
Chronic Obstructive Pulmonary Disease: Epidemiology, Biomarkers, and Paving the Way to Lung Cancer

 , ,
, ,  and
and 
Abstract

{kind=link}
{kind=link}
1. Background
2. Epidemiology of COPD
3. Risk Factors for Developing COPD
4. Phenotypes: Many Faces of COPD
5. Diagnostic Tests of COPD
6. Progression, Molecular Players, and Biomarkers of COPD
6.1. Circulating Biomarkers of COPD
6.2. Sputum Biomarkers of COPD
6.3. Biomarkers of the Inflammatory Microenvironment in COPD
6.4. Extracellular Vesicles and COPD
6.5. Purinergic Signaling in COPD
6.6. Galectins in COPD
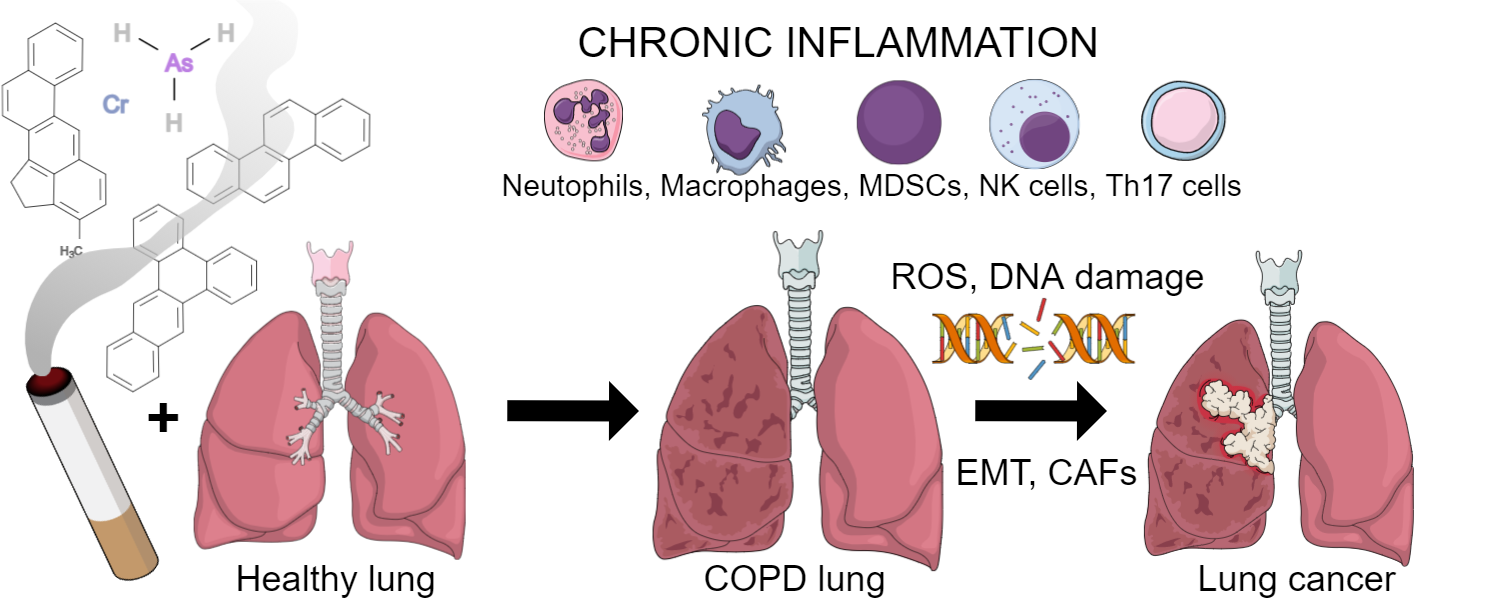
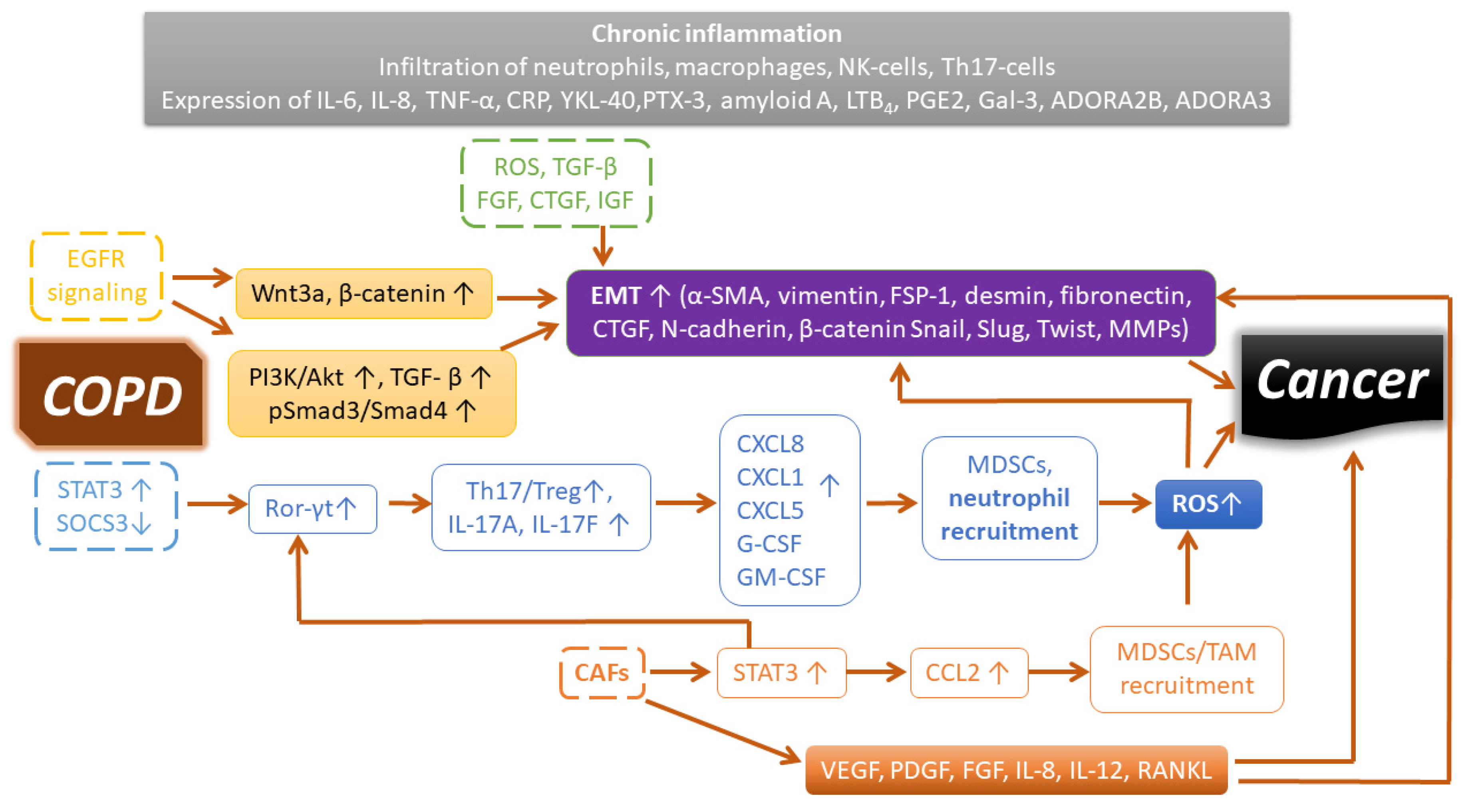
7. COPD: The Soil of Lung Cancer
7.1. Genetic and Epigenetic Factors Associated with Lung Cancer in COPD
7.2. Chronic Inflammatory Microenvironment of COPD
7.3. Cancer-Associated Fibroblasts and COPD
7.4. Lung Microbiota and COPD
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Mouronte-Roibas, C.; Leiro-Fernandez, V.; Fernandez-Villar, A.; Botana-Rial, M.; Ramos-Hernandez, C.; Ruano-Ravina, A. COPD, emphysema and the onset of lung cancer. A systematic review. Cancer Lett. 2016, 382, 240–244. [Google Scholar] [CrossRef]
- Bogos, K.; Kiss, Z.; Galffy, G.; Tamasi, L.; Ostoros, G.; Muller, V.; Urban, L.; Bittner, N.; Sarosi, V.; Vastag, A.; et al. Lung Cancer in Hungary. J. Thorac. Oncol. 2020, 15, 692–699. [Google Scholar] [CrossRef] [PubMed]
- Barta, J.A.; Powell, C.A.; Wisnivesky, J.P. Global Epidemiology of Lung Cancer. Ann. Glob Health 2019, 85. [Google Scholar] [CrossRef] [PubMed]
- Lortet-Tieulent, J.; Soerjomataram, I.; Ferlay, J.; Rutherford, M.; Weiderpass, E.; Bray, F. International trends in lung cancer incidence by histological subtype: Adenocarcinoma stabilizing in men but still increasing in women. Lung Cancer 2014, 84, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Travis, W.D. Pathology of lung cancer. Clin. Chest Med. 2011, 32, 669–692. [Google Scholar] [CrossRef]
- Peters, E.N.; Warren, G.W.; Sloan, J.A.; Marshall, J.R. Tobacco assessment in completed lung cancer treatment trials. Cancer 2016, 122, 3260–3262. [Google Scholar] [CrossRef]
- Gomes, M.; Teixeira, A.L.; Coelho, A.; Araujo, A.; Medeiros, R. The role of inflammation in lung cancer. Adv. Exp. Med. Biol. 2014, 816, 1–23. [Google Scholar] [CrossRef]
- Goldstraw, P.; Chansky, K.; Crowley, J.; Rami-Porta, R.; Asamura, H.; Eberhardt, W.E.; Nicholson, A.G.; Groome, P.; Mitchell, A.; Bolejack, V.; et al. The IASLC Lung Cancer Staging Project: Proposals for Revision of the TNM Stage Groupings in the Forthcoming (Eighth) Edition of the TNM Classification for Lung Cancer. J. Thorac. Oncol. 2016, 11, 39–51. [Google Scholar] [CrossRef]
- Yoshida, T.; Tuder, R.M. Pathobiology of cigarette smoke-induced chronic obstructive pulmonary disease. Physiol. Rev. 2007, 87, 1047–1082. [Google Scholar] [CrossRef]
- Almagro, P.; Martinez-Camblor, P.; Soriano, J.B.; Marin, J.M.; Alfageme, I.; Casanova, C.; Esteban, C.; Soler-Cataluna, J.J.; de-Torres, J.P.; Celli, B.R.; et al. Finding the best thresholds of FEV1 and dyspnea to predict 5-year survival in COPD patients: The COCOMICS study. PLoS ONE 2014, 9, e89866. [Google Scholar] [CrossRef] [PubMed]
- WHO. The Top 10 Causes of Death. Published by World Health Organization (WHO). 9 December 2020. Available online: https://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death (accessed on 28 June 2021).
- Laniado-Laborin, R. Smoking and chronic obstructive pulmonary disease (COPD). Parallel epidemics of the 21 century. Int. J. Environ. Res. Public Health 2009, 6, 209–224. [Google Scholar] [CrossRef] [PubMed]
- Durham, A.L.; Adcock, I.M. The relationship between COPD and lung cancer. Lung Cancer 2015, 90, 121–127. [Google Scholar] [CrossRef]
- Proctor, R.N. The history of the discovery of the cigarette-lung cancer link: Evidentiary traditions, corporate denial, global toll. Tob. Control. 2012, 21, 87–91. [Google Scholar] [CrossRef]
- Oh, J.Y.; Sin, D.D. Lung inflammation in COPD: Why does it matter? F1000 Med. Rep. 2012, 4, 23. [Google Scholar] [CrossRef]
- Raviv, S.; Hawkins, K.A.; DeCamp, M.M., Jr.; Kalhan, R. Lung cancer in chronic obstructive pulmonary disease: Enhancing surgical options and outcomes. Am. J. Respir Crit. Care Med. 2011, 183, 1138–1146. [Google Scholar] [CrossRef]
- Balkwill, F.; Mantovani, A. Inflammation and cancer: Back to Virchow? Lancet 2001, 357, 539–545. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Szebeni, G.J.; Vizler, C.; Kitajka, K.; Puskas, L.G. Inflammation and Cancer: Extra- and Intracellular Determinants of Tumor-Associated Macrophages as Tumor Promoters. Mediators Inflamm. 2017, 2017, 9294018. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef]
- Bremnes, R.M.; Al-Shibli, K.; Donnem, T.; Sirera, R.; Al-Saad, S.; Andersen, S.; Stenvold, H.; Camps, C.; Busund, L.T. The role of tumor-infiltrating immune cells and chronic inflammation at the tumor site on cancer development, progression, and prognosis: Emphasis on non-small cell lung cancer. J. Thorac. Oncol. 2011, 6, 824–833. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The three Es of cancer immunoediting. Annu. Rev. Immunol. 2004, 22, 329–360. [Google Scholar] [CrossRef] [PubMed]
- Shankaran, V.; Ikeda, H.; Bruce, A.T.; White, J.M.; Swanson, P.E.; Old, L.J.; Schreiber, R.D. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature 2001, 410, 1107–1111. [Google Scholar] [CrossRef]
- Postmus, P.E.; Kerr, K.M.; Oudkerk, M.; Senan, S.; Waller, D.A.; Vansteenkiste, J.; Escriu, C.; Peters, S.; Committee, E.G. Early and locally advanced non-small-cell lung cancer (NSCLC): ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2017, 28, iv1–iv21. [Google Scholar] [CrossRef] [PubMed]
- Fruh, M.; De Ruysscher, D.; Popat, S.; Crino, L.; Peters, S.; Felip, E.; Group, E.G.W. Small-cell lung cancer (SCLC): ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2013, 24 (Suppl. 6), vi99–vi105. [Google Scholar] [CrossRef]
- Planchard, D.; Popat, S.; Kerr, K.; Novello, S.; Smit, E.F.; Faivre-Finn, C.; Mok, T.S.; Reck, M.; Van Schil, P.E.; Hellmann, M.D.; et al. Metastatic non-small cell lung cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2018, 29, iv192–iv237. [Google Scholar] [CrossRef]
- Xie, M.; Liu, X.; Cao, X.; Guo, M.; Li, X. Trends in prevalence and incidence of chronic respiratory diseases from 1990 to 2017. Respir Res. 2020, 21, 49. [Google Scholar] [CrossRef]
- GBD Chronic Respiratory Disease Collaborators. Prevalence and attributable health burden of chronic respiratory diseases, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet Respir. Med. 2020, 8, 585–596. [Google Scholar] [CrossRef]
- Agarwal, A.K.; Raja, A.; Brown, B.D. Chronic Obstructive Pulmonary Disease. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. Available online: https://www.ncbi.nlm.nih.gov/books/NBK559281/ (accessed on 28 June 2021).
- Atsou, K.; Chouaid, C.; Hejblum, G. Variability of the chronic obstructive pulmonary disease key epidemiological data in Europe: Systematic review. BMC Med. 2011, 9, 7. [Google Scholar] [CrossRef]
- Rycroft, C.E.; Heyes, A.; Lanza, L.; Becker, K. Epidemiology of chronic obstructive pulmonary disease: A literature review. Int. J. Chron. Obstruct Pulmon Dis. 2012, 7, 457–494. [Google Scholar] [CrossRef]
- Halbert, R.J.; Natoli, J.L.; Gano, A.; Badamgarav, E.; Buist, A.S.; Mannino, D.M. Global burden of COPD: Systematic review and meta-analysis. Eur. Respir. J. 2006, 28, 523–532. [Google Scholar] [CrossRef]
- Terzikhan, N.; Verhamme, K.M.; Hofman, A.; Stricker, B.H.; Brusselle, G.G.; Lahousse, L. Prevalence and incidence of COPD in smokers and non-smokers: The Rotterdam Study. Eur. J. Epidemiol. 2016, 31, 785–792. [Google Scholar] [CrossRef] [PubMed]
- Raherison, C.; Girodet, P.O. Epidemiology of COPD. Eur. Respir. Rev. 2009, 18, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Niu, S.R.; Yang, G.H.; Chen, Z.M.; Wang, J.L.; Wang, G.H.; He, X.Z.; Schoepff, H.; Boreham, J.; Pan, H.C.; Peto, R. Emerging tobacco hazards in China: 2. Early mortality results from a prospective study. BMJ 1998, 317, 1423–1424. [Google Scholar] [CrossRef] [PubMed]
- Mannino, D.M.; Buist, A.S.; Petty, T.L.; Enright, P.L.; Redd, S.C. Lung function and mortality in the United States: Data from the First National Health and Nutrition Examination Survey follow up study. Thorax 2003, 58, 388–393. [Google Scholar] [CrossRef]
- Marsh, S.; Aldington, S.; Shirtcliffe, P.; Weatherall, M.; Beasley, R. Smoking and COPD: What really are the risks? Eur. Respir. J. 2006, 28, 883–884. [Google Scholar] [CrossRef]
- Salvi, S.S.; Barnes, P.J. Chronic obstructive pulmonary disease in non-smokers. Lancet 2009, 374, 733–743. [Google Scholar] [CrossRef]
- Salvi, S.; Barnes, P.J. Is exposure to biomass smoke the biggest risk factor for COPD globally? Chest 2010, 138, 3–6. [Google Scholar] [CrossRef]
- Zeng, G.; Sun, B.; Zhong, N. Non-smoking-related chronic obstructive pulmonary disease: A neglected entity? Respirology 2012, 17, 908–912. [Google Scholar] [CrossRef]
- Postma, D.S.; Bush, A.; van den Berge, M. Risk factors and early origins of chronic obstructive pulmonary disease. Lancet 2015, 385, 899–909. [Google Scholar] [CrossRef]
- Fullerton, D.G.; Bruce, N.; Gordon, S.B. Indoor air pollution from biomass fuel smoke is a major health concern in the developing world. Trans. R Soc. Trop. Med. Hyg. 2008, 102, 843–851. [Google Scholar] [CrossRef]
- Ingebrigtsen, T.; Thomsen, S.F.; Vestbo, J.; van der Sluis, S.; Kyvik, K.O.; Silverman, E.K.; Svartengren, M.; Backer, V. Genetic influences on Chronic Obstructive Pulmonary Disease-A twin study. Respir. Med. 2010, 104, 1890–1895. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.J.; Cho, M.H.; Castaldi, P.J.; Hersh, C.P.; Silverman, E.K.; Laird, N.M. Heritability of chronic obstructive pulmonary disease and related phenotypes in smokers. Am. J. Respir. Crit. Care Med. 2013, 188, 941–947. [Google Scholar] [CrossRef] [PubMed]
- Pillai, S.G.; Ge, D.; Zhu, G.; Kong, X.; Shianna, K.V.; Need, A.C.; Feng, S.; Hersh, C.P.; Bakke, P.; Gulsvik, A.; et al. A genome-wide association study in chronic obstructive pulmonary disease (COPD): Identification of two major susceptibility loci. PLoS Genet. 2009, 5, e1000421. [Google Scholar] [CrossRef] [PubMed]
- Wilk, J.B.; Chen, T.H.; Gottlieb, D.J.; Walter, R.E.; Nagle, M.W.; Brandler, B.J.; Myers, R.H.; Borecki, I.B.; Silverman, E.K.; Weiss, S.T.; et al. A genome-wide association study of pulmonary function measures in the Framingham Heart Study. PLoS Genet. 2009, 5, e1000429. [Google Scholar] [CrossRef] [PubMed]
- Cho, M.H.; Boutaoui, N.; Klanderman, B.J.; Sylvia, J.S.; Ziniti, J.P.; Hersh, C.P.; DeMeo, D.L.; Hunninghake, G.M.; Litonjua, A.A.; Sparrow, D.; et al. Variants in FAM13A are associated with chronic obstructive pulmonary disease. Nat. Genet. 2010, 42, 200–202. [Google Scholar] [CrossRef]
- Cho, M.H.; Castaldi, P.J.; Wan, E.S.; Siedlinski, M.; Hersh, C.P.; Demeo, D.L.; Himes, B.E.; Sylvia, J.S.; Klanderman, B.J.; Ziniti, J.P.; et al. A genome-wide association study of COPD identifies a susceptibility locus on chromosome 19q13. Hum. Mol. Genet. 2012, 21, 947–957. [Google Scholar] [CrossRef] [PubMed]
- Stoller, J.K.; Aboussouan, L.S. Alpha1-antitrypsin deficiency. Lancet 2005, 365, 2225–2236. [Google Scholar] [CrossRef]
- Flenley, D.C. Chronic obstructive pulmonary disease. Dis. Mon. 1988, 34, 537–599. [Google Scholar] [CrossRef]
- Kim, V.; Crapo, J.; Zhao, H.; Jones, P.W.; Silverman, E.K.; Comellas, A.; Make, B.J.; Criner, G.J.; Investigators, C.O. Comparison between an alternative and the classic definition of chronic bronchitis in COPDGene. Ann. Am. Thorac. Soc. 2015, 12, 332–339. [Google Scholar] [CrossRef] [PubMed]
- Han, M.K.; Agusti, A.; Calverley, P.M.; Celli, B.R.; Criner, G.; Curtis, J.L.; Fabbri, L.M.; Goldin, J.G.; Jones, P.W.; Macnee, W.; et al. Chronic obstructive pulmonary disease phenotypes: The future of COPD. Am. J. Respir. Crit. Care Med. 2010, 182, 598–604. [Google Scholar] [CrossRef] [PubMed]
- Soler-Cataluna, J.J.; Cosio, B.; Izquierdo, J.L.; Lopez-Campos, J.L.; Marin, J.M.; Aguero, R.; Baloira, A.; Carrizo, S.; Esteban, C.; Galdiz, J.B.; et al. Consensus document on the overlap phenotype COPD-asthma in COPD. Arch. Bronconeumol. 2012, 48, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Halpin, D.M.; Decramer, M.; Celli, B.; Kesten, S.; Liu, D.; Tashkin, D.P. Exacerbation frequency and course of COPD. Int. J. Chron. Obstruct. Pulmon. Dis. 2012, 7, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Fishman, A.; Martinez, F.; Naunheim, K.; Piantadosi, S.; Wise, R.; Ries, A.; Weinmann, G.; Wood, D.E.; National Emphysema Treatment Trial Research Group. A randomized trial comparing lung-volume-reduction surgery with medical therapy for severe emphysema. N. Engl. J. Med. 2003, 348, 2059–2073. [Google Scholar] [CrossRef]
- Pinto, L.M.; Alghamdi, M.; Benedetti, A.; Zaihra, T.; Landry, T.; Bourbeau, J. Derivation and validation of clinical phenotypes for COPD: A systematic review. Respir. Res. 2015, 16, 50. [Google Scholar] [CrossRef]
- Koskela, J.; Kilpelainen, M.; Kupiainen, H.; Mazur, W.; Sintonen, H.; Boezen, M.; Lindqvist, A.; Postma, D.; Laitinen, T. Co-morbidities are the key nominators of the health related quality of life in mild and moderate COPD. BMC Pulm. Med. 2014, 14, 102. [Google Scholar] [CrossRef]
- Mirza, S.; Benzo, R. Chronic Obstructive Pulmonary Disease Phenotypes: Implications for Care. Mayo Clin. Proc. 2017, 92, 1104–1112. [Google Scholar] [CrossRef]
- Lahousse, L.; Ziere, G.; Verlinden, V.J.; Zillikens, M.C.; Uitterlinden, A.G.; Rivadeneira, F.; Tiemeier, H.; Joos, G.F.; Hofman, A.; Ikram, M.A.; et al. Risk of Frailty in Elderly With COPD: A Population-Based Study. J. Gerontol. A Biol. Sci. Med. Sci. 2016, 71, 689–695. [Google Scholar] [CrossRef]
- Mittal, N.; Raj, R.; Islam, E.A.; Nugent, K. The Frequency of Frailty in Ambulatory Patients With Chronic Lung Diseases. J. Prim. Care Community Health 2016, 7, 10–15. [Google Scholar] [CrossRef]
- Laurin, C.; Moullec, G.; Bacon, S.L.; Lavoie, K.L. Impact of anxiety and depression on chronic obstructive pulmonary disease exacerbation risk. Am. J. Respir. Crit. Care Med. 2012, 185, 918–923. [Google Scholar] [CrossRef] [PubMed]
- Van Buul, A.R.; Kasteleyn, M.J.; Chavannes, N.H.; Taube, C. Association between morning symptoms and physical activity in COPD: A systematic review. Eur. Respir. Rev. 2017, 26. [Google Scholar] [CrossRef]
- Fazleen, A.; Wilkinson, T. Early COPD: Current evidence for diagnosis and management. Ther. Adv. Respir. Dis. 2020, 14. [Google Scholar] [CrossRef]
- Halpin, D.M.G.; Criner, G.J.; Papi, A.; Singh, D.; Anzueto, A.; Martinez, F.J.; Agusti, A.A.; Vogelmeier, C.F. Global Initiative for the Diagnosis, Management, and Prevention of Chronic Obstructive Lung Disease. The 2020 GOLD Science Committee Report on COVID-19 and Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2021, 203, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Bhatta, L.; Leivseth, L.; Mai, X.M.; Henriksen, A.H.; Carslake, D.; Chen, Y.; Langhammer, A.; Brumpton, B.M. GOLD Classifications, COPD Hospitalization, and All-Cause Mortality in Chronic Obstructive Pulmonary Disease: The HUNT Study. Int. J. Chron. Obstruct. Pulmon. Dis. 2020, 15, 225–233. [Google Scholar] [CrossRef]
- Besa, V.; Teschler, H.; Kurth, I.; Khan, A.M.; Zarogoulidis, P.; Baumbach, J.I.; Sommerwerck, U.; Freitag, L.; Darwiche, K. Exhaled volatile organic compounds discriminate patients with chronic obstructive pulmonary disease from healthy subjects. Int. J. Chron. Obstruct. Pulmon. Dis. 2015, 10, 399–406. [Google Scholar] [CrossRef]
- Harvey, B.G.; Strulovici-Barel, Y.; Kaner, R.J.; Sanders, A.; Vincent, T.L.; Mezey, J.G.; Crystal, R.G. Risk of COPD with obstruction in active smokers with normal spirometry and reduced diffusion capacity. Eur. Respir. J. 2015, 46, 1589–1597. [Google Scholar] [CrossRef]
- Cukic, V. The changes of arterial blood gases in COPD during four-year period. Med. Arch. 2014, 68, 14–18. [Google Scholar] [CrossRef]
- Frantz, S.; Nihlen, U.; Dencker, M.; Engstrom, G.; Lofdahl, C.G.; Wollmer, P. Impulse oscillometry may be of value in detecting early manifestations of COPD. Respir. Med. 2012, 106, 1116–1123. [Google Scholar] [CrossRef]
- Lipworth, B.J.; Jabbal, S. What can we learn about COPD from impulse oscillometry? Respir. Med. 2018, 139, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Miravitlles, M.; Dirksen, A.; Ferrarotti, I.; Koblizek, V.; Lange, P.; Mahadeva, R.; McElvaney, N.G.; Parr, D.; Piitulainen, E.; Roche, N.; et al. European Respiratory Society statement: Diagnosis and treatment of pulmonary disease in alpha1-antitrypsin deficiency. Eur. Respir. J. 2017, 50. [Google Scholar] [CrossRef]
- Greulich, T.; Nell, C.; Hohmann, D.; Grebe, M.; Janciauskiene, S.; Koczulla, A.R.; Vogelmeier, C.F. The prevalence of diagnosed alpha1-antitrypsin deficiency and its comorbidities: Results from a large population-based database. Eur. Respir. J. 2017, 49. [Google Scholar] [CrossRef]
- Jeffery, P.K. Remodeling and inflammation of bronchi in asthma and chronic obstructive pulmonary disease. Proc. Am. Thorac. Soc. 2004, 1, 176–183. [Google Scholar] [CrossRef]
- Vestbo, J.; Edwards, L.D.; Scanlon, P.D.; Yates, J.C.; Agusti, A.; Bakke, P.; Calverley, P.M.; Celli, B.; Coxson, H.O.; Crim, C.; et al. Changes in forced expiratory volume in 1 second over time in COPD. N. Engl. J. Med. 2011, 365, 1184–1192. [Google Scholar] [CrossRef] [PubMed]
- Vestbo, J.; Agusti, A.; Wouters, E.F.; Bakke, P.; Calverley, P.M.; Celli, B.; Coxson, H.; Crim, C.; Edwards, L.D.; Locantore, N.; et al. Should we view chronic obstructive pulmonary disease differently after ECLIPSE? A clinical perspective from the study team. Am. J. Respir. Crit. Care Med. 2014, 189, 1022–1030. [Google Scholar] [CrossRef]
- Baughman, P.; Marott, J.L.; Lange, P.; Martin, C.J.; Shankar, A.; Petsonk, E.L.; Hnizdo, E. Combined effect of lung function level and decline increases morbidity and mortality risks. Eur. J. Epidemiol. 2012, 27, 933–943. [Google Scholar] [CrossRef] [PubMed]
- Haruna, A.; Muro, S.; Nakano, Y.; Ohara, T.; Hoshino, Y.; Ogawa, E.; Hirai, T.; Niimi, A.; Nishimura, K.; Chin, K.; et al. CT scan findings of emphysema predict mortality in COPD. Chest 2010, 138, 635–640. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, C.; Guest, P.J.; Hill, S.L.; Stockley, R.A. Physiological and radiological characterisation of patients diagnosed with chronic obstructive pulmonary disease in primary care. Thorax 2000, 55, 635–642. [Google Scholar] [CrossRef]
- Patel, I.S.; Vlahos, I.; Wilkinson, T.M.; Lloyd-Owen, S.J.; Donaldson, G.C.; Wilks, M.; Reznek, R.H.; Wedzicha, J.A. Bronchiectasis, exacerbation indices, and inflammation in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2004, 170, 400–407. [Google Scholar] [CrossRef]
- Williams, M.C.; Murchison, J.T.; Edwards, L.D.; Agusti, A.; Bakke, P.; Calverley, P.M.; Celli, B.; Coxson, H.O.; Crim, C.; Lomas, D.A.; et al. Coronary artery calcification is increased in patients with COPD and associated with increased morbidity and mortality. Thorax 2014, 69, 718–723. [Google Scholar] [CrossRef]
- Hackler, L., Jr.; Dorman, G.; Kele, Z.; Urge, L.; Darvas, F.; Puskas, L.G. Development of chemically modified glass surfaces for nucleic acid, protein and small molecule microarrays. Mol. Divers. 2003, 7, 25–36. [Google Scholar] [CrossRef]
- Kalman, J.; Palotas, A.; Juhasz, A.; Rimanoczy, A.; Hugyecz, M.; Kovacs, Z.; Galsi, G.; Szabo, Z.; Pakaski, M.; Feher, L.Z.; et al. Impact of venlafaxine on gene expression profile in lymphocytes of the elderly with major depression--evolution of antidepressants and the role of the "neuro-immune" system. Neurochem. Res. 2005, 30, 1429–1438. [Google Scholar] [CrossRef]
- Darvas, F.; Dorman, G.; Krajcsi, P.; Puskas, L.G.; Kovari, Z.; Lorincz, Z.; Urge, L. Recent advances in chemical genomics. Curr. Med. Chem. 2004, 11, 3119–3145. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, S.; Mariani, T.J. Array of hope: Expression profiling identifies disease biomarkers and mechanism. Biochem. Soc. Trans. 2009, 37, 855–862. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Alfoldi, R.; Balog, J.A.; Farago, N.; Halmai, M.; Kotogany, E.; Neuperger, P.; Nagy, L.I.; Feher, L.Z.; Szebeni, G.J.; Puskas, L.G. Single Cell Mass Cytometry of Non-Small Cell Lung Cancer Cells Reveals Complexity of In vivo And Three-Dimensional Models over the Petri-dish. Cells 2019, 8, 1093. [Google Scholar] [CrossRef] [PubMed]
- Rong, B.; Fu, T.; Gao, W.; Li, M.; Rong, C.; Liu, W.; Liu, H. Reduced Serum Concentration of CC16 Is Associated with Severity of Chronic Obstructive Pulmonary Disease and Contributes to the Diagnosis and Assessment of the Disease. Int. J. Chron. Obstruct. Pulmon. Dis. 2020, 15, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Pouwels, S.D.; Klont, F.; Kwiatkowski, M.; Wiersma, V.R.; Faiz, A.; van den Berge, M.; Horvatovich, P.; Bischoff, R.; Ten Hacken, N.H.T. Cigarette Smoking Acutely Decreases Serum Levels of the Chronic Obstructive Pulmonary Disease Biomarker sRAGE. Am. J. Respir. Crit. Care Med. 2018, 198, 1456–1458. [Google Scholar] [CrossRef]
- Li, D.; Wu, Y.; Guo, S.; Qin, J.; Feng, M.; An, Y.; Zhang, J.; Li, Y.; Xiong, S.; Zhou, H.; et al. Circulating syndecan-1 as a novel biomarker relates to lung function, systemic inflammation, and exacerbation in COPD. Int. J. Chron. Obstruct. Pulmon. Dis. 2019, 14, 1933–1941. [Google Scholar] [CrossRef]
- Stockley, R.A.; Halpin, D.M.G.; Celli, B.R.; Singh, D. Chronic Obstructive Pulmonary Disease Biomarkers and Their Interpretation. Am. J. Respir. Crit. Care Med. 2019, 199, 1195–1204. [Google Scholar] [CrossRef]
- Shaw, J.G.; Vaughan, A.; Dent, A.G.; O’Hare, P.E.; Goh, F.; Bowman, R.V.; Fong, K.M.; Yang, I.A. Biomarkers of progression of chronic obstructive pulmonary disease (COPD). J. Thorac. Dis. 2014, 6, 1532–1547. [Google Scholar] [CrossRef]
- Yonchuk, J.G.; Silverman, E.K.; Bowler, R.P.; Agusti, A.; Lomas, D.A.; Miller, B.E.; Tal-Singer, R.; Mayer, R.J. Circulating soluble receptor for advanced glycation end products (sRAGE) as a biomarker of emphysema and the RAGE axis in the lung. Am. J. Respir. Crit. Care Med. 2015, 192, 785–792. [Google Scholar] [CrossRef]
- Shahriary, A.; Panahi, Y.; Shirali, S.; Rahmani, H. Relationship of serum levels of interleukin 6, interleukin 8, and C-reactive protein with forced expiratory volume in first second in patients with mustard lung and chronic obstructive pulmonary diseases: Systematic review and meta-analysis. Postepy Dermatol. Alergol. 2017, 34, 192–198. [Google Scholar] [CrossRef]
- Lopez-Campos, J.L.; Arellano, E.; Calero, C.; Delgado, A.; Marquez, E.; Cejudo, P.; Ortega, F.; Rodriguez-Panadero, F.; Montes-Worboys, A. Determination of inflammatory biomarkers in patients with COPD: A comparison of different assays. BMC Med. Res. Methodol. 2012, 12, 40. [Google Scholar] [CrossRef]
- Lai, T.; Wu, D.; Chen, M.; Cao, C.; Jing, Z.; Huang, L.; Lv, Y.; Zhao, X.; Lv, Q.; Wang, Y.; et al. YKL-40 expression in chronic obstructive pulmonary disease: Relation to acute exacerbations and airway remodeling. Respir. Res. 2016, 17, 31. [Google Scholar] [CrossRef]
- Han, S.S.; Lee, W.H.; Hong, Y.; Kim, W.J.; Yang, J.; Lim, M.N.; Lee, S.J.; Kwon, J.W. Comparison of serum biomarkers between patients with asthma and with chronic obstructive pulmonary disease. J. Asthma 2016, 53, 583–588. [Google Scholar] [CrossRef]
- Sin, D.D.; Miller, B.E.; Duvoix, A.; Man, S.F.; Zhang, X.; Silverman, E.K.; Connett, J.E.; Anthonisen, N.A.; Wise, R.A.; Tashkin, D.; et al. Serum PARC/CCL-18 concentrations and health outcomes in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2011, 183, 1187–1192. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Qin, J.; He, J.; Shen, Y.; Wang, H.; Li, Y.; Zeng, Q.; Dong, J.; An, Y.; Xiong, S.; et al. Serum Endostatin Is a Novel Marker for COPD Associated with Lower Lung Function, Exacerbation and Systemic Inflammation. Int. J. Chron. Obstruct. Pulmon. Dis. 2020, 15, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Ronnow, S.R.; Sand, J.M.B.; Langholm, L.L.; Manon-Jensen, T.; Karsdal, M.A.; Tal-Singer, R.; Miller, B.E.; Vestbo, J.; Leeming, D.J. Type IV collagen turnover is predictive of mortality in COPD: A comparison to fibrinogen in a prospective analysis of the ECLIPSE cohort. Respir. Res. 2019, 20, 63. [Google Scholar] [CrossRef] [PubMed]
- Poznanski, M.; Brzezianska-Lasota, E.; Kiszalkiewicz, J.; Kurnatowska, I.; Kroczynska-Bednarek, J.; Pekala-Wojciechowska, A.; Pietras, T.; Antczak, A. Serum levels and gene expression of pentraxin 3 are elevated in COPD. Adv. Med. Sci. 2019, 64, 85–89. [Google Scholar] [CrossRef]
- Akiki, Z.; Fakih, D.; Jounblat, R.; Chamat, S.; Waked, M.; Holmskov, U.; Sorensen, G.L.; Nadif, R.; Salameh, P. Surfactant protein D, a clinical biomarker for chronic obstructive pulmonary disease with excellent discriminant values. Exp. Ther. Med. 2016, 11, 723–730. [Google Scholar] [CrossRef]
- Paone, G.; Conti, V.; Vestri, A.; Leone, A.; Puglisi, G.; Benassi, F.; Brunetti, G.; Schmid, G.; Cammarella, I.; Terzano, C. Analysis of sputum markers in the evaluation of lung inflammation and functional impairment in symptomatic smokers and COPD patients. Dis. Markers 2011, 31, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Koutsokera, A.; Kostikas, K.; Nicod, L.P.; Fitting, J.W. Pulmonary biomarkers in COPD exacerbations: A systematic review. Respir. Res. 2013, 14, 111. [Google Scholar] [CrossRef] [PubMed]
- Celejewska-Wojcik, N.; Kania, A.; Gorka, K.; Nastalek, P.; Wojcik, K.; Gielicz, A.; Mastalerz, L.; Sanak, M.; Sladek, K. Eicosanoids and Eosinophilic Inflammation of Airways in Stable COPD. Int. J. Chron. Obstruct. Pulmon. Dis. 2021, 16, 1415–1424. [Google Scholar] [CrossRef]
- Di Stefano, A.; Capelli, A.; Lusuardi, M.; Balbo, P.; Vecchio, C.; Maestrelli, P.; Mapp, C.E.; Fabbri, L.M.; Donner, C.F.; Saetta, M. Severity of airflow limitation is associated with severity of airway inflammation in smokers. Am. J. Respir. Crit. Care Med. 1998, 158, 1277–1285. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Brightling, C.E. Eosinophilic airway inflammation in COPD. Int. J. Chron. Obstruct. Pulmon. Dis. 2006, 1, 39–47. [Google Scholar] [CrossRef]
- Barnes, P.J. Therapeutic approaches to asthma-chronic obstructive pulmonary disease overlap syndromes. J. Allergy Clin. Immunol. 2015, 136, 531–545. [Google Scholar] [CrossRef] [PubMed]
- Christenson, S.A.; Steiling, K.; van den Berge, M.; Hijazi, K.; Hiemstra, P.S.; Postma, D.S.; Lenburg, M.E.; Spira, A.; Woodruff, P.G. Asthma-COPD overlap. Clinical relevance of genomic signatures of type 2 inflammation in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2015, 191, 758–766. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.H.; Park, H.Y.; Kang, D.; Cho, J.; Kwon, S.O.; Park, J.H.; Lee, J.S.; Oh, Y.M.; Sin, D.D.; Kim, W.J.; et al. Serial blood eosinophils and clinical outcome in patients with chronic obstructive pulmonary disease. Respir. Res. 2018, 19, 134. [Google Scholar] [CrossRef]
- Dweik, R.A.; Comhair, S.A.; Gaston, B.; Thunnissen, F.B.; Farver, C.; Thomassen, M.J.; Kavuru, M.; Hammel, J.; Abu-Soud, H.M.; Erzurum, S.C. NO chemical events in the human airway during the immediate and late antigen-induced asthmatic response. Proc. Natl. Acad. Sci. USA 2001, 98, 2622–2627. [Google Scholar] [CrossRef] [PubMed]
- Jo, Y.S.; Choe, J.; Shin, S.H.; Koo, H.K.; Lee, W.Y.; Kim, Y.I.; Ra, S.W.; Yoo, K.H.; Jung, K.S.; Park, H.Y.; et al. Exhaled Nitric Oxide in Patients with Stable Chronic Obstructive Pulmonary Disease: Clinical Implications of the Use of Inhaled Corticosteroids. Tuberc. Respir. Dis. 2020, 83, 42–50. [Google Scholar] [CrossRef]
- Antus, B.; Barta, I.; Horvath, I.; Csiszer, E. Relationship between exhaled nitric oxide and treatment response in COPD patients with exacerbations. Respirology 2010, 15, 472–477. [Google Scholar] [CrossRef]
- Montuschi, P.; Kharitonov, S.A.; Ciabattoni, G.; Barnes, P.J. Exhaled leukotrienes and prostaglandins in COPD. Thorax 2003, 58, 585–588. [Google Scholar] [CrossRef]
- Kirkham, S.; Kolsum, U.; Rousseau, K.; Singh, D.; Vestbo, J.; Thornton, D.J. MUC5B is the major mucin in the gel phase of sputum in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2008, 178, 1033–1039. [Google Scholar] [CrossRef]
- Manzel, L.J.; Shi, L.; O’Shaughnessy, P.T.; Thorne, P.S.; Look, D.C. Inhibition by cigarette smoke of nuclear factor-kappaB-dependent response to bacteria in the airway. Am. J. Respir. Cell Mol. Biol. 2011, 44, 155–165. [Google Scholar] [CrossRef]
- Yang, J.; Zuo, W.L.; Fukui, T.; Chao, I.; Gomi, K.; Lee, B.; Staudt, M.R.; Kaner, R.J.; Strulovici-Barel, Y.; Salit, J.; et al. Smoking-Dependent Distal-to-Proximal Repatterning of the Adult Human Small Airway Epithelium. Am. J. Respir. Crit. Care Med. 2017, 196, 340–352. [Google Scholar] [CrossRef] [PubMed]
- Sohal, S.S.; Eapen, M.S.; Ward, C.; Walters, E.H. Epithelial-Mesenchymal Transition: A Necessary New Therapeutic Target in Chronic Obstructive Pulmonary Disease? Am. J. Respir. Crit. Care Med. 2017, 196, 393–394. [Google Scholar] [CrossRef] [PubMed]
- Willis, B.C.; Borok, Z. TGF-beta-induced EMT: Mechanisms and implications for fibrotic lung disease. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 293, L525–L534. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, M.Q.; Walters, E.H.; Shukla, S.D.; Weston, S.; Muller, H.K.; Ward, C.; Sohal, S.S. beta-catenin, Twist and Snail: Transcriptional regulation of EMT in smokers and COPD, and relation to airflow obstruction. Sci. Rep. 2017, 7, 10832. [Google Scholar] [CrossRef] [PubMed]
- Eapen, M.S.; Sharma, P.; Gaikwad, A.V.; Lu, W.; Myers, S.; Hansbro, P.M.; Sohal, S.S. Epithelial-mesenchymal transition is driven by transcriptional and post transcriptional modulations in COPD: Implications for disease progression and new therapeutics. Int. J. Chron. Obstruct. Pulmon. Dis. 2019, 14, 1603–1610. [Google Scholar] [CrossRef] [PubMed]
- Milara, J.; Peiro, T.; Serrano, A.; Cortijo, J. Epithelial to mesenchymal transition is increased in patients with COPD and induced by cigarette smoke. Thorax 2013, 68, 410–420. [Google Scholar] [CrossRef]
- Wang, N.; Wang, Q.; Du, T.; Gabriel, A.N.A.; Wang, X.; Sun, L.; Li, X.; Xu, K.; Jiang, X.; Zhang, Y. The Potential Roles of Exosomes in Chronic Obstructive Pulmonary Disease. Front. Med. 2020, 7, 618506. [Google Scholar] [CrossRef] [PubMed]
- Holtzman, J.; Lee, H. Emerging role of extracellular vesicles in the respiratory system. Exp. Mol. Med. 2020, 52, 887–895. [Google Scholar] [CrossRef] [PubMed]
- Pastor, L.; Vera, E.; Marin, J.M.; Sanz-Rubio, D. Extracellular Vesicles from Airway Secretions: New Insights in Lung Diseases. Int. J. Mol. Sci. 2021, 22, 583. [Google Scholar] [CrossRef] [PubMed]
- O’Farrell, H.E.; Yang, I.A. Extracellular vesicles in chronic obstructive pulmonary disease (COPD). J. Thorac. Dis. 2019, 11, S2141–S2154. [Google Scholar] [CrossRef]
- Fujii, S.; Hara, H.; Araya, J.; Takasaka, N.; Kojima, J.; Ito, S.; Minagawa, S.; Yumino, Y.; Ishikawa, T.; Numata, T.; et al. Insufficient autophagy promotes bronchial epithelial cell senescence in chronic obstructive pulmonary disease. Oncoimmunology 2012, 1, 630–641. [Google Scholar] [CrossRef]
- Moon, H.G.; Kim, S.H.; Gao, J.; Quan, T.; Qin, Z.; Osorio, J.C.; Rosas, I.O.; Wu, M.; Tesfaigzi, Y.; Jin, Y. CCN1 secretion and cleavage regulate the lung epithelial cell functions after cigarette smoke. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 307, L326–337. [Google Scholar] [CrossRef]
- Mohan, A.; Agarwal, S.; Clauss, M.; Britt, N.S.; Dhillon, N.K. Extracellular vesicles: Novel communicators in lung diseases. Respir. Res. 2020, 21, 175. [Google Scholar] [CrossRef] [PubMed]
- Genschmer, K.R.; Russell, D.W.; Lal, C.; Szul, T.; Bratcher, P.E.; Noerager, B.D.; Abdul Roda, M.; Xu, X.; Rezonzew, G.; Viera, L.; et al. Activated PMN Exosomes: Pathogenic Entities Causing Matrix Destruction and Disease in the Lung. Cell 2019, 176, 113–126.e115. [Google Scholar] [CrossRef]
- Kadota, T.; Fujita, Y.; Yoshioka, Y.; Araya, J.; Kuwano, K.; Ochiya, T. Extracellular Vesicles in Chronic Obstructive Pulmonary Disease. Int. J. Mol. Sci. 2016, 17, 1801. [Google Scholar] [CrossRef]
- Kubo, H. Extracellular Vesicles in Lung Disease. Chest 2018, 153, 210–216. [Google Scholar] [CrossRef]
- Trappe, A.; Donnelly, S.C.; McNally, P.; Coppinger, J.A. Role of extracellular vesicles in chronic lung disease. Thorax 2021. [Google Scholar] [CrossRef]
- Le, T.T.; Berg, N.K.; Harting, M.T.; Li, X.; Eltzschig, H.K.; Yuan, X. Purinergic Signaling in Pulmonary Inflammation. Front. Immunol. 2019, 10, 1633. [Google Scholar] [CrossRef]
- Esther, C.R., Jr.; Lazaar, A.L.; Bordonali, E.; Qaqish, B.; Boucher, R.C. Elevated airway purines in COPD. Chest 2011, 140, 954–960. [Google Scholar] [CrossRef] [PubMed]
- Pelleg, A.; Schulman, E.S.; Barnes, P.J. Extracellular Adenosine 5’-Triphosphate in Obstructive Airway Diseases. Chest 2016, 150, 908–915. [Google Scholar] [CrossRef] [PubMed]
- Karmouty-Quintana, H.; Xia, Y.; Blackburn, M.R. Adenosine signaling during acute and chronic disease states. J. Mol. Med. 2013, 91, 173–181. [Google Scholar] [CrossRef]
- Collum, S.D.; Molina, J.G.; Hanmandlu, A.; Bi, W.; Pedroza, M.; Mertens, T.C.J.; Wareing, N.; Wei, W.; Wilson, C.; Sun, W.; et al. Adenosine and hyaluronan promote lung fibrosis and pulmonary hypertension in combined pulmonary fibrosis and emphysema. Dis. Model. Mech. 2019, 12. [Google Scholar] [CrossRef] [PubMed]
- Karmouty-Quintana, H.; Weng, T.; Garcia-Morales, L.J.; Chen, N.Y.; Pedroza, M.; Zhong, H.; Molina, J.G.; Bunge, R.; Bruckner, B.A.; Xia, Y.; et al. Adenosine A2B receptor and hyaluronan modulate pulmonary hypertension associated with chronic obstructive pulmonary disease. Am. J. Respir. Cell Mol. Biol. 2013, 49, 1038–1047. [Google Scholar] [CrossRef] [PubMed]
- Boo, H.J.; Park, S.J.; Noh, M.; Min, H.Y.; Jeong, L.S.; Lee, H.Y. LJ-2698, an Adenosine A3 Receptor Antagonist, Alleviates Elastase-Induced Pulmonary Emphysema in Mice. Biomol. Ther. 2020, 28, 250–258. [Google Scholar] [CrossRef]
- Johannes, L.; Jacob, R.; Leffler, H. Galectins at a glance. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef]
- Ion, G.; Fajka-Boja, R.; Kovacs, F.; Szebeni, G.; Gombos, I.; Czibula, A.; Matko, J.; Monostori, E. Acid sphingomyelinase mediated release of ceramide is essential to trigger the mitochondrial pathway of apoptosis by galectin-1. Cell Signal 2006, 18, 1887–1896. [Google Scholar] [CrossRef]
- Kovacs-Solyom, F.; Blasko, A.; Fajka-Boja, R.; Katona, R.L.; Vegh, L.; Novak, J.; Szebeni, G.J.; Krenacs, L.; Uher, F.; Tubak, V.; et al. Mechanism of tumor cell-induced T-cell apoptosis mediated by galectin-1. Immunol. Lett. 2010, 127, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Szebeni, G.J.; Kriston-Pal, E.; Blazso, P.; Katona, R.L.; Novak, J.; Szabo, E.; Czibula, A.; Fajka-Boja, R.; Hegyi, B.; Uher, F.; et al. Identification of galectin-1 as a critical factor in function of mouse mesenchymal stromal cell-mediated tumor promotion. PLoS ONE 2012, 7, e41372. [Google Scholar] [CrossRef] [PubMed]
- Kathiriya, J.J.; Nakra, N.; Nixon, J.; Patel, P.S.; Vaghasiya, V.; Alhassani, A.; Tian, Z.; Allen-Gipson, D.; Dave, V. Galectin-1 inhibition attenuates profibrotic signaling in hypoxia-induced pulmonary fibrosis. Cell. Death Discov. 2017, 3, 17010. [Google Scholar] [CrossRef]
- Pilette, C.; Colinet, B.; Kiss, R.; Andre, S.; Kaltner, H.; Gabius, H.J.; Delos, M.; Vaerman, J.P.; Decramer, M.; Sibille, Y. Increased galectin-3 expression and intra-epithelial neutrophils in small airways in severe COPD. Eur. Respir. J. 2007, 29, 914–922. [Google Scholar] [CrossRef]
- Nishi, Y.; Sano, H.; Kawashima, T.; Okada, T.; Kuroda, T.; Kikkawa, K.; Kawashima, S.; Tanabe, M.; Goto, T.; Matsuzawa, Y.; et al. Role of galectin-3 in human pulmonary fibrosis. Allergol. Int. 2007, 56, 57–65. [Google Scholar] [CrossRef]
- Feng, W.; Wu, X.; Li, S.; Zhai, C.; Wang, J.; Shi, W.; Li, M. Association of Serum Galectin-3 with the Acute Exacerbation of Chronic Obstructive Pulmonary Disease. Med. Sci. Monit. 2017, 23, 4612–4618. [Google Scholar] [CrossRef]
- Sundqvist, M.; Andelid, K.; Ekberg-Jansson, A.; Bylund, J.; Karlsson-Bengtsson, A.; Linden, A. Systemic Galectin-3 in Smokers with Chronic Obstructive Pulmonary Disease and Chronic Bronchitis: The Impact of Exacerbations. Int. J. Chron. Obstruct. Pulmon. Dis. 2021, 16, 367–377. [Google Scholar] [CrossRef]
- Horio, Y.; Ichiyasu, H.; Kojima, K.; Saita, N.; Migiyama, Y.; Iriki, T.; Fujii, K.; Niki, T.; Hirashima, M.; Kohrogi, H. Protective effect of Galectin-9 in murine model of lung emphysema: Involvement of neutrophil migration and MMP-9 production. PLoS ONE 2017, 12, e0180742. [Google Scholar] [CrossRef] [PubMed]
- Parris, B.A.; O’Farrell, H.E.; Fong, K.M.; Yang, I.A. Chronic obstructive pulmonary disease (COPD) and lung cancer: Common pathways for pathogenesis. J. Thorac. Dis. 2019, 11, S2155–S2172. [Google Scholar] [CrossRef] [PubMed]
- Refaee, T.; Wu, G.; Ibrahim, A.; Halilaj, I.; Leijenaar, R.T.H.; Rogers, W.; Gietema, H.A.; Hendriks, L.E.L.; Lambin, P.; Woodruff, H.C. The Emerging Role of Radiomics in COPD and Lung Cancer. Respiration 2020, 99, 99–107. [Google Scholar] [CrossRef]
- Papi, A.; Casoni, G.; Caramori, G.; Guzzinati, I.; Boschetto, P.; Ravenna, F.; Calia, N.; Petruzzelli, S.; Corbetta, L.; Cavallesco, G.; et al. COPD increases the risk of squamous histological subtype in smokers who develop non-small cell lung carcinoma. Thorax 2004, 59, 679–681. [Google Scholar] [CrossRef]
- Young, R.P.; Hopkins, R.J.; Christmas, T.; Black, P.N.; Metcalf, P.; Gamble, G.D. COPD prevalence is increased in lung cancer, independent of age, sex and smoking history. Eur. Respir. J. 2009, 34, 380–386. [Google Scholar] [CrossRef]
- Young, R.P.; Hopkins, R.J.; Gamble, G.D.; Etzel, C.; El-Zein, R.; Crapo, J.D. Genetic evidence linking lung cancer and COPD: A new perspective. Appl. Clin. Genet. 2011, 4, 99–111. [Google Scholar] [CrossRef]
- Cui, K.; Ge, X.; Ma, H. Four SNPs in the CHRNA3/5 alpha-neuronal nicotinic acetylcholine receptor subunit locus are associated with COPD risk based on meta-analyses. PLoS ONE 2014, 9, e102324. [Google Scholar] [CrossRef]
- Young, R.P.; Hopkins, R.J.; Hay, B.A.; Whittington, C.F.; Epton, M.J.; Gamble, G.D. FAM13A locus in COPD is independently associated with lung cancer-evidence of a molecular genetic link between COPD and lung cancer. Appl. Clin. Genet. 2011, 4, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Young, R.P.; Whittington, C.F.; Hopkins, R.J.; Hay, B.A.; Epton, M.J.; Black, P.N.; Gamble, G.D. Chromosome 4q31 locus in COPD is also associated with lung cancer. Eur. Respir. J. 2010, 36, 1375–1382. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Choi, S.; Kwon, S.Y.; Lee, Y.; Lee, J.K.; Heo, E.Y.; Chung, H.S.; Kim, D.K. A Genome-Wide Association Study in Early COPD: Identification of One Major Susceptibility Loci. Int. J. Chron. Obstruct. Pulmon. Dis. 2020, 15, 2967–2975. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yang, L.; Deng, J.; Wang, B.; Yang, X.; Yang, R.; Cheng, M.; Fang, W.; Qiu, F.; Zhang, X.; et al. Genetic variant in the 3’-untranslated region of VEGFR1 gene influences chronic obstructive pulmonary disease and lung cancer development in Chinese population. Mutagenesis 2014, 29, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Mateu-Jimenez, M.; Curull, V.; Rodriguez-Fuster, A.; Aguilo, R.; Sanchez-Font, A.; Pijuan, L.; Gea, J.; Barreiro, E. Profile of epigenetic mechanisms in lung tumors of patients with underlying chronic respiratory conditions. Clin. Epigenetics 2018, 10, 7. [Google Scholar] [CrossRef]
- Denkceken, T.; Pala, E. Investigation of key miRNAs and potential mechanisms in non-small cell lung cancer development from chronic obstructive pulmonary disease. Gen. Physiol. Biophys. 2020, 39, 69–77. [Google Scholar] [CrossRef]
- Barnes, P.J. Inflammatory mechanisms in patients with chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2016, 138, 16–27. [Google Scholar] [CrossRef]
- Hogg, J.C.; Chu, F.; Utokaparch, S.; Woods, R.; Elliott, W.M.; Buzatu, L.; Cherniack, R.M.; Rogers, R.M.; Sciurba, F.C.; Coxson, H.O.; et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N. Engl. J. Med. 2004, 350, 2645–2653. [Google Scholar] [CrossRef] [PubMed]
- Tian, W.; Jiang, X.; Kim, D.; Guan, T.; Nicolls, M.R.; Rockson, S.G. Leukotrienes in Tumor-Associated Inflammation. Front Pharmacol 2020, 11, 1289. [Google Scholar] [CrossRef]
- Drakatos, P.; Lykouras, D.; Sampsonas, F.; Karkoulias, K.; Spiropoulos, K. Targeting leukotrienes for the treatment of COPD? Inflamm. Allergy Drug Targets 2009, 8, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Iacona, J.R.; Monteleone, N.J.; Lutz, C.S. miR-146a suppresses 5-lipoxygenase activating protein (FLAP) expression and Leukotriene B4 production in lung cancer cells. Oncotarget 2018, 9, 26751–26769. [Google Scholar] [CrossRef] [PubMed]
- Dong, R.; Xie, L.; Zhao, K.; Zhang, Q.; Zhou, M.; He, P. Cigarette smoke-induced lung inflammation in COPD mediated via LTB4/BLT1/SOCS1 pathway. Int. J. Chron. Obstruct. Pulmon. Dis. 2016, 11, 31–41. [Google Scholar] [CrossRef]
- Cazzola, M.; Boveri, B.; Carlucci, P.; Santus, P.; DiMarco, F.; Centanni, S.; Allegra, L. Lung function improvement in smokers suffering from COPD with zafirlukast, a CysLT(1)-receptor antagonist. Pulm. Pharmacol. Ther. 2000, 13, 301–305. [Google Scholar] [CrossRef]
- Thomsen, M.; Dahl, M.; Lange, P.; Vestbo, J.; Nordestgaard, B.G. Inflammatory biomarkers and comorbidities in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2012, 186, 982–988. [Google Scholar] [CrossRef]
- Gwilt, C.R.; Donnelly, L.E.; Rogers, D.F. The non-neuronal cholinergic system in the airways: An unappreciated regulatory role in pulmonary inflammation? Pharmacol. Ther. 2007, 115, 208–222. [Google Scholar] [CrossRef]
- Calzetta, L.; Coppola, A.; Ritondo, B.L.; Matino, M.; Chetta, A.; Rogliani, P. The Impact of Muscarinic Receptor Antagonists on Airway Inflammation: A Systematic Review. Int. J. Chron. Obstruct. Pulmon. Dis. 2021, 16, 257–279. [Google Scholar] [CrossRef]
- Szebeni, G.J.; Vizler, C.; Nagy, L.I.; Kitajka, K.; Puskas, L.G. Pro-Tumoral Inflammatory Myeloid Cells as Emerging Therapeutic Targets. Int. J. Mol. Sci. 2016, 17, 1958. [Google Scholar] [CrossRef]
- Strauss, L.; Sangaletti, S.; Consonni, F.M.; Szebeni, G.; Morlacchi, S.; Totaro, M.G.; Porta, C.; Anselmo, A.; Tartari, S.; Doni, A.; et al. RORC1 Regulates Tumor-Promoting "Emergency" Granulo-Monocytopoiesis. Cancer Cell 2015, 28, 253–269. [Google Scholar] [CrossRef]
- Chu, S.; Zhong, X.; Zhang, J.; Lao, Q.; He, Z.; Bai, J. The expression of Foxp3 and ROR gamma t in lung tissues from normal smokers and chronic obstructive pulmonary disease patients. Int. Immunopharmacol. 2011, 11, 1780–1788. [Google Scholar] [CrossRef]
- Capone, A.; Volpe, E. Transcriptional Regulators of T Helper 17 Cell Differentiation in Health and Autoimmune Diseases. Front. Immunol. 2020, 11, 348. [Google Scholar] [CrossRef] [PubMed]
- Vaguliene, N.; Zemaitis, M.; Lavinskiene, S.; Miliauskas, S.; Sakalauskas, R. Local and systemic neutrophilic inflammation in patients with lung cancer and chronic obstructive pulmonary disease. BMC Immunol. 2013, 14, 36. [Google Scholar] [CrossRef]
- Le Rouzic, O.; Pichavant, M.; Frealle, E.; Guillon, A.; Si-Tahar, M.; Gosset, P. Th17 cytokines: Novel potential therapeutic targets for COPD pathogenesis and exacerbations. Eur. Respir. J. 2017, 50. [Google Scholar] [CrossRef] [PubMed]
- Ponce-Gallegos, M.A.; Ramirez-Venegas, A.; Falfan-Valencia, R. Th17 profile in COPD exacerbations. Int. J. Chron. Obstruct. Pulmon. Dis. 2017, 12, 1857–1865. [Google Scholar] [CrossRef]
- Silva, L.E.F.; Lourenco, J.D.; Silva, K.R.; Santana, F.P.R.; Kohler, J.B.; Moreira, A.R.; Velosa, A.P.P.; Prado, C.M.; Vieira, R.P.; Aun, M.V.; et al. Th17/Treg imbalance in COPD development: Suppressors of cytokine signaling and signal transducers and activators of transcription proteins. Sci. Rep. 2020, 10, 15287. [Google Scholar] [CrossRef]
- Armstrong, D.; Chang, C.Y.; Lazarus, D.R.; Corry, D.; Kheradmand, F. Lung Cancer Heterogeneity in Modulation of Th17/IL17A Responses. Front. Oncol. 2019, 9, 1384. [Google Scholar] [CrossRef] [PubMed]
- Peng, D.H.; Rodriguez, B.L.; Diao, L.; Gaudreau, P.O.; Padhye, A.; Konen, J.M.; Ochieng, J.K.; Class, C.A.; Fradette, J.J.; Gibson, L.; et al. Th17 cells contribute to combination MEK inhibitor and anti-PD-L1 therapy resistance in KRAS/p53 mutant lung cancers. Nat. Commun. 2021, 12, 2606. [Google Scholar] [CrossRef]
- Saha, S.P.; Bhalla, D.K.; Whayne, T.F., Jr.; Gairola, C. Cigarette smoke and adverse health effects: An overview of research trends and future needs. Int. J. Angiol. 2007, 16, 77–83. [Google Scholar] [CrossRef]
- Fantozzi, A.; Gruber, D.C.; Pisarsky, L.; Heck, C.; Kunita, A.; Yilmaz, M.; Meyer-Schaller, N.; Cornille, K.; Hopfer, U.; Bentires-Alj, M.; et al. VEGF-mediated angiogenesis links EMT-induced cancer stemness to tumor initiation. Cancer Res. 2014, 74, 1566–1575. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.D.; Wright, J.L.; Churg, A. Transforming growth factor-beta1 drives airway remodeling in cigarette smoke-exposed tracheal explants. Am. J. Respir. Cell Mol. Biol. 2005, 33, 387–393. [Google Scholar] [CrossRef]
- Saito, A.; Horie, M.; Nagase, T. TGF-beta Signaling in Lung Health and Disease. Int. J. Mol. Sci. 2018, 19, 2460. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhou, F.; ten Dijke, P. Signaling interplay between transforming growth factor-beta receptor and PI3K/AKT pathways in cancer. Trends Biochem. Sci. 2013, 38, 612–620. [Google Scholar] [CrossRef]
- Huang, F.; Shi, Q.; Li, Y.; Xu, L.; Xu, C.; Chen, F.; Wang, H.; Liao, H.; Chang, Z.; Liu, F.; et al. HER2/EGFR-AKT Signaling Switches TGFbeta from Inhibiting Cell Proliferation to Promoting Cell Migration in Breast Cancer. Cancer Res. 2018, 78, 6073–6085. [Google Scholar] [CrossRef] [PubMed]
- Sohal, S.S.; Hansbro, P.M.; Walters, E.H. Epithelial Mesenchymal Transition in Chronic Obstructive Pulmonary Disease, a Precursor for Epithelial Cancers: Understanding and Translation to Early Therapy. Ann. Am. Thorac. Soc. 2017, 14, 1491–1492. [Google Scholar] [CrossRef]
- Romero-Palacios, P.J.; Alcazar-Navarrete, B.; Diaz Mochon, J.J.; de Miguel-Perez, D.; Lopez Hidalgo, J.L.; Garrido-Navas, M.D.C.; Quero Valenzuela, F.; Lorente, J.A.; Serrano, M.J. Liquid biopsy beyond of cancer: Circulating pulmonary cells as biomarkers of COPD aggressivity. Crit. Rev. Oncol. Hematol. 2019, 136, 31–36. [Google Scholar] [CrossRef]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef]
- Nishioka, M.; Venkatesan, N.; Dessalle, K.; Mogas, A.; Kyoh, S.; Lin, T.Y.; Nair, P.; Baglole, C.J.; Eidelman, D.H.; Ludwig, M.S.; et al. Fibroblast-epithelial cell interactions drive epithelial-mesenchymal transition differently in cells from normal and COPD patients. Respir. Res. 2015, 16, 72. [Google Scholar] [CrossRef]
- Courtney, J.M.; Spafford, P.L. The Role of Epithelial-Mesenchymal Transition in Chronic Obstructive Pulmonary Disease. Cells Tissues Organs 2017, 203, 99–104. [Google Scholar] [CrossRef]
- Gascard, P.; Tlsty, T.D. Carcinoma-associated fibroblasts: Orchestrating the composition of malignancy. Genes Dev. 2016, 30, 1002–1019. [Google Scholar] [CrossRef]
- Meads, M.B.; Gatenby, R.A.; Dalton, W.S. Environment-mediated drug resistance: A major contributor to minimal residual disease. Nat. Rev. Cancer 2009, 9, 665–674. [Google Scholar] [CrossRef]
- Patsouras, D.; Papaxoinis, K.; Kostakis, A.; Safioleas, M.C.; Lazaris, A.C.; Nicolopoulou-Stamati, P. Fibroblast activation protein and its prognostic significance in correlation with vascular endothelial growth factor in pancreatic adenocarcinoma. Mol. Med. Rep. 2015, 11, 4585–4590. [Google Scholar] [CrossRef]
- Yang, X.; Lin, Y.; Shi, Y.; Li, B.; Liu, W.; Yin, W.; Dang, Y.; Chu, Y.; Fan, J.; He, R. FAP Promotes Immunosuppression by Cancer-Associated Fibroblasts in the Tumor Microenvironment via STAT3-CCL2 Signaling. Cancer Res. 2016, 76, 4124–4135. [Google Scholar] [CrossRef]
- Shi, J.; Hou, Z.; Yan, J.; Qiu, W.; Liang, L.; Meng, M.; Li, L.; Wang, X.; Xie, Y.; Jiang, L.; et al. The prognostic significance of fibroblast activation protein-alpha in human lung adenocarcinoma. Ann. Transl. Med. 2020, 8, 224. [Google Scholar] [CrossRef]
- Wang, L.C.; Lo, A.; Scholler, J.; Sun, J.; Majumdar, R.S.; Kapoor, V.; Antzis, M.; Cotner, C.E.; Johnson, L.A.; Durham, A.C.; et al. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol. Res. 2014, 2, 154–166. [Google Scholar] [CrossRef]
- Loeffler, M.; Kruger, J.A.; Niethammer, A.G.; Reisfeld, R.A. Targeting tumor-associated fibroblasts improves cancer chemotherapy by increasing intratumoral drug uptake. J. Clin. Investig. 2006, 116, 1955–1962. [Google Scholar] [CrossRef]
- D’Anna, C.; Cigna, D.; Costanzo, G.; Bruno, A.; Ferraro, M.; Di Vincenzo, S.; Bianchi, L.; Bini, L.; Gjomarkaj, M.; Pace, E. Cigarette smoke alters the proteomic profile of lung fibroblasts. Mol. Biosyst. 2015, 11, 1644–1652. [Google Scholar] [CrossRef]
- Woldhuis, R.R.; Heijink, I.H.; van den Berge, M.; Timens, W.; Oliver, B.G.G.; de Vries, M.; Brandsma, C.A. COPD-derived fibroblasts secrete higher levels of senescence-associated secretory phenotype proteins. Thorax 2020. [Google Scholar] [CrossRef]
- Woldhuis, R.R.; de Vries, M.; Timens, W.; van den Berge, M.; Demaria, M.; Oliver, B.G.G.; Heijink, I.H.; Brandsma, C.A. Link between increased cellular senescence and extracellular matrix changes in COPD. Am. J. Physiol. Lung Cell Mol. Physiol. 2020, 319, L48–L60. [Google Scholar] [CrossRef]
- Charlson, E.S.; Bittinger, K.; Haas, A.R.; Fitzgerald, A.S.; Frank, I.; Yadav, A.; Bushman, F.D.; Collman, R.G. Topographical continuity of bacterial populations in the healthy human respiratory tract. Am. J. Respir. Crit. Care Med. 2011, 184, 957–963. [Google Scholar] [CrossRef]
- Mammen, M.J.; Sethi, S. COPD and the microbiome. Respirology 2016, 21, 590–599. [Google Scholar] [CrossRef]
- Wang, Z.; Bafadhel, M.; Haldar, K.; Spivak, A.; Mayhew, D.; Miller, B.E.; Tal-Singer, R.; Johnston, S.L.; Ramsheh, M.Y.; Barer, M.R.; et al. Lung microbiome dynamics in COPD exacerbations. Eur. Respir. J. 2016, 47, 1082–1092. [Google Scholar] [CrossRef]
- Wang, Z.; Maschera, B.; Lea, S.; Kolsum, U.; Michalovich, D.; Van Horn, S.; Traini, C.; Brown, J.R.; Hessel, E.M.; Singh, D. Airway host-microbiome interactions in chronic obstructive pulmonary disease. Respir. Res. 2019, 20, 113. [Google Scholar] [CrossRef]
- Bowerman, K.L.; Rehman, S.F.; Vaughan, A.; Lachner, N.; Budden, K.F.; Kim, R.Y.; Wood, D.L.A.; Gellatly, S.L.; Shukla, S.D.; Wood, L.G.; et al. Disease-associated gut microbiome and metabolome changes in patients with chronic obstructive pulmonary disease. Nat. Commun. 2020, 11, 5886. [Google Scholar] [CrossRef]
- Liu, N.N.; Ma, Q.; Ge, Y.; Yi, C.X.; Wei, L.Q.; Tan, J.C.; Chu, Q.; Li, J.Q.; Zhang, P.; Wang, H. Microbiome dysbiosis in lung cancer: From composition to therapy. NPJ Precis. Oncol. 2020, 4, 33. [Google Scholar] [CrossRef]
- Ramirez-Labrada, A.G.; Isla, D.; Artal, A.; Arias, M.; Rezusta, A.; Pardo, J.; Galvez, E.M. The Influence of Lung Microbiota on Lung Carcinogenesis, Immunity, and Immunotherapy. Trends Cancer 2020, 6, 86–97. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szalontai, K.; Gémes, N.; Furák, J.; Varga, T.; Neuperger, P.; Balog, J.Á.; Puskás, L.G.; Szebeni, G.J. Chronic Obstructive Pulmonary Disease: Epidemiology, Biomarkers, and Paving the Way to Lung Cancer. J. Clin. Med. 2021, 10, 2889. https://doi.org/10.3390/jcm10132889
Szalontai K, Gémes N, Furák J, Varga T, Neuperger P, Balog JÁ, Puskás LG, Szebeni GJ. Chronic Obstructive Pulmonary Disease: Epidemiology, Biomarkers, and Paving the Way to Lung Cancer. Journal of Clinical Medicine. 2021; 10(13):2889. https://doi.org/10.3390/jcm10132889
Chicago/Turabian StyleSzalontai, Klára, Nikolett Gémes, József Furák, Tünde Varga, Patrícia Neuperger, József Á. Balog, László G. Puskás, and Gábor J. Szebeni. 2021. "Chronic Obstructive Pulmonary Disease: Epidemiology, Biomarkers, and Paving the Way to Lung Cancer" Journal of Clinical Medicine 10, no. 13: 2889. https://doi.org/10.3390/jcm10132889
APA StyleSzalontai, K., Gémes, N., Furák, J., Varga, T., Neuperger, P., Balog, J. Á., Puskás, L. G., & Szebeni, G. J. (2021). Chronic Obstructive Pulmonary Disease: Epidemiology, Biomarkers, and Paving the Way to Lung Cancer. Journal of Clinical Medicine, 10(13), 2889. https://doi.org/10.3390/jcm10132889