised than that of isolated cells. Channels 2021, 15, 239–252. [Google Scholar] [CrossRef]
- Li, W.; Han, J.L.; Entcheva, E. Syncytium cell growth increases Kir2.1 contribution in human iPSC-cardiomyocytes. Am. J. Physiol. Circ. Physiol. 2020, 319, H1112–H1122. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Attribute | Finding |
|---|---|
| Demographic | |
| Gender | Female |
| Race/Ethnicity | South Asian |
| Presentation (age [yrs]: symptoms) | 22: anterior precordial T wave inversions noted on ECG. 32: dizziness, palpations |
| Age at diagnosis (yrs) | 45 |
| Age at last follow-up (yrs) | 51 |
| 2010 Task Force Criteria Fulfillment [26] | |
| ECG Repolarization Abnm. | Major: Inverted T waves in right precordial leads (V1, V2, and V3) |
| ECG Depolarization Abnm. | None |
| Arrhythmia | Minor: >500 PVCs per 24 h on Holter monitor |
| Structural | Major: Regional RV dyskinesia + RV EDVi > 100 ml/m2 |
| Family History | Major: ARVC confirmed in a first-degree relative who meets current Task Force criteria |
| Cardiac MRI (at last follow-up) | |
| RV EDVi (ml/m2) | 118 |
| RV EDVi / LV EDVi | 2.62 |
| RV wall-motion Abnm. | Regional dyskinesia, aneurysms |
| LV wall-motion Abnm. | None |
| RVEF (%) | 44 |
| LVEF (%) | 57 |
| Delayed enhancement | Extensive delayed enhancement in RV free wall and inferior wall |
| Events | |
| Sustained ventricular arrythmia | No |
| Cardiac transplant | No |
| Death | No |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hawthorne, R.N.; Blazeski, A.; Lowenthal, J.; Kannan, S.; Teuben, R.; DiSilvestre, D.; Morrissette-McAlmon, J.; Saffitz, J.E.; Boheler, K.R.; James, C.A.; et al. Altered Electrical, Biomolecular, and Immunologic Phenotypes in a Novel Patient-Derived Stem Cell Model of Desmoglein-2 Mutant ARVC. J. Clin. Med. 2021, 10, 3061. https://doi.org/10.3390/jcm10143061
Hawthorne RN, Blazeski A, Lowenthal J, Kannan S, Teuben R, DiSilvestre D, Morrissette-McAlmon J, Saffitz JE, Boheler KR, James CA, et al. Altered Electrical, Biomolecular, and Immunologic Phenotypes in a Novel Patient-Derived Stem Cell Model of Desmoglein-2 Mutant ARVC. Journal of Clinical Medicine. 2021; 10(14):3061. https://doi.org/10.3390/jcm10143061
Chicago/Turabian StyleHawthorne, Robert N., Adriana Blazeski, Justin Lowenthal, Suraj Kannan, Roald Teuben, Deborah DiSilvestre, Justin Morrissette-McAlmon, Jeffrey E. Saffitz, Kenneth R. Boheler, Cynthia A. James, and et al. 2021. "Altered Electrical, Biomolecular, and Immunologic Phenotypes in a Novel Patient-Derived Stem Cell Model of Desmoglein-2 Mutant ARVC" Journal of Clinical Medicine 10, no. 14: 3061. https://doi.org/10.3390/jcm10143061
APA StyleHawthorne, R. N., Blazeski, A., Lowenthal, J., Kannan, S., Teuben, R., DiSilvestre, D., Morrissette-McAlmon, J., Saffitz, J. E., Boheler, K. R., James, C. A., Chelko, S. P., Tomaselli, G., & Tung, L. (2021). Altered Electrical, Biomolecular, and Immunologic Phenotypes in a Novel Patient-Derived Stem Cell Model of Desmoglein-2 Mutant ARVC. Journal of Clinical Medicine, 10(14), 3061. https://doi.org/10.3390/jcm10143061