
Autoimmune Brainstem Encephalitis: An Illustrative Case and a Review of the Literature

 ,
,  ,
,  and
and
Abstract
:1. Introduction
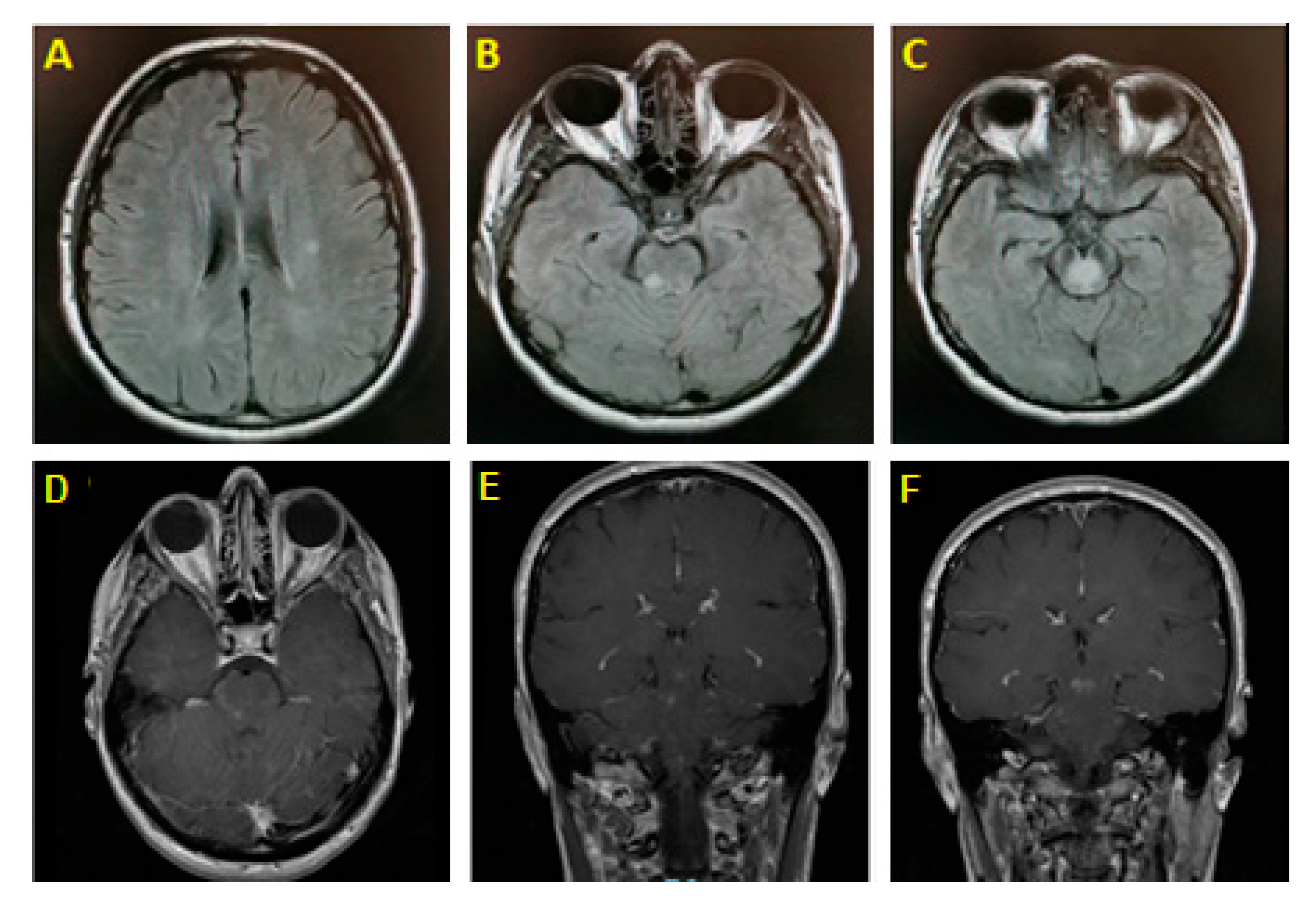
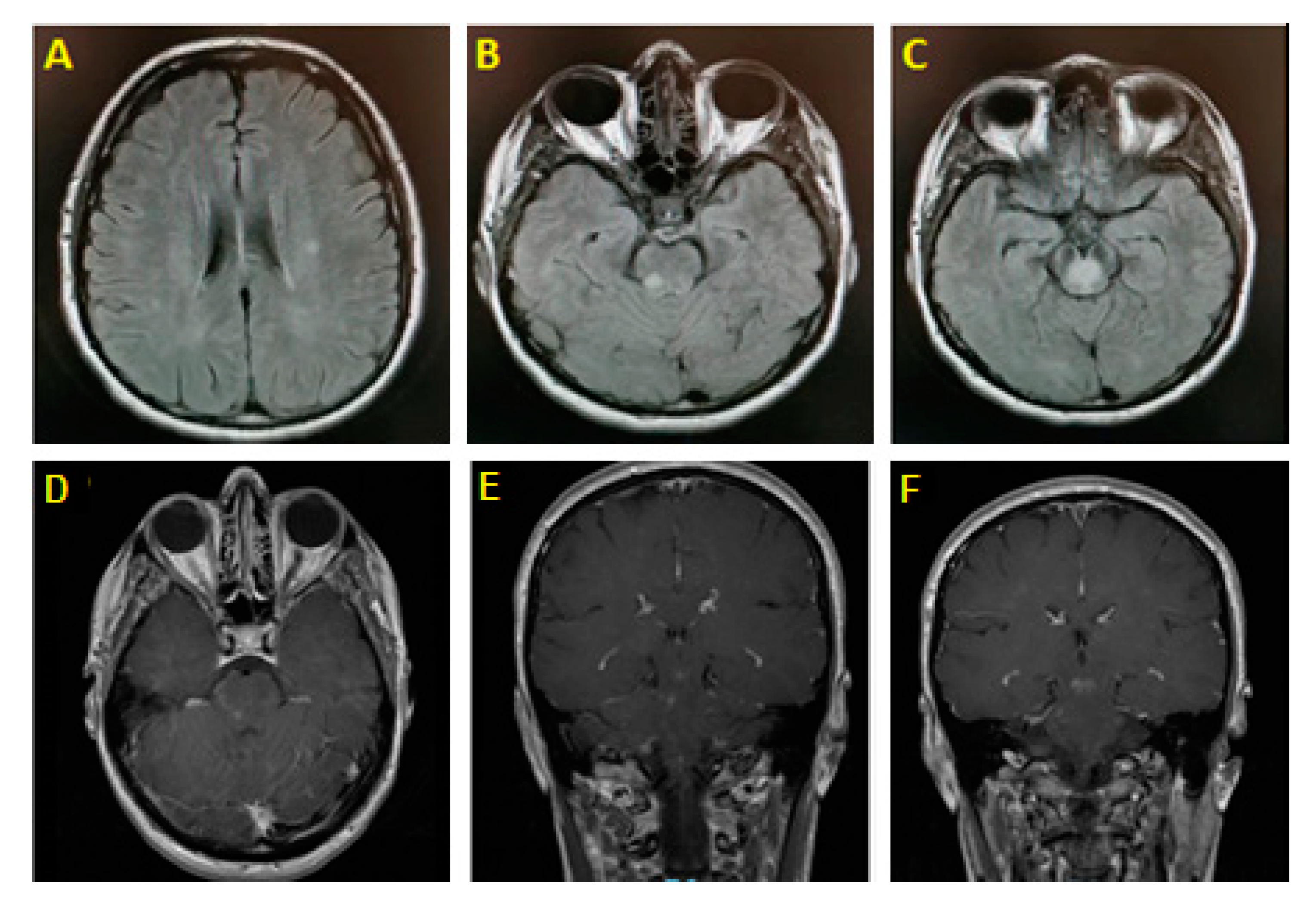
2. Illustrative Case
3. Etiology of Autoimmune Brainstem Encephalitis
3.1. Multiple Sclerosis
3.2. Neuromyelitis Optica Spectrum Disorder and MOG-Antibodies-Associated Disease
3.3. Autoimmune Glial Fibrillary Acidic Protein Astrocytopathy
3.4. Acute Disseminated Encephalomyelitis/Acute Hemorrhagic Leukoencephalitis
3.5. Bickerstaff Brainstem Encephalitis
3.6. CLIPPERS
3.7. Connective Tissue Disease and Vasculitis
3.8. Paraneoplastic Syndromes
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A

{kind=link}
| Disease | Characteristic Features | Brain MRI Findings | Antibodies | Treatment | |
|---|---|---|---|---|---|
| MS [9,13] | Nonspecific symptoms | Periventricular white matter lesions, Dawson fingers, DIT + DIS, pontine predilection | - | DMDs | |
| NMOSD [14,15,22,30] | Intractable vomiting, hiccups, optic neuritis, myelitis | LETM, optic nerve lesions, BS lesions | AQP4-Ab | 1st line: steroids 2nd line: Azathioprine, Mycophenolate mofetil, Methotrexate, Mitoxantrone, Cyclophosphamide, and monoclonal antibodies | |
| Anti-MOG [17,27] | Bilateral optic neuritis, LETM, BS encephalitis | Poorly demarcated, ‘fluffy’ lesions of grey and white matter Non-enhancing hyperintensities in ventral grey matter of spinal cord Anterior optic nerve involvement sparing the chiasm | MOG-IgG | Acute attack: steroids/PLEX Relapse: DMDs, immunosuppressants | |
| GFAP Astrocytopathy [31] | Drug-resistant seizures, meningoencephalomyelitis, psychiatric symptoms | Radial linear periventricular gadolinium enhancement | GFAP-IgG | Acute: steroids Long-term: mycophenolate, azathioprine, rituximab, cyclophosphamide | |
| ADEM/AHLE [9,34,35,36] | Post-viral presentation, fever, meningismus | Asymmetric ill-defined lesions, ventral midbrain | MOG-IgG | Eradicate pathogen Steroids, IVIg, PLEX | |
| Bickerstaff BS Encephalitis [16,40,48] | Ataxia, impaired consciousness and ophthalmoplegia | Unremarkable, occasional T2 BS abnormalities | Anti-GQ1b ganglioside antibodies | IVIg, steroids, PLEX | |
| CLIPPERS [50,51,55] | Drastic clinical and radiological response to steroid | Punctate patchy enhancing pontine lesions | - | Steroids Steroid-sparing as add on | |
| Behcet’s Disease [60] | Oro-genital ulcers, uveitis, skin lesions | Lesions in posterior midbrain-diencephalic junction, sparing the red nucleus | - | Steroids Immunosuppressants | |
| Sjögren Syndrome [65] | PNS involvement, Sicca Symptoms, MS-like presentation after age 50 | Nonspecific, MS-like lesions | Anti-SSA Anti-SSB | Steroids + immunosuppressants, Tocilizumab | |
| Systemic Lupus Erythematosus [66] | Rash, Neuropsychiatric features, young women | Non-specific | ANA, Anti-DNA antibodies | Immunosuppressants | |
| Paraneoplastic Syndromes[71,72,73,75,79,80] | Anti-Hu | Small cell lung carcinoma Medullary dysfunction | Normal | Anti-Hu antibodies | Immunotherapy and treatment of underlying tumor |
| Anti-Ma2 | Testicular germ-cell tumors Midbrain dysfunction | T2-Hyperintense lesions in superior colliculi & periaqueductal region | Anti-Ma2 antibodies | ||
| Anti-Ri | Breast/Ovarian cancers Opsoclonus-Myoclonus | Normal | Anti-Ri antibodies | ||
| KLHL11 | Testicular germ cell tumor Hearing loss and tinnitus | T2/FLAIR abnormalities in BS and limbic system | KLHL11 IgG | Immunotherapy and treatment of underlying tumor | |
| LUZP4 | Germ cell tumor (seminoma) Lower motor neuron involvement, encephalitis, polyradiculopathy | T2/FLAIR abnormalities | LUZP4 IgG | Immunotherapy and treatment of underlying tumor | |
References
- Tan, I.L.; Mowry, E.M.; Steele, S.U.; Pardo, C.A.; McArthur, J.C.; Nath, A.; Venkatesan, A. Brainstem encephalitis: Etiologies, treatment, and predictors of outcome. J. Neurol. 2013, 260, 2312–2319. [Google Scholar] [CrossRef] [PubMed]
- Compston, A.; Coles, A. Multiple sclerosis. Lancet 2008, 372, 1502–1517. [Google Scholar] [CrossRef]
- Habek, M. Evaluation of brainstem involvement in multiple sclerosis. Expert Rev. Neurother. 2013, 13, 299–311. [Google Scholar] [CrossRef]
- Sastre-Garriga, J.; Tintore, M.; Nos, C.; Tur, C.; Rio, J.; Tellez, N.; Castillo, J.; Horga, A.; Perkal, H.; Comabella, M.; et al. Clinical features of CIS of the brainstem/cerebellum of the kind seen in MS. J. Neurol. 2010, 257, 742–746. [Google Scholar] [CrossRef]
- Freiha, J.; Riachi, N.; Chalah, M.A.; Zoghaib, R.; Ayache, S.S.; Ahdab, R. Paroxysmal symptoms in multiple sclerosis—A review of the literature. J. Clin. Med. 2020, 9, 3100. [Google Scholar] [CrossRef]
- Tortorella, C.; Direnzo, V.; D’Onghia, M.; Trojano, M. Brainstem PML lesion mimicking MS plaque in a 402 natalizumab-treated MS patient. Neurology 2013, 81, 1470–1471. [Google Scholar] [CrossRef] [Green Version]
- Hodel, J.; Darchis, C.; Outteryck, O.; Verclytte, S.; Deramecourt, V.; Lacour, A.; Zins, M.; Pruvo, J.-P.; Vermersch, P.; Leclerc, X. Punctate pattern: A promising imaging marker for the diagnosis of natalizumab-associated PML. Neurology 2016, 86, 1516–1523. [Google Scholar] [CrossRef]
- Tecellioglu, M.; Kamisli, O.; Kamisli, S.; Erdogmus, U.A.; Özcan, C. Listeria monocytogenes rhombencephalitis in a patient with multiple sclerosis during fingolimod therapy. Mult. Scler. Relat. Disord. 2019, 27, 409–411. [Google Scholar] [CrossRef]
- Lu, Z.; Zhang, B.; Qiu, W.; Kang, Z.; Shen, L.; Long, Y.; Huang, J.; Hu, X. Comparative brain stem lesions on MRI of acute disseminated encephalomyelitis, neuromyelitis optica, and multiple sclerosis. PLoS ONE 2011, 6, e22766. [Google Scholar] [CrossRef]
- Tintore, M.; Rovira, A.; Arrambide, G.; Mitjana, R.; Río, J.; Auger, C.; Nos, C.; Edo, M.C.; Castilló, J.; Horga, A.; et al. Brainstem lesions in clinically isolated syndromes. Neurology 2010, 75, 1933–1938. [Google Scholar] [CrossRef] [PubMed]
- Mitsutake, A.; Sato, T.; Katsumata, J.; Nakamoto, F.K.; Seki, T.; Maekawa, R.; Hideyama, T.; Shimizu, J.; Shiio, Y. Tumefactive multiple sclerosis which initially presented with brainstem encephalitis with a long-term follow-up. Mult. Scler. Relat. Disord. 2019, 32, 23–26. [Google Scholar] [CrossRef]
- Hauser, S.L.; Cree, B.A.C. Treatment of multiple sclerosis: A review. Am. J. Med. 2020, 133, 1380–1390.e1382. [Google Scholar] [CrossRef] [PubMed]
- Gholamzad, M.; Ebtekar, M.; Ardestani, M.S.; Azimi, M.; Mahmodi, Z.; Mousavi, M.J.; Aslani, S. A comprehensive review on the treatment approaches of multiple sclerosis: Currently and in the future. Inflamm. Res. 2019, 68, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Lana-Peixoto, M.A.; Talim, N. Neuromyelitis optica spectrum disorder and anti-MOG Syndromes. Biomedicines 2019, 7, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamid, S.H.M.; Whittam, D.; Mutch, K.; Linaker, S.; Solomon, T.; Das, K.; Bhojak, M.; Jacob, A. What proportion of AQP4-IgG-negative NMO spectrum disorder patients are MOG-IgG positive? A cross sectional study of 132 patients. J. Neurol. 2017, 264, 2088–2094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reindl, M.; Waters, P. Myelin oligodendrocyte glycoprotein antibodies in neurological disease. Nat. Rev. Neurol. 2019, 15, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Hegen, H.; Reindl, M. Recent developments in MOG-IgG associated neurological disorders. Adv. Neurol. Disord. 2020, 13, 1756286420945135. [Google Scholar]
- Hor, J.Y.; Asgari, N.; Nakashima, I.; Broadley, S.A.; Leite, M.I.; Kissani, N.; Jacob, A.; Marignier, R.; Weinshenker, B.G.; Paul, F.; et al. Epidemiology of neuromyelitis optica spectrum disorder and its prevalence and incidence worldwide. Front. Neurol. 2020, 11, 501. [Google Scholar] [CrossRef]
- Kim, S.H.; Mealy, M.A.; Levy, M.; Schmidt, F.; Ruprecht, K.; Paul, F.; Ringelstein, M.; Aktas, O.; Hartung, H.P.; Asgari, N.; et al. Racial differences in neuromyelitis optica spectrum disorder. Neurology 2018, 91, e2089–e2099. [Google Scholar] [CrossRef] [Green Version]
- Kremer, L.; Mealy, M.; Jacob, A.; Nakashima, I.; Cabre, P.; Bigi, S.; Paul, F.; Jarius, S.; Aktas, O.; Elsone, L.; et al. Brainstem manifestations in neuromyelitis optica: A multicenter study of 258 patients. Mult. Scler. 2014, 20, 843–847. [Google Scholar] [CrossRef]
- Apiwattanakul, M.; Popescu, B.F.; Matiello, M.; Weinshenker, B.G.; Lucchinetti, C.F.; Lennon, V.A.; McKeon, A.; Carpenter, A.F.; Miller, G.M.; Pittock, S.J. Intractable vomiting as the initial presentation of neuromyelitis optica. Ann. Neurol. 2010, 68, 757–761. [Google Scholar] [CrossRef]
- Iorio, R.; Lucchinetti, C.F.; Lennon, V.A.; Farrugia, G.; Pasricha, P.J.; Weinshenker, B.G.; Pittock, S.J. Intractable nausea and vomiting from autoantibodies against a brain water channel. Clin. Gastroenterol. Hepatol. 2013, 11, 240–245. [Google Scholar] [CrossRef] [Green Version]
- Prabhu, M.M.; Agrawal, U. Intractable vomiting and hiccups: An atypical presentation of neuromyelitis optica. Cureus 2019, 11, e6245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Lee, H.-S.; Baek, S.-H. Paroxysmal pruritus as the first relapsing symptom of neuromyelitis optica. Neurol. Asia 2010, 15, 185–187. [Google Scholar]
- Elsone, L.; Townsend, T.; Mutch, K.; Das, K.; Boggild, M.; Nurmikko, T.; Jacob, A. Neuropathic pruritus (itch) in neuromyelitis optica. Mult. Scler. 2013, 19, 475–479. [Google Scholar] [CrossRef] [PubMed]
- Cobo-Calvo, A.; Vukusic, S.; Marignier, R. Clinical spectrum of central nervous system myelin oligodendrocyte glycoprotein autoimmunity in adults. Curr. Opin. Neurol. 2019, 32, 459–466. [Google Scholar] [CrossRef]
- Jarius, S.; Kleiter, I.; Ruprecht, K.; Asgari, N.; Pitarokoili, K.; Borisow, N.; Hummert, M.W.; Trebst, C.; Pache, F.; Winkelmann, A.; et al. MOG-IgG in NMO and related disorders: A multicenter study of 50 patients. Part 3: Brainstem involvement-frequency, presentation and outcome. J. Neuroinflamm. 2016, 13, 281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wingerchuk, D.M.; Banwell, B.; Bennett, J.L.; Cabre, P.; Carroll, W.; Chitnis, T.; de Seze, J.; Fujihara, K.; Greenberg, B.; Jacob, A.; et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology 2015, 85, 177–189. [Google Scholar] [CrossRef]
- Wu, Y.; Zhong, L.; Geng, J. Neuromyelitis optica spectrum disorder: Pathogenesis, treatment, and experimental models. Mult. Scler. Relat. Disord. 2019, 27, 412–418. [Google Scholar] [CrossRef] [PubMed]
- Akaishi, T.; Nakashima, I. Efficiency of antibody therapy in demyelinating diseases. Int. Immunol. 2017, 29, 327–335. [Google Scholar] [CrossRef] [Green Version]
- Ding, J.; Ren, K.; Wu, J.; Li, H.; Sun, T.; Yan, Y.; Guo, J. Overlapping syndrome of MOG-IgG-associated disease and autoimmune GFAP astrocytopathy. J. Neurol. 2020, 267, 2589–2593. [Google Scholar] [CrossRef]
- Kunchok, A.; Zekeridou, A.; McKeon, A. Autoimmune glial fibrillary acidic protein astrocytopathy. Curr. Opin. Neurol. 2019, 32, 452–458. [Google Scholar] [CrossRef]
- Li, X.L.; Han, J.; Zhao, H.T.; Long, Y.M.; Zhang, B.W.; Wang, H.Y. Autoimmune glial fibrillary acidic protein astrocytopathy with lesions distributed predominantly in the entire spinal cord. Ther. Adv. Neurol. Disord. 2020, 13, 1756286420909973. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, S.; Mohr, A.; Knauth, M.; Wildemann, B.; Storch-Hagenlocher, B. Acute disseminated encephalomyelitis: A follow-up study of 40 adult patients. Neurology 2001, 56, 1313–1318. [Google Scholar] [CrossRef] [PubMed]
- Hardy, T.A.; Reddel, S.W.; Barnett, M.H.; Palace, J.; Lucchinetti, C.F.; Weinshenker, B.G. Atypical inflammatory demyelinating syndromes of the CNS. Lancet Neurol. 2016, 15, 967–981. [Google Scholar] [CrossRef]
- Baumann, M.; Sahin, K.; Lechner, C.; Hennes, E.M.; Schanda, K.; Mader, S.; Karenfort, M.; Selch, C.; Hausler, M.; Eisenkolbl, A.; et al. Clinical and neuroradiological differences of paediatric acute disseminating encephalomyelitis with and without antibodies to the myelin oligodendrocyte glycoprotein. J. Neurol. Neurosurg. Psychiatry 2015, 86, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Atlas, S.W.; Grossman, R.I.; Goldberg, H.I.; Hackney, D.B.; Bilaniuk, L.T.; Zimmerman, R.A. MR diagnosis of acute disseminated encephalomyelitis. J. Comput. Assist. Tomogr. 1986, 10, 798–801. [Google Scholar] [CrossRef] [PubMed]
- Alper, G.; Sreedher, G.; Zuccoli, G. Isolated brain stem lesion in children: Is it acute disseminated encephalomyelitis or not? AJNR Am. J. Neuroradiol. 2013, 34, 217–220. [Google Scholar] [CrossRef] [Green Version]
- Atherton, D.S.; Perez, S.R.; Gundacker, N.D.; Franco, R.; Han, X. Acute disseminated encephalomyelitis presenting as a brainstem encephalitis. Clin. Neurol. Neurosurg. 2016, 143, 76–79. [Google Scholar] [CrossRef]
- Shahrizaila, N.; Yuki, N. Bickerstaff brainstem encephalitis and Fisher syndrome: Anti-GQ1b antibody syndrome. J. Neurol. Neurosurg. Psychiatry 2013, 84, 576–583. [Google Scholar] [CrossRef]
- Horton, E.; Krishnamoorthy, S.; Reynolds, L. Bickerstaff’s encephalitis. BMJ Case Rep. 2014, 2014, bcr2014205336. [Google Scholar] [CrossRef]
- Graus, F.; Titulaer, M.J.; Balu, R.; Benseler, S.; Bien, C.G.; Cellucci, T.; Cortese, I.; Dale, C.R.; Gelfand, M.J.; Geschwind, M.; et al. A clinical approach to diagnosis of autoimmune encephalitis. Lancet Neurol. 2016, 15, 391–404. [Google Scholar] [CrossRef] [Green Version]
- Kuwabara, S.; Misawa, S.; Mori, M. Bickerstaff brainstem encephalitis: More common than we think? J. Neurol. Neurosurg. Psychiatry 2013, 84, 1184. [Google Scholar] [CrossRef]
- Wakerley, B.R.; Soon, D.; Chan, Y.C.; Yuki, N. Atypical Bickerstaff brainstem encephalitis: Ataxic hypersomnolence without ophthalmoplegia. J. Neurol. Neurosurg. Psychiatry 2013, 84, 1206–1207. [Google Scholar] [CrossRef]
- Ito, M.; Kuwabara, S.; Odaka, M.; Misawa, S.; Koga, M.; Hirata, K.; Yuki, N. Bickerstaff′s brainstem encephalitis and Fisher syndrome form a continuous spectrum: Clinical analysis of 581 cases. J. Neurol. 2008, 255, 674–682. [Google Scholar] [CrossRef] [PubMed]
- Kurihara, M.; Bannai, T.; Otsuka, J.; Kawabe Matsukawa, M.; Terao, Y.; Shimizu, J.; Tsuji, S. Optic neuropathy and decorticate-like posture as presenting symptoms of Bickerstaff′s brainstem encephalitis: A case report and literature review. Clin. Neurol. Neurosurg. 2018, 173, 159–162. [Google Scholar] [CrossRef]
- Zeiner, P.S.; Brandhofe, A.; Müller-Eschner, M.; Steinmetz, H.; Pfeilschifter, W. Area postrema syndrome as frequent feature of Bickerstaff brainstem encephalitis. Ann. Clin. Transl. Neurol. 2018, 5, 1534–1542. [Google Scholar] [CrossRef]
- Koga, M.; Kusunoki, S.; Kaida, K.; Uehara, R.; Nakamura, Y.; Kohriyama, T.; Kanda, T. Nationwide survey of patients in Japan with Bickerstaff brainstem encephalitis: Epidemiological and clinical characteristics. J. Neurol. Neurosurg. Psychiatry 2012, 83, 1210–1215. [Google Scholar] [CrossRef]
- Yoshikawa, K.; Kuwahara, M.; Morikawa, M.; Kusunoki, S. Bickerstaff brainstem encephalitis with or without anti-GQ1b antibody. Neurol. Neuroimmunol. Neuroinflamm. 2020, 7, e889. [Google Scholar] [CrossRef] [PubMed]
- Dudesek, A.; Rimmele, F.; Tesar, S.; Kolbaske, S.; Rommer, P.S.; Benecke, R.; Zettl, U.K. CLIPPERS: Chronic lymphocytic inflammation with pontine perivascular enhancement responsive to steroids. Review of an increasingly recognized entity within the spectrum of inflammatory central nervous system disorders. Clin. Exp. Immunol. 2014, 175, 385–396. [Google Scholar] [CrossRef] [PubMed]
- Pittock, S.J.; Debruyne, J.; Krecke, K.N.; Giannini, C.; van den Ameele, J.; De Herdt, V.; McKeon, A.; Fealey, R.D.; Weinshenker, B.G.; Aksamit, A.J.; et al. Chronic lymphocytic inflammation with pontine perivascular enhancement responsive to steroids (CLIPPERS). Brain 2010, 133, 2626–2634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zalewski, N.L.; Tobin, W.O. CLIPPERS. Curr. Neurol. Neurosci. Rep. 2017, 17, 65. [Google Scholar] [CrossRef]
- Taieb, G.; Mulero, P.; Psimaras, D.; van Oosten, B.W.; Seebach, J.D.; Marignier, R.; Pico, F.; Rigau, V.; Ueno, Y.; Duflos, C.; et al. CLIPPERS and its mimics: Evaluation of new criteria for the diagnosis of CLIPPERS. J. Neurol. Neurosurg. Psychiatry 2019, 90, 1027–1038. [Google Scholar] [CrossRef]
- Tobin, W.O.; Guo, Y.; Krecke, K.N.; Parisi, J.E.; Lucchinetti, C.F.; Pittock, S.J.; Mandrekar, J.; Dubey, D.; Debruyne, J.; Keegan, B.M. Diagnostic criteria for chronic lymphocytic inflammation with pontine perivascular enhancement responsive to steroids (CLIPPERS). Brain 2017, 140, 2415–2425. [Google Scholar] [CrossRef] [Green Version]
- Taieb, G.; Allou, T.; Labauge, P. Therapeutic Approaches in CLIPPERS. Curr. Treat. Options Neurol. 2017, 19, 17. [Google Scholar] [CrossRef]
- Rempe, T.; Becktepe, J.S.; Metz, I.; Brück, W.; Stürner, K.H.; Deuschl, G.; Berg, D.; Baron, R.; Zeuner, R.; Leypoldt, F. A case of CLIPPERS syndrome responsive to tocilizumab. Neurol. Neuroimmunol. Neuroinflamm. 2019, 6, e545. [Google Scholar] [CrossRef] [Green Version]
- De Graaff, H.J.; Wattjes, M.P.; Rozemuller-Kwakkel, A.J.; Petzold, A.; Killestein, J. Fatal B-cell lymphoma following chronic lymphocytic inflammation with pontine perivascular enhancement responsive to steroids. JAMA Neurol. 2013, 70, 915–918. [Google Scholar] [CrossRef] [Green Version]
- Akman-Demir, G.; Serdaroglu, P.; Tasçi, B. Clinical patterns of neurological involvement in Behçet’s disease: Evaluation of 200 patients. The Neuro-Behçet study group. Brain 1999, 122, 2171–2182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moragas, M.; Martínez-Yélamos, S.; Majós, C.; Fernández-Viladrich, P.; Rubio, F.; Arbizu, T. Rhombencephalitis: A series of 97 patients. Medicine 2011, 90, 256–261. [Google Scholar] [CrossRef]
- Kidd, D.; Steuer, A.; Denman, A.M.; Rudge, P. Neurological complications in Behçet’s syndrome. Brain 1999, 122, 2183–2194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujimori, J.; Takahashi, T.; Matsumoto, Y.; Fujihara, K.; Takai, Y.; Misu, T.; Nakashima, I. Two Japanese cases of anti-MOG antibody-associated encephalitis that mimicked neuro-Behçet’s disease. J. Neuroimmunol. 2019, 334, 577002. [Google Scholar] [CrossRef] [PubMed]
- Hajj-Ali, R.A.; Calabrese, L.H. Central nervous system vasculitis. Curr. Opin. Rheumatol. 2009, 21, 10–18. [Google Scholar] [CrossRef] [Green Version]
- Matsui, Y.; Takenouchi, T.; Narabayashi, A.; Ohara, K.; Nakahara, T.; Takahashi, T. Childhood Sjögren syndrome presenting as acute brainstem encephalitis. Brain Dev. 2016, 38, 158–162. [Google Scholar] [CrossRef]
- Chen, J.; Wang, L.; He, L.; Yi, X.; Yan, Z. Teaching NeuroImages: Primary Sjögren syndrome presenting as isolated lesion of medulla oblongata. Neurology 2015, 84, e5–e6. [Google Scholar] [CrossRef] [Green Version]
- Delalande, S.; de Seze, J.; Fauchais, A.L.; Hachulla, E.; Stojkovic, T.; Ferriby, D.; Dubucquoi, S.; Pruvo, J.P.; Vermersch, P.; Hatron, P.Y. Neurologic manifestations in primary Sjögren syndrome: A study of 82 patients. Medicine 2004, 83, 280–291. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Sharma, N.; Sharma, A.; Mahi, S.; Bhalla, A.; Varma, S. A case of systemic lupus erythematosus with extensive brain stem involvement. Clin. Rheumatol. 2009, 28 (Suppl. S1), S69–S71. [Google Scholar] [CrossRef] [PubMed]
- Delèvaux, I.; André, M.; Marroun, I.; Lamaison, D.; Piette, J.C.; Aumaître, O. Intractable hiccup as the initial presenting feature of systemic lupus erythematosus. Lupus 2005, 14, 406–408. [Google Scholar] [CrossRef]
- Shahmohammadi, S.; Doosti, R.; Shahmohammadi, A.; Mohammadianinejad, S.E.; Sahraian, M.A.; Azimi, A.R.; Harirchian, M.H.; Asgari, N.; Naser Moghadasi, A. Autoimmune diseases associated with neuromyelitis optica spectrum disorders: A literature review. Mult. Scler. Relat. Disord. 2019, 27, 350–363. [Google Scholar] [CrossRef]
- Zhong, Y.H.; Zhong, Z.G.; Zhou, Z.; Ma, Z.Y.; Qiu, M.Y.; Peng, F.H.; Zhang, W.X. Comparisons of presentations and outcomes of neuromyelitis optica patients with and without Sjögren’s Syndrome. Neurol. Sci. 2017, 38, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Marino, A.; Narula, S.; Lerman, M.A. First pediatric patient with neuromyelitis optica and Sjögren syndrome successfully treated with tocilizumab. Pediatr. Neurol. 2017, 73, e5–e6. [Google Scholar] [CrossRef]
- Dubey, D.; Wilson, M.R.; Clarkson, B.; Giannini, C.; Gandhi, M.; Cheville, J.; Lennon, V.A.; Eggers, S.; Devine, M.F.; Mandel-Brehm, C.; et al. Expanded Clinical Phenotype, Oncological Associations, and Immunopathologic Insights of Paraneoplastic Kelch-like Protein-11 Encephalitis. JAMA Neurol. 2020, 77, 1420–1429. [Google Scholar] [CrossRef]
- Dubey, D.; Kryzer, T.; Guo, Y.; Clarkson, B.; Cheville, J.C.; Costello, B.A.; Leibovich, B.C.; Algeciras-Schimnich, A.; Lucchinnetti, C.; Hammami, M.B.; et al. Leucine Zipper 4 Autoantibody: A Novel Germ Cell Tumor and Paraneoplastic Biomarker. Ann Neurol. 2021, 89, 1001–1010. [Google Scholar] [CrossRef]
- Mandel-Brehm, C.; Dubey, D.; Kryzer, T.J.; O′Donovan, B.D.; Tran, B.; Vazquez, S.E.; Sample, H.A.; Zorn, K.C.; Khan, L.M.; Bledsoe, I.O.; et al. Kelch-like Protein 11 Antibodies in Seminoma-Associated Paraneoplastic Encephalitis. N. Engl. J. Med. 2019, 381, 47–54. [Google Scholar] [CrossRef]
- Graus, F.; Vogrig, A.; Muñiz-Castrillo, S.; Antoine, J.G.; Desestret, V.; Dubey, D.; Giometto, B.; Irani, S.R.; Joubert, B.; Leypoldt, F.; et al. Updated Diagnostic Criteria for Paraneoplastic Neurologic Syndromes. Neurol. Neuroimmunol Neuroinflamm. 2021, 8, e1014. [Google Scholar] [CrossRef] [PubMed]
- Dalmau, J.; Rosenfeld, M.R. Paraneoplastic syndromes of the CNS. Lancet Neurol. 2008, 7, 327–340. [Google Scholar] [CrossRef] [Green Version]
- Saiz, A.; Bruna, J.; Stourac, P.; Vigliani, M.C.; Giometto, B.; Grisold, W.; Honnorat, J.; Psimaras, D.; Voltz, R.; Graus, F. Anti-Hu-associated brainstem encephalitis. J. Neurol. Neurosurg. Psychiatry 2009, 80, 404–407. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.S.; Higgins, M.J.; Patel, B.M.; Larson, J.S.; Rady, M.Y. Paraneoplastic coma and acquired central alveolar hypoventilation as a manifestation of brainstem encephalitis in a patient with ANNA-1 antibody and small-cell lung cancer. Neurocrit. Care 2006, 4, 137–139. [Google Scholar] [CrossRef]
- Dalmau, J.; Graus, F.; Villarejo, A.; Posner, J.B.; Blumenthal, D.; Thiessen, B.; Saiz, A.; Meneses, P.; Rosenfeld, M.R. Clinical analysis of anti-Ma2-associated encephalitis. Brain 2004, 127, 1831–1844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pittock, S.J.; Lucchinetti, C.F.; Lennon, V.A. Anti-neuronal nuclear autoantibody type 2: Paraneoplastic accompaniments. Ann. Neurol. 2003, 53, 580–587. [Google Scholar] [CrossRef] [PubMed]
- Sutton, I.J.; Barnett, M.H.; Watson, J.D.; Ell, J.J.; Dalmau, J. Paraneoplastic brainstem encephalitis and anti-Ri antibodies. J. Neurol. 2002, 249, 1597–1598. [Google Scholar] [CrossRef]
- Lambeck, J.; Hieber, M.; Dreßing, A.; Niesen, W.D. Central pontine myelinosis and osmotic demyelination syndrome. Dtsch. Ärzteblatt Int. 2019, 116, 600–606. [Google Scholar]
- Biousse, V.; Newman, N.J.; Chang, G.Y. Brainstem involvement in hypertensive encephalopathy: Clinical and radiological findings. Neurology 2004, 63, 1759–1760. [Google Scholar] [CrossRef] [PubMed]
- Vasan, S.; Kumar, A. Wernicke encephalopathy. In Statpearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zoghaib, R.; Sreij, A.; Maalouf, N.; Freiha, J.; Kikano, R.; Riachi, N.; Chalah, M.A.; Ayache, S.S.; Ahdab, R. Autoimmune Brainstem Encephalitis: An Illustrative Case and a Review of the Literature. J. Clin. Med. 2021, 10, 2970. https://doi.org/10.3390/jcm10132970
Zoghaib R, Sreij A, Maalouf N, Freiha J, Kikano R, Riachi N, Chalah MA, Ayache SS, Ahdab R. Autoimmune Brainstem Encephalitis: An Illustrative Case and a Review of the Literature. Journal of Clinical Medicine. 2021; 10(13):2970. https://doi.org/10.3390/jcm10132970
Chicago/Turabian StyleZoghaib, Romy, Ali Sreij, Nancy Maalouf, Joumana Freiha, Raghid Kikano, Naji Riachi, Moussa A. Chalah, Samar S. Ayache, and Rechdi Ahdab. 2021. "Autoimmune Brainstem Encephalitis: An Illustrative Case and a Review of the Literature" Journal of Clinical Medicine 10, no. 13: 2970. https://doi.org/10.3390/jcm10132970
APA StyleZoghaib, R., Sreij, A., Maalouf, N., Freiha, J., Kikano, R., Riachi, N., Chalah, M. A., Ayache, S. S., & Ahdab, R. (2021). Autoimmune Brainstem Encephalitis: An Illustrative Case and a Review of the Literature. Journal of Clinical Medicine, 10(13), 2970. https://doi.org/10.3390/jcm10132970