
The Outcomes of Cochlear Implantation in Usher Syndrome: A Systematic Review

 , , ,
, , , 

Abstract
1. Introduction
1.1. Clinical Presentation
1.1.1. USH1
1.1.2. USH2
1.1.3. USH3
1.2. Etiology and Epidemiology
1.3. Diagnosis
1.4. Current Use of Cochlear Implantation
1.5. Current Management
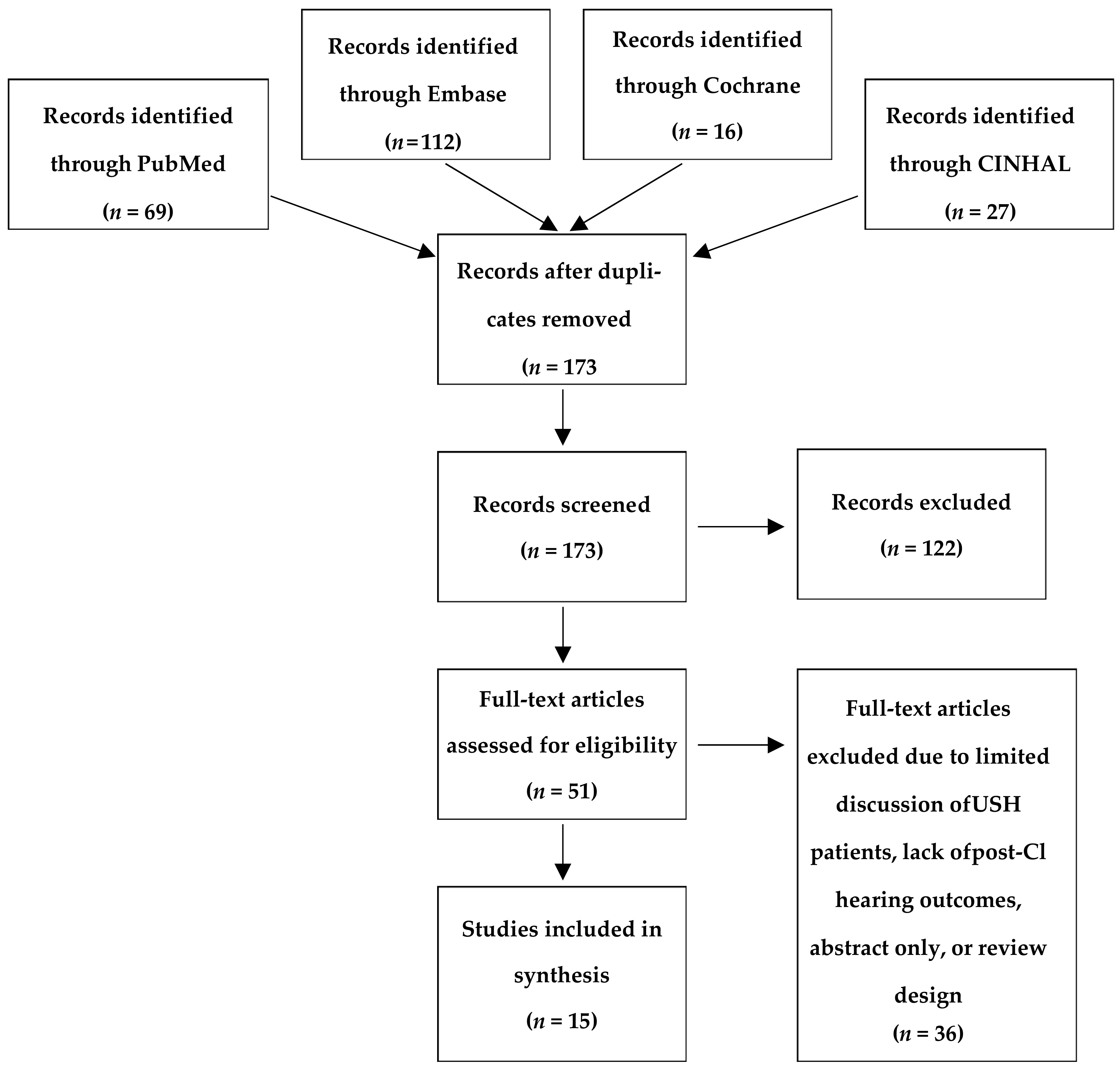
2. Materials and Methods
2.1. Search Strategy
2.2. Eligibility of Studies
3. Results
3.1. Multi-Patient Studies

{kind=link}
| References | USH Subtype | Pre-CI Hearing Measurement | Average Age at CI (Years) | Average Duration of Implant Use at Time of Audiological Evaluation (Years) | Post-CI Hearing Measurement | Other Findings |
|---|---|---|---|---|---|---|
| Alzhrani et al. [36], 2018 | Not Mentioned (n = 9) | Not mentioned | 5.3 (3–7.6) | 3 | Measurement 3 years post CI. All scored the maximum of five on the SIR, and most of them had a CAP score of 9. Only one subject failed to exceed a CAP score of 5, although he haEHLd a very high SIR score of 5 Average PTA = 23.67 | |
| Broomfield et al. [37], 2013 | Not Mentioned [37], (congentially deaf from birth, likely USH1) (n = 9) | [37] Not Mentioned | Early = 2.7 Late = 12.73 | 10.3 (4.8–11.5) | Early: SRS = 5.5 Late: SRS = 4 | Outcomes are often excellent but can be variable even within the same syndrome groups |
| Damen et al. [34], 2016 | USH1 (n = 14) | >110 dB at 0.5, 1, and 2 kHz | Children = 12.4 ± 2.9 Adults = 30.7 ± 6.8 | 9.2 (3.0–15.7) | CI in USH1 children resulted in EHL of 84.4 dB HL | |
| Hartel et al. [32], 2017 | USH2 (n = 8) | Average phoneme score = 41% | 59 | 4.4 (1–19) | Average phoneme score = 87% | Postoperative quality of life and speech production improvements is greater in those with post-lingual deafness than those with pre-lingual deafness (USH1), as determined by NCIQ. |
| Hoshino et al. [39], 2017 | USH1 (n = 10) | Average PTA HA = 103 dB HL | 18.9 (5–49) | 11.4 (1–27) | Average PTA CI = 35 dB | 3 patients improved sentence recognition and 5 patients were able to improve in speech detection. Late implantation limits speech perception |
| Jatana et al. [1], 2013 | USH1 (n = 3) | Not mentioned. | Mean age of CI for patients born before 1992 * = 4.3 (3.3–7.1) | 7.8 (1–15.6) | 92.3% were able to achieve some level of open-set speech perception on age-appropriate testing | All but 2 children in the current series were able to develop some open-set speech discrimination, and 69.2% were using oral or primarily oral communication by time of last follow-up. |
| USH unspecified (n = 23) | Mean age of CI for patients born after 1992 * = 1 (0.5–11.6) | |||||
| Liu et al. [7], 2008 | USH1 (n = 9) | Hearing threshold = 110 dB | 5.4 years (2–11) | 1.5 (1–2) | Hearing threshold at 0.5 kHZ = 46 | All patients showed post-implantation improvements. |
| Loundon et al. [40], 2003 | USH1 (n = 11) | Closed set perception scores = 0% | 9.3 (1.5–44) | 4.4 (0.8–9) | Closed set perception score = 84% (mean) | Although all patients perceived no closed or open set words prior to implantation, children implanted before 9 years of age had the best perceptive results. |
| USH3 (n = 3) | ||||||
| Unspecified (n = 1) | Open set perception scores = 0% | |||||
| Pennings et al. [31], 2006 | USH1 (n = 14) | Not mentioned. | 10 (3.5–30.4) | 11.4 (2–28) | Significant reduction in EHL in 5 of 7 patients implanted before age 10, with a mean EHL of 84 dB HL. The mean EHL increased for those implanted at later ages. | Cochlear implantation within the first two decades of life results in improvement in 93% of patients, all of whom had profound hearing loss at baseline. |
| Pietola et al. [35], 2012 | USH3 (n = 19) | Hearing threshold = 110 ± 8 dB HL (PTA = 0.5–4 kHz) and Hearing aid threshold = 58 ± 11 dB HL | 41 ± 17 | 1 (0.5–1.5) | Hearing threshold = 34 ± 9 dB HL | All patients used hearing aids preoperatively, and all benefited from CI as evidenced by improvements in PTA post-CI and the Glasglow Benefit Hearing and speech discrimination, age at implantation and the change in the hearing ability varied significantly after implantation. |
3.2. Case Reports
4. Discussion
Limitations
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jatana, K.R.; Thomas, D.; Weber, L.; Mets, M.B.; Silverman, J.B.; Young, N.M. Usher syndrome: Characteristics and outcomes of pediatric cochlear implant recipients. Otol. Neurotol. 2013, 34, 484–489. [Google Scholar] [CrossRef] [PubMed]
- Mathur, P.D.; Yang, J. Usher syndrome and non-syndromic deafness: Functions of different whirlin isoforms in the cochlea, vestibular organs, and retina. Hear. Res. 2019, 375, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Moller, C.G.; Kimberling, W.J.; Davenport, S.L.; Priluck, I.; White, V.; Biscone-Halterman, K.; Odkvist, L.M.; Brookhouser, P.E.; Lund, G.; Grissom, T.J. Usher syndrome: An otoneurologic study. Laryngoscope 1989, 99, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.J.; Berlin, C.I.; Hejtmancik, J.F.; Keats, B.J.; Kimberling, W.J.; Lewis, R.A.; Moller, C.G.; Pelias, M.Z.; Tranebjaerg, L. Clinical diagnosis of the Usher syndromes. Usher Syndrome Consortium. Am. J. Med. Genet. 1994, 50, 32–38. [Google Scholar] [CrossRef]
- Yan, D.; Liu, X.Z. Genetics and pathological mechanisms of Usher syndrome. J. Hum. Genet. 2010, 55, 327–335. [Google Scholar] [CrossRef]
- Kumar, A.; Fishman, G.; Torok, N. Vestibular and auditory function in Usher’s syndrome. Ann. Otol. Rhinol. Laryngol. 1984, 93, 600–608. [Google Scholar] [CrossRef]
- Liu, X.Z.; Angeli, S.I.; Rajput, K.; Yan, D.; Hodges, A.V.; Eshraghi, A.; Telischi, F.F.; Balkany, T.J. Cochlear implantation in individuals with Usher type 1 syndrome. Int. J. Pediatr. Otorhinolaryngol. 2008, 72, 841–847. [Google Scholar] [CrossRef]
- Pakarinen, L.; Karjalainen, S.; Simola, K.O.; Laippala, P.; Kaitalo, H. Usher’s syndrome type 3 in Finland. Laryngoscope 1995, 105, 613–617. [Google Scholar] [CrossRef]
- Cejas, I.; Hoffman, M.F.; Quittner, A.L. Outcomes and benefits of pediatric cochlear implantation in children with additional disabilities: A review and report of family influences on outcomes. Pediatr. Health Med. Ther. 2015, 6, 45–63. [Google Scholar] [CrossRef]
- Plantinga, R.F.; Kleemola, L.; Huygen, P.L.; Joensuu, T.; Sankila, E.M.; Pennings, R.J.; Cremers, C.W. Serial audiometry and speech recognition findings in Finnish Usher syndrome type III patients. Audiol. Neurotol. 2005, 10, 79–89. [Google Scholar] [CrossRef]
- Adato, A.; Vreugde, S.; Joensuu, T.; Avidan, N.; Hamalainen, R.; Belenkiy, O.; Olender, T.; Bonne-Tamir, B.; Ben-Asher, E.; Espinos, C.; et al. USH3A transcripts encode clarin-1, a four-transmembrane-domain protein with a possible role in sensory synapses. Eur. J. Hum. Genet. 2002, 10, 339–350. [Google Scholar] [CrossRef]
- Petit, C. Usher syndrome: From genetics to pathogenesis. Annu. Rev. Genom. Hum. Genet. 2001, 2, 271–297. [Google Scholar] [CrossRef]
- Mathur, P.; Yang, J. Usher syndrome: Hearing loss, retinal degeneration and associated abnormalities. Biochim. Biophys. Acta 2015, 1852, 406–420. [Google Scholar] [CrossRef]
- Koffler, T.; Ushakov, K.; Avraham, K.B. Genetics of Hearing Loss: Syndromic. Otolaryngol. Clin. North Am. 2015, 48, 1041–1061. [Google Scholar] [CrossRef]
- Toms, M.; Pagarkar, W.; Moosajee, M. Usher syndrome: Clinical features, molecular genetics and advancing therapeutics. Ther. Adv. Ophthalmol. 2020, 12, 2515841420952194. [Google Scholar] [CrossRef]
- Reference, G.H. Usher Syndrome. Genetics Home Reference: Your Guide to Understanding Genetic Conditions. Available online: https://ghr.nlm.nih.gov/condition/usher-syndrome#statistics (accessed on 10 November 2019).
- Seeliger, M.W.; Fischer, M.D.; Pfister, M. Usher Syndrome: Clinical Features, Diagnostic Options, and Therapeutic Prospects. Ophthalmologe 2009, 106, 505–511. [Google Scholar] [CrossRef]
- Dedhia, K.; Graham, E.; Park, A. Hearing Loss and Failed Newborn Hearing Screen. Clin. Perinatol. 2018, 45, 629–643. [Google Scholar] [CrossRef]
- Konrádsson, K.; Magnusson, M.; Andréasson, S. Perform vestibular test among all small deaf children! Early detection of Usher syndrome improves the possibilities of communication in the event of later deaf-blindness. Lakartidningen 1998, 95, 379–381. [Google Scholar]
- Abeshi, A.; Bruson, A.; Beccari, T.; Dundar, M.; Colombo, L.; Bertelli, M. Genetic testing for Usher syndrome. EuroBiotech J. 2017, 1, 108–110. [Google Scholar] [CrossRef]
- Wagenaar, M.; van Aarem, A.; Huygen, P.; Pieke-Dahl, S.; Kimberling, W.; Cremers, C. Hearing impairment related to age in Usher syndrome types 1B and 2A. Arch. Otolaryngol. Head Neck Surg. 1999, 125, 441–445. [Google Scholar] [CrossRef][Green Version]
- Khalaileh, A.; Abu-Diab, A.; Ben-Yosef, T.; Raas-Rothschild, A.; Lerer, I.; Alswaiti, Y.; Chowers, I.; Banin, E.; Sharon, D.; Khateb, S. The Genetics of Usher Syndrome in the Israeli and Palestinian Populations. Investig. Ophthalmol. Vis. Sci. 2018, 59, 1095–1104. [Google Scholar] [CrossRef]
- Ivanova, M.E.; Trubilin, V.N.; Atarshchikov, D.S.; Demchinsky, A.M.; Strelnikov, V.V.; Tanas, A.S.; Orlova, O.M.; Machalov, A.S.; Overchenko, K.V.; Markova, T.V.; et al. Genetic screening of Russian Usher syndrome patients toward selection for gene therapy. Ophthalmic Genet. 2018, 39, 706–713. [Google Scholar] [CrossRef]
- Sun, T.; Xu, K.; Ren, Y.; Xie, Y.; Zhang, X.; Tian, L.; Li, Y. Comprehensive Molecular Screening in Chinese Usher Syndrome Patients. Investig. Ophthalmol. Vis. Sci. 2018, 59, 1229–1237. [Google Scholar] [CrossRef]
- Eppsteiner, R.W.; Shearer, A.E.; Hildebrand, M.S.; Deluca, A.P.; Ji, H.; Dunn, C.C.; Black-Ziegelbein, E.A.; Casavant, T.L.; Braun, T.A.; Scheetz, T.E.; et al. Prediction of cochlear implant performance by genetic mutation: The spiral ganglion hypothesis. Hear. Res. 2012, 292, 51–58. [Google Scholar] [CrossRef]
- Cheng, A.K.; Rubin, H.R.; Powe, N.R.; Mellon, N.K.; Francis, H.W.; Niparko, J.K. Cost-utility analysis of the cochlear implant in children. JAMA 2000, 284, 850–856. [Google Scholar] [CrossRef]
- D’Alessandro, H.D.; Sennaroglu, G.; Yucel, E.; Belgin, E.; Mancini, P. Binaural squelch and head shadow effects in children with unilateral cochlear implants and contralateral hearing aids. Acta Otorhinolaryngol. Ital. 2015, 35, 343–349. [Google Scholar] [CrossRef]
- Ching, T.Y.C.; Dillon, H.; Button, L.; Seeto, M.; Van Buynder, P.; Marnane, V.; Cupples, L.; Leigh, G. Age at Intervention for Permanent Hearing Loss and 5-Year Language Outcomes. Pediatrics 2017, 140, e20164274. [Google Scholar] [CrossRef]
- Ching, T.Y.C.; Dillon, H.; Leigh, G.; Cupples, L. Learning from the Longitudinal Outcomes of Children with Hearing Impairment (LOCHI) study: Summary of 5-year findings and implications. Int. J. Audiol. 2018, 57, S105–S111. [Google Scholar] [CrossRef]
- Lentz, J.; Keats, B.J. Usher Syndrome Type I. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Pennings, R.J.; Damen, G.W.; Snik, A.F.; Hoefsloot, L.; Cremers, C.W.; Mylanus, E.A. Audiologic performance and benefit of cochlear implantation in Usher syndrome type I. Laryngoscope 2006, 116, 717–722. [Google Scholar] [CrossRef]
- Hartel, B.P.; van Nierop, J.W.I.; Huinck, W.J.; Rotteveel, L.J.C.; Mylanus, E.A.M.; Snik, A.F.; Kunst, H.P.M.; Pennings, R.J.E. Cochlear Implantation in Patients with Usher Syndrome Type IIa Increases Performance and Quality of Life. Otol. Neurotol. 2017, 38, e120–e127. [Google Scholar] [CrossRef]
- Alsanosi, A.A. Simultaneous bilateral cochlear implantation in a five-month-old child with Usher syndrome. J. Laryngol. Otol. 2015, 129, 919–922. [Google Scholar] [CrossRef] [PubMed]
- Damen, G.W.; Pennings, R.J.; Snik, A.F.; Mylanus, E.A. Quality of life and cochlear implantation in Usher syndrome type I. Laryngoscope 2006, 116, 723–728. [Google Scholar] [CrossRef] [PubMed]
- Pietola, L.; Aarnisalo, A.A.; Abdel-Rahman, A.; Vastinsalo, H.; Isosomppi, J.; Lopponen, H.; Kentala, E.; Johansson, R.; Valtonen, H.; Vasama, J.-P.; et al. Speech recognition and communication outcomes with cochlear implantation in Usher syndrome type 3. Otol. Neurotol. 2012, 33, 38–41. [Google Scholar] [CrossRef] [PubMed]
- Alzhrani, F.; Alhussini, R.; Hudeib, R.; Alkaff, T.; Islam, T.; Alsanosi, A. The outcome of cochlear implantation among children with genetic syndromes. Eur. Arch. Otorhinolaryngol. 2018, 275, 365–369. [Google Scholar] [CrossRef]
- Broomfield, S.J.; Bruce, I.A.; Henderson, L.; Ramsden, R.T.; Green, K.M. Cochlear implantation in children with syndromic deafness. Int. J. Pediatr. Otorhinolaryngol. 2013, 77, 1312–1316. [Google Scholar] [CrossRef]
- Henricson, C.; Wass, M.; Lidestam, B.; Moller, C.; Lyxell, B. Cognitive skills in children with Usher syndrome type 1 and cochlear implants. Int. J. Pediatr. Otorhinolaryngol. 2012, 76, 1449–1457. [Google Scholar] [CrossRef]
- Hoshino, A.C.; Echegoyen, A.; Goffi-Gomez, M.V.; Tsuji, R.K.; Bento, R.F. Outcomes of Late Implantation in Usher Syndrome Patients. Int. Arch. Otorhinolaryngol. 2017, 21, 140–143. [Google Scholar] [CrossRef]
- Loundon, N.; Marlin, S.; Busquet, D.; Denoyelle, F.; Roger, G.; Renaud, F.; Garabedian, E.N. Usher syndrome and cochlear implantation. Otol. Neurotol. 2003, 24, 216–221. [Google Scholar] [CrossRef]
- Ruiz, A.P.; Gomez, J.M.G. Labyrinthitis ossificans in a cochlear implant patient with Usher syndrome. Otol. Neurotol. 2013, 34, e10–e11. [Google Scholar] [CrossRef]
- Derinsu, U.; Ciprut, A. Cochlear ımplantatıon ın a patıent wıth usher’s syndrome. Marmara Med. J. 2002, 15, 258–261. [Google Scholar]
- Shiomi, Y.; Naito, Y.; Hirano, S.; Fujiki, N.; Honjo, I. Cortical activity of a patient with usher’s syndrome using a cochlear implant. Am. J. Otolaryngol. 1997, 18, 412–414. [Google Scholar] [CrossRef]
- Eshraghi, A.A.; Nazarian, R.; Telischi, F.F.; Rajguru, S.M.; Truy, E.; Gupta, C. The cochlear implant: Historical aspects and future prospects. Anat. Rec. 2012, 295, 1967–1980. [Google Scholar] [CrossRef]
- Geers, A.E.; Nicholas, J.G. Enduring advantages of early cochlear implantation for spoken language development. J. Speech Lang. Hear. Res. 2013, 56, 643–655. [Google Scholar] [CrossRef]
- Connor, C.M.; Craig, H.K.; Raudenbush, S.W.; Heavner, K.; Zwolan, T.A. The age at which young deaf children receive cochlear implants and their vocabulary and speech-production growth: Is there an added value for early implantation? Ear Hear. 2006, 27, 628–644. [Google Scholar] [CrossRef]
- Hayes, H.; Geers, A.E.; Treiman, R.; Moog, J.S. Receptive vocabulary development in deaf children with cochlear implants: Achievement in an intensive auditory-oral educational setting. Ear Hear. 2009, 30, 128–135. [Google Scholar] [CrossRef]
- Sharma, A.; Dorman, M.F.; Spahr, A.J. Rapid development of cortical auditory evoked potentials after early cochlear implantation. Neuroreport 2002, 13, 1365–1368. [Google Scholar] [CrossRef]
- Well, D.; Blanchard, S.; Kaplan, J.; Guilford, P.; Gibson, F.; Walsh, J.; Mburu, P.; Varela, A.; Levilliers, J.; Weston, M.D.; et al. Defective myosin VIIA gene responsible for Usher syndrome type 1B. Nature 1995, 374, 60–61. [Google Scholar] [CrossRef]
- Jacobs, E.; Langereis, M.C.; Frijns, J.H.; Free, R.H.; Goedegebure, A.; Smits, C.; Stokroos, R.J.; Ariens-Meijer, S.A.; Mylanus, E.A.; Vermeulen, A.M. Benefits of simultaneous bilateral cochlear implantation on verbal reasoning skills in prelingually deaf children. Res. Dev. Disabil. 2016, 58, 104–113. [Google Scholar] [CrossRef]
- Ramsden, J.D.; Papaioannou, V.; Gordon, K.A.; James, A.L.; Papsin, B.C. Parental and program’s decision making in paediatric simultaneous bilateral cochlear implantation: Who says no and why? Int. J. Pediatr. Otorhinolaryngol. 2009, 73, 1325–1328. [Google Scholar] [CrossRef]
- Gordon, K.A.; Valero, J.; Papsin, B.C. Binaural processing in children using bilateral cochlear implants. Neuroreport 2007, 18, 613–617. [Google Scholar] [CrossRef]
- Wahlqvist, M.; Möller, C.; Möller, K.; Danermark, B. Implications of Deafblindness: The Physical and Mental Health and Social Trust of Persons with Usher Syndrome Type 3. J. Vis. Impair. Blind. 2018, 110, 245–256. [Google Scholar] [CrossRef]
- Friesen, L.; Shannon, R.V.; Baskent, D.; Wang, X. Speech Recognition in Noise as a Function of the Number of Spectral Channels: Comparison of Acoustic Hearing and Cochlear Implants. J. Acoust. Soc. Am. 2001, 110, 1150–1163. [Google Scholar] [CrossRef]
- Shah, V.; Mittal, R.; Shahal, D.; Sinha, P.; Bulut, E.; Mittal, J.; Eshraghi, A.A. Evaluating the Efficacy of Taurodeoxycholic Acid in Providing Otoprotection Using an in vitro Model of Electrode Insertion Trauma. Front. Mol. Neurosci. 2020, 13, 113. [Google Scholar] [CrossRef]
- Eshraghi, A.A.; Shahal, D.; Davies, C.; Mittal, J.; Shah, V.; Bulut, E.; Garnham, C.; Sinha, P.; Mishra, D.; Marwede, H.; et al. Evaluating the Efficacy of L-N-acetylcysteine and Dexamethasone in Combination to Provide Otoprotection for Electrode Insertion Trauma. J. Clin. Med. 2020, 9, 716. [Google Scholar] [CrossRef]
- Eshraghi, A.A.; Roell, J.; Shaikh, N.; Telischi, F.F.; Bauer, B.; Guardiola, M.; Bas, E.; Van De Water, T.; Rivera, I.; Mittal, J. A novel combination of drug therapy to protect residual hearing post cochlear implant surgery. Acta Otolaryngol. 2016, 136, 420–424. [Google Scholar] [CrossRef]

| Clinical Subtype | Genetic Subtype | Gene-Function | Hearing Loss | Vestibular Function | Ocular Manifestations |
|---|---|---|---|---|---|
| Usher type 1 | USH1A | Withdrawn | Profound congenital HL | Abnormal or absent vestibular sense | Early onset RP |
| USH1B | MYO7A—Motor protein | ||||
| USH1C | USH1C—Scaffold protein | ||||
| USH1D | CDH23—Cell adhesion protein | ||||
| USH1E | Unknown | ||||
| USH1F | PCDH15—Cell to cell adhesion protein | ||||
| USH1G | USH1G—Scaffold Protein with Ankyrin repeat and SAM Domain | ||||
| USH1H | Unknown | ||||
| Usher type 2 | USH2A | Usherin—Transmembrane protein | Prelingual onset of moderate to severe high-frequency sloping HL | Normal | Onset of RP in 2nd decade of life |
| USH2B | ADGRV1—Transmembrane receptor protein | ||||
| USH2C | VLGR1—Transmembrane receptor protein | ||||
| USH2D | Whirlin—Scaffold Protein | ||||
| Usher type 3 | USH3A | USH3A—Clarin 1 Transmembrane Protein | Variable onset of progressive HL | Variable | Variable onset |
| Database | Search |
|---|---|
| PubMed | (“Usher Syndromes”[Mesh] or usher[tw] or ushers[tw] or usher’ [tw] or Graefe-Usher[tw] or Hallgren[tw] or (Retinitis Pigmentosa Deafness Syndrome[tw]) or ((“Retinitis Pigmentosa”[Mesh:NoExp] or Retinitis Pigmentosa[tw]) AND (“Deafness”[Mesh:NoExp] or deaf*[tw])) or USH1B or USH1C or USH1D or USH1E or USH1F or USH1G or USH1H or USH2A or USH2C) AND (“Cochlear Implants”[Mesh] Or (“Cochlea”[Mesh] or “Ear, Inner”[Mesh] or auditory[tw] or cochlea[tw] or cochlear[tw] or cochleas[tw] or intracochlear[tw] or intra-cochlear[tw] or inner ear[tw] or hearing[tw]) AND (“Prostheses and Implants”[Mesh] or Prosthes*[tw] or prosthetic[tw] or Implant[tw] or Impants[tw] or Implantation[tw] or Implanted[tw] or Device[tw] or Devices[tw] or artificial[tw]) |
| Embase | (‘cochlea prosthesis’/exp OR ‘cochlea prosthesis’:ti,ab OR ‘nucleus hybrid l24’:tn,ti,ab OR ‘artificial cochlea implant’:tn,ti,ab OR ‘auditory prostheses’:tn,ti,ab OR ‘cochlea implant’:tn,ti,ab OR ‘cochlea prosthesis’:tn,ti,ab OR ‘cochlear implant’:tn,ti,ab OR ‘cochlear implants’:tn,ti,ab OR ‘cochlear prostheses’:tn,ti,ab OR ‘cochlear prosthesis’:tn,ti,ab OR ‘hearing prosthesis’:tn,ti,ab OR ‘prosthesis, cochlea’:tn,ti,ab) AND (‘usher syndrome’/de OR ‘usher’ OR ‘ushers’ OR ‘usher$s’ OR ‘graefe-usher’ OR hallgren) |
| CINAHL | ((usher’s syndrome or usher syndrome or usher) AND (cochlear implant or cochlear implants or cochlear implantation)) OR ((MH “Usher’s Syndrome”) AND (MH “Cochlear Implant”)) OR ((MH “Usher’s Syndrome”) AND (MH “Prostheses and Implants”)) OR (AB usher syndrome AND AB cochlear implant)) |
| References | USH Subtype | Pre-Operative Hearing | Age at CI | Post-Operative Outcome Measures |
|---|---|---|---|---|
| Alsanosi [33], 2015 | Unspecified | Congenital profound bilateral deafness | 5 months | PTA (25 dB HL at most frequencies), questionnaires, and CAP testing 6–8 months post-op |
| Derinsu & Ciprut [42], 2002 | Unspecified | Post-lingual profound deafness | 52 years | PTA (30–35 dB HL across most frequencies) and serial speech perception tests 6 months-4 years post-op |
| Ruiz & Garcia [41], 2013 | USH2 | Post-lingual deafness with CI in R ear 8 months prior | 34 years | PTA (26 and 35 dB HL in left and right, respectively) at 16–25 months post-op |
| Shiomi et al. [43], 1997 | USH3 | Post-lingual deafness | 35 years | Vowel and consonant identification and cortical activation 3 months post-op |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Davies, C.; Bergman, J.; Misztal, C.; Ramchandran, R.; Mittal, J.; Bulut, E.; Shah, V.; Mittal, R.; Eshraghi, A.A. The Outcomes of Cochlear Implantation in Usher Syndrome: A Systematic Review. J. Clin. Med. 2021, 10, 2915. https://doi.org/10.3390/jcm10132915
Davies C, Bergman J, Misztal C, Ramchandran R, Mittal J, Bulut E, Shah V, Mittal R, Eshraghi AA. The Outcomes of Cochlear Implantation in Usher Syndrome: A Systematic Review. Journal of Clinical Medicine. 2021; 10(13):2915. https://doi.org/10.3390/jcm10132915
Chicago/Turabian StyleDavies, Camron, Jenna Bergman, Carly Misztal, Renuka Ramchandran, Jeenu Mittal, Erdogan Bulut, Viraj Shah, Rahul Mittal, and Adrien A. Eshraghi. 2021. "The Outcomes of Cochlear Implantation in Usher Syndrome: A Systematic Review" Journal of Clinical Medicine 10, no. 13: 2915. https://doi.org/10.3390/jcm10132915
APA StyleDavies, C., Bergman, J., Misztal, C., Ramchandran, R., Mittal, J., Bulut, E., Shah, V., Mittal, R., & Eshraghi, A. A. (2021). The Outcomes of Cochlear Implantation in Usher Syndrome: A Systematic Review. Journal of Clinical Medicine, 10(13), 2915. https://doi.org/10.3390/jcm10132915