
Targeting the Complement Cascade in the Pathophysiology of COVID-19 Disease


Abstract
:1. Introduction
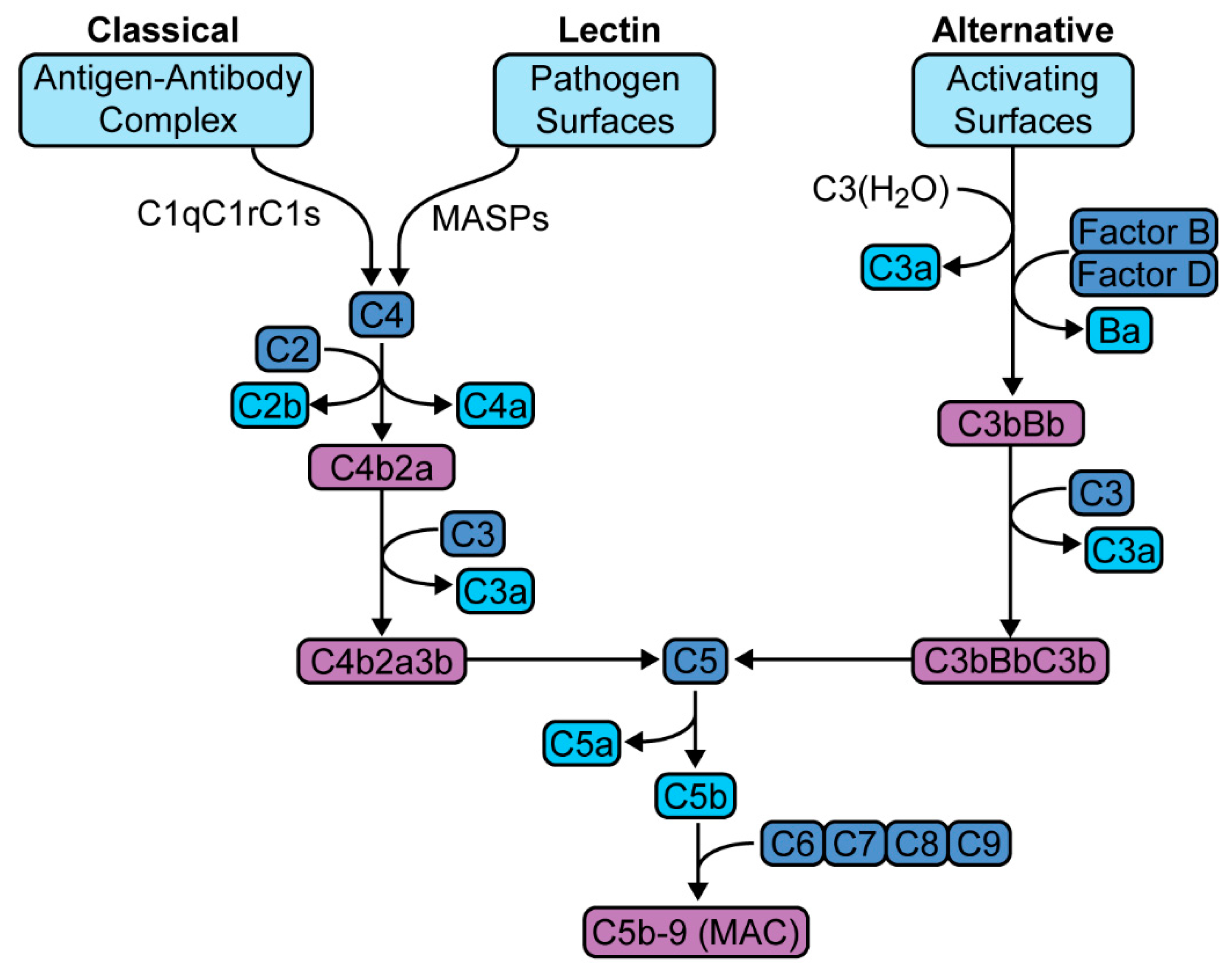
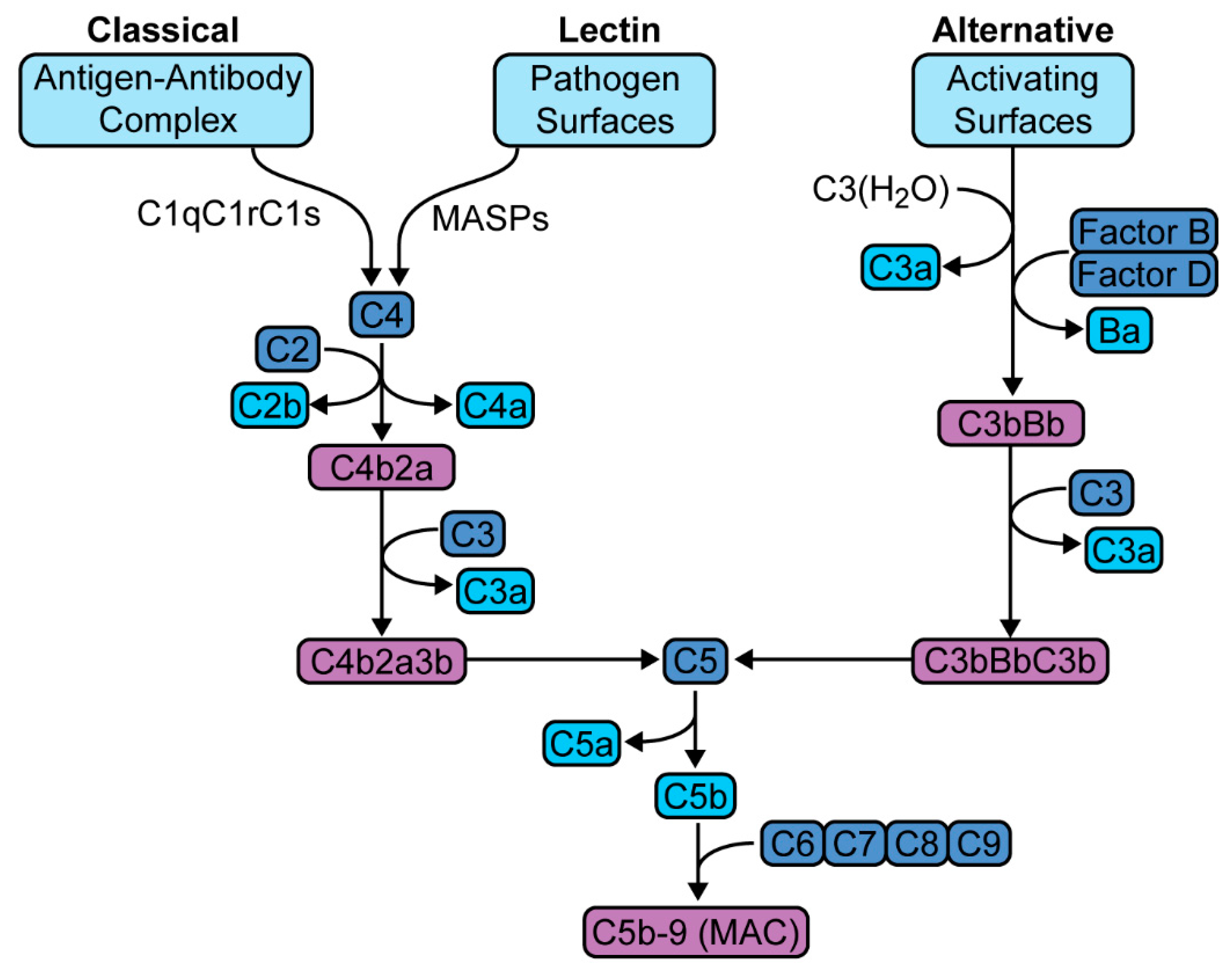
2. The Complement Cascade
3. Methodology and Literature Search
4. Complement in SARS-CoV-1, MERS-CoV
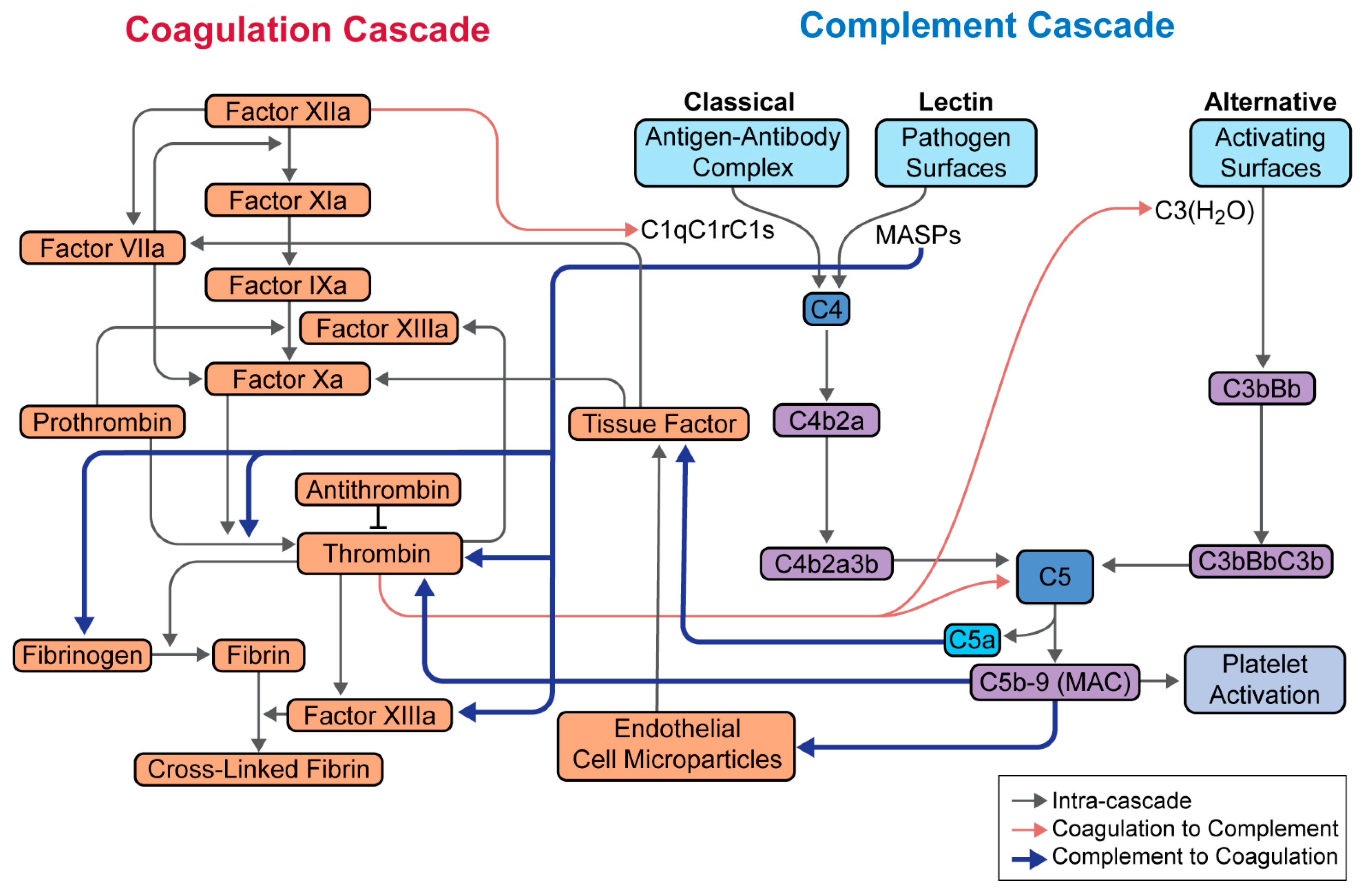
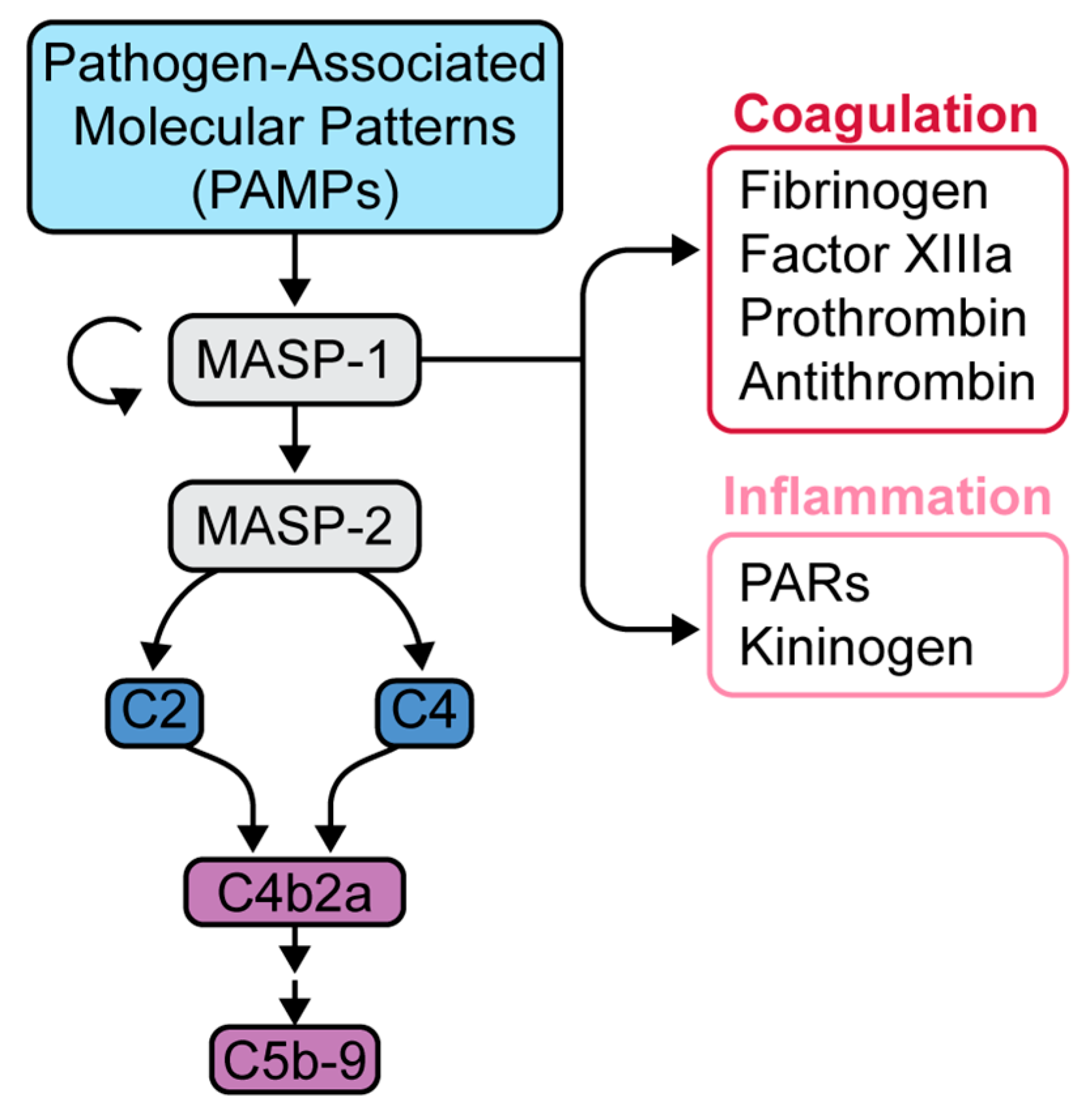
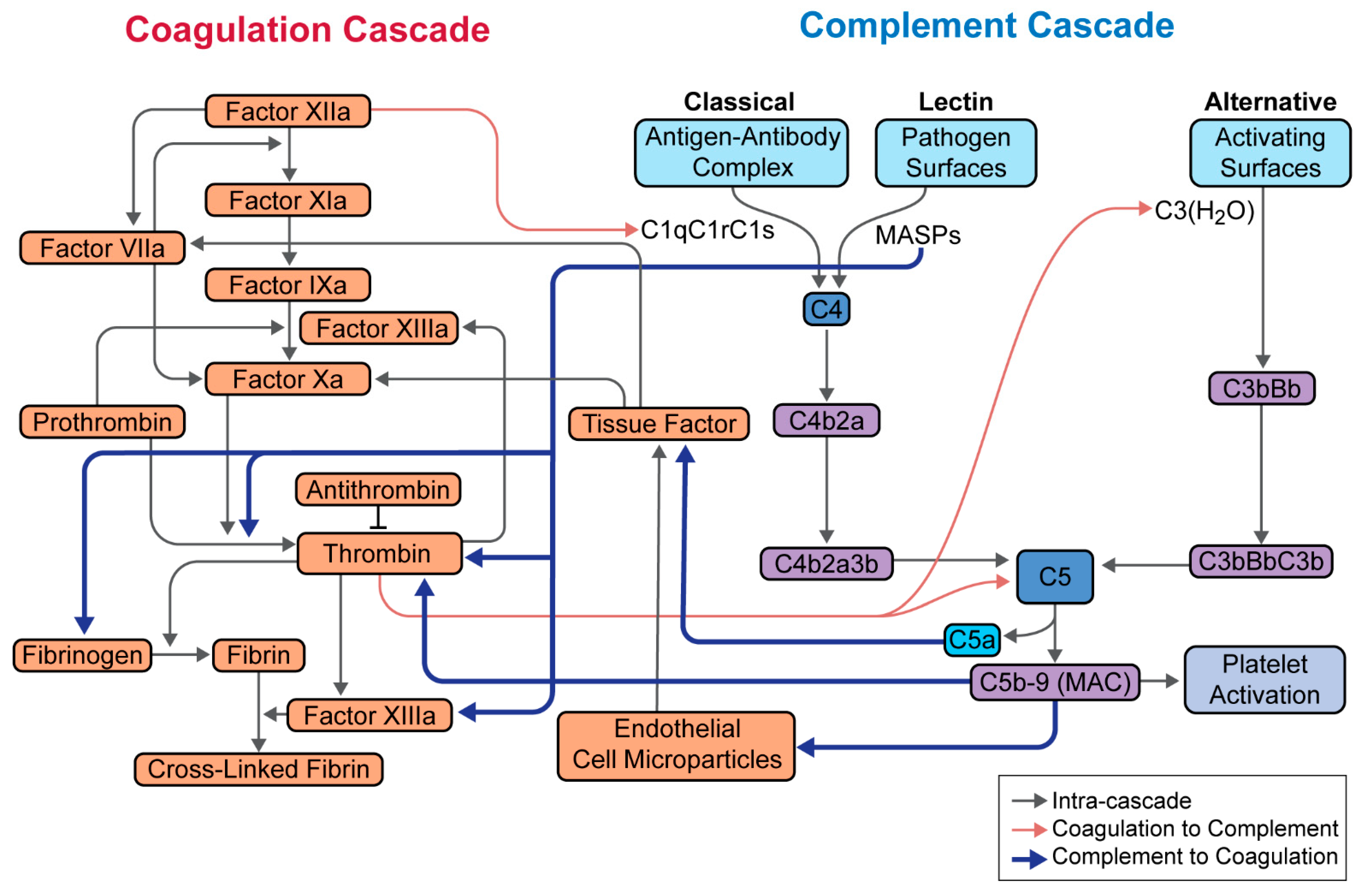
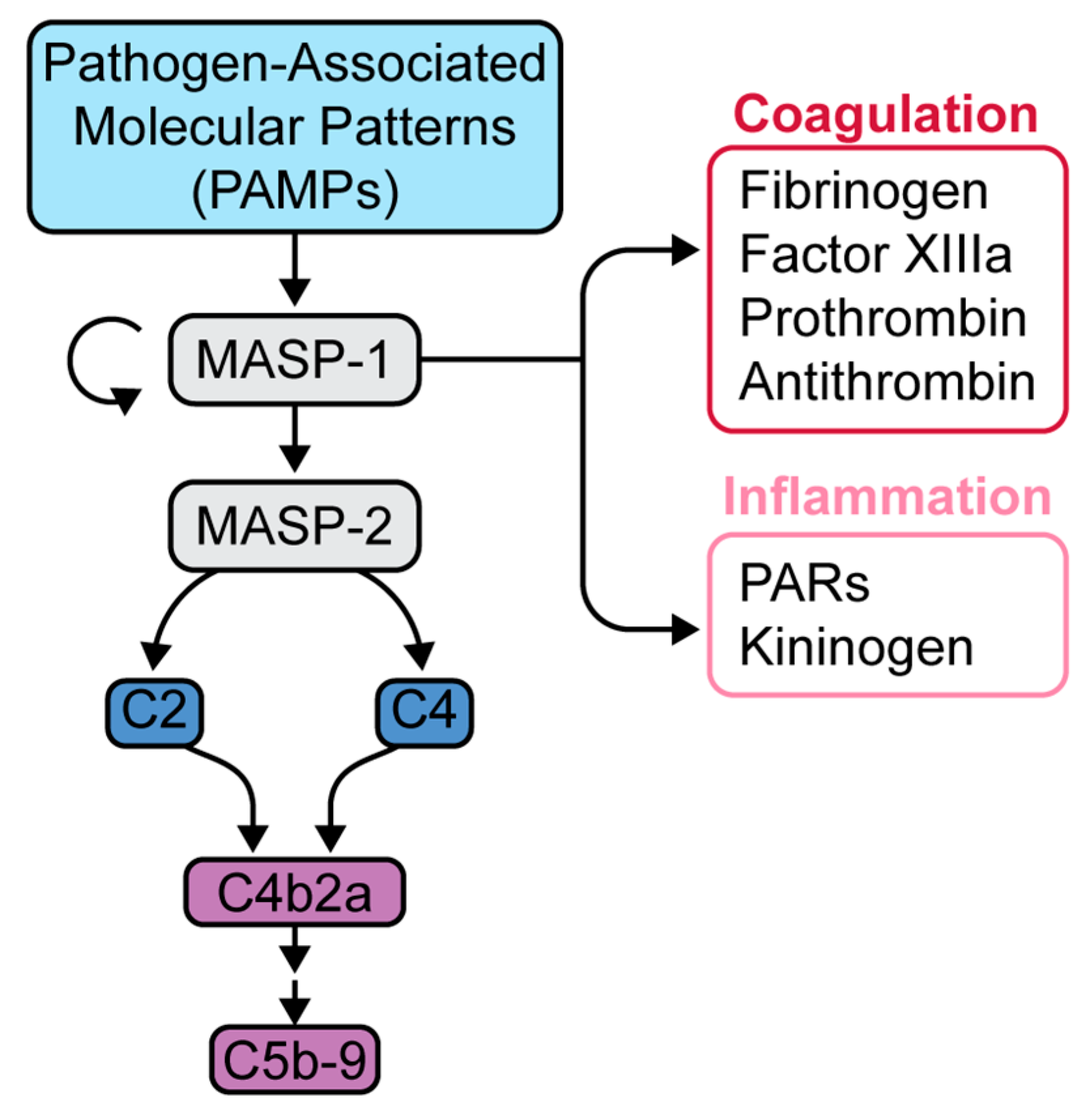
5. Complement in SARS-CoV-2
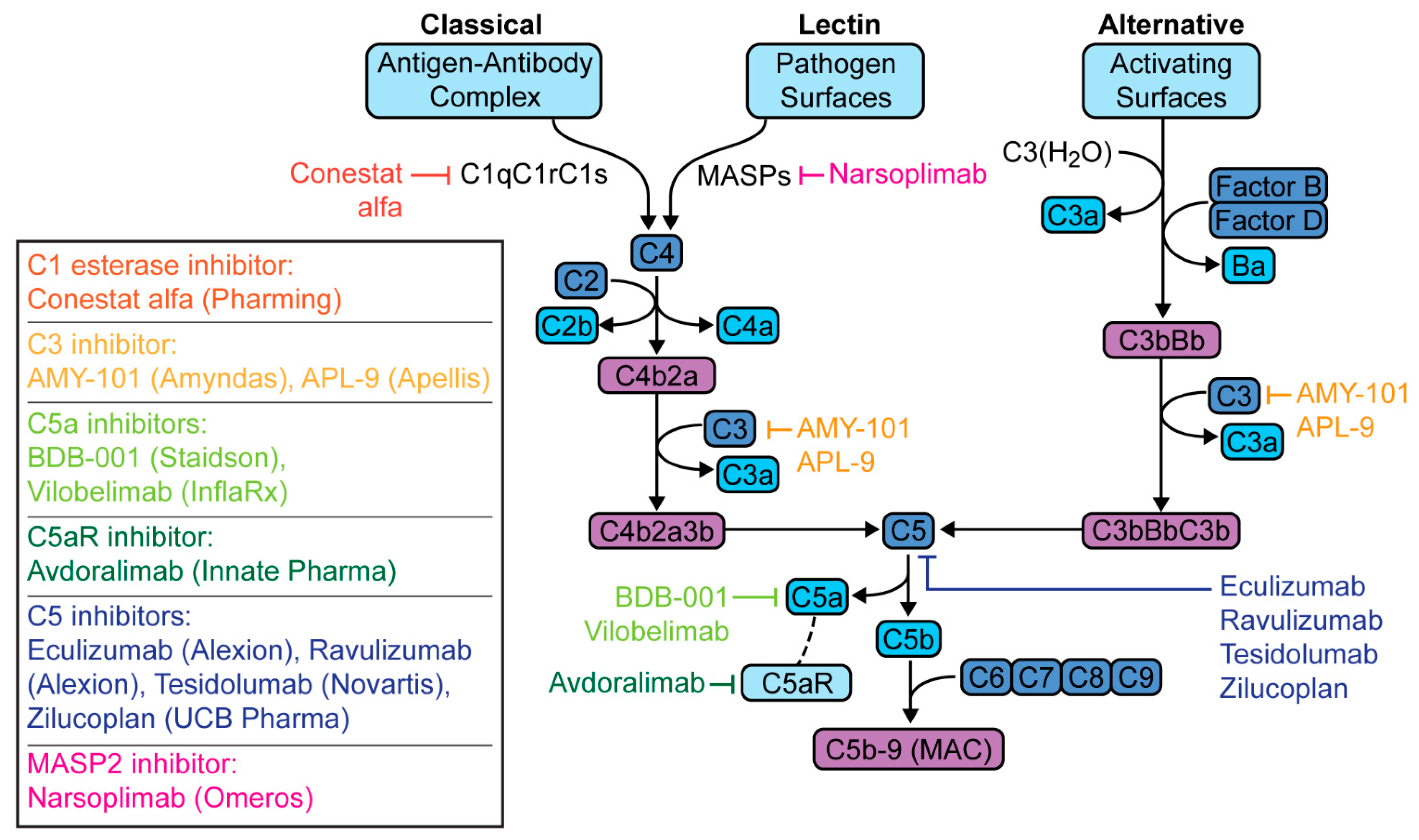
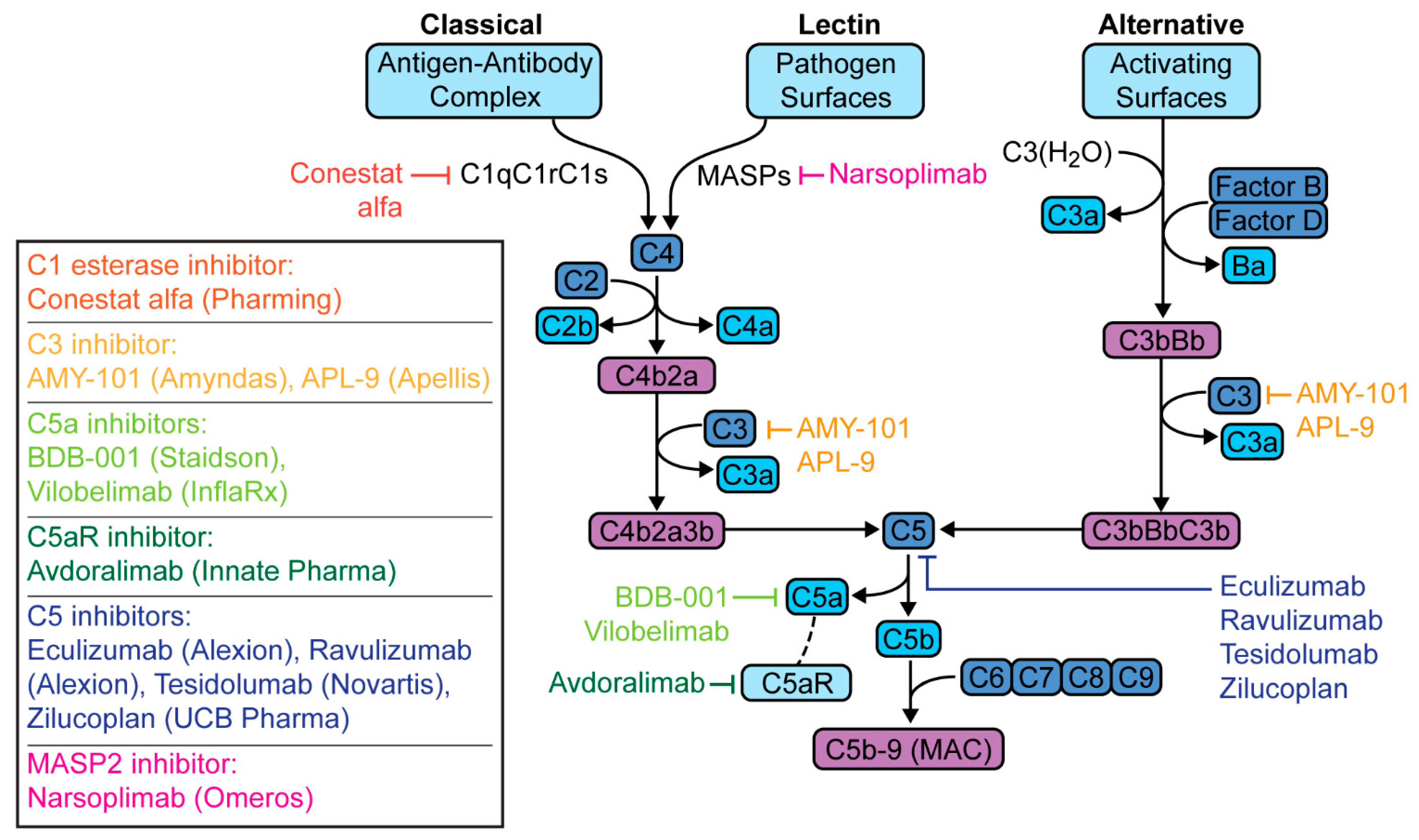
6. Targeted Complement Therapies in COVID-19 Pneumonia
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chaplin, H. Review: The burgeoning history of the complement system 1888–2005. Immunohematology 2005, 21, 85–93. [Google Scholar]
- Ghebrehiwet, B. The complement system: An evolution in progress. F1000Research 2016, 5, 2840. [Google Scholar] [CrossRef] [Green Version]
- Tegla, C.A.; Cudrici, C.; Patel, S.; Trippe, R.; Rus, V.; Niculescu, F.; Rus, H. Membrane attack by complement: The assembly and biology of terminal complement complexes. Immunol. Res. 2011, 51, 45–60. [Google Scholar] [CrossRef] [Green Version]
- Dunkelberger, J.R.; Song, W.-C. Complement and its role in innate and adaptive immune responses. Cell Res. 2010, 20, 34–50. [Google Scholar] [CrossRef] [Green Version]
- Stoermer, K.A.; Morrison, T.E. Complement and viral pathogenesis. Virology 2011, 411, 362–373. [Google Scholar] [CrossRef] [Green Version]
- Agrawal, P.; Nawadkar, R.; Ojha, H.; Kumar, J.; Sahu, A. Complement Evasion Strategies of Viruses: An Overview. Front. Microbiol. 2017, 8, 1117. [Google Scholar] [CrossRef] [Green Version]
- Bernet, J.; Mullick, J.; Singh, A.K.; Sahu, A. Viral mimicry of the complement system. J. Biosci. 2003, 28, 249–264. [Google Scholar] [CrossRef]
- Zhang, J.; Li, G.; Liu, X.; Wang, Z.; Liu, W.; Ye, X. Influenza A virus M1 blocks the classical complement pathway through interacting with C1qA. J. Gen. Virol. 2009, 90, 2751–2758. [Google Scholar] [CrossRef]
- Liszewski, M.K.; Bertram, P.; Leung, M.K.; Hauhart, R.; Zhang, L.; Atkinson, J.P. Smallpox Inhibitor of Complement Enzymes (SPICE): Regulation of Complement Activation on Cells and Mechanism of Its Cellular Attachment. J. Immunol. 2008, 181, 4199–4207. [Google Scholar] [CrossRef] [Green Version]
- Cedzyński, M.; Thielens, N.M.; Mollnes, T.E.; Vorup-Jensen, T. Editorial: The Role of Complement in Health and Disease. Front. Immunol. 2019, 10, 1869. [Google Scholar] [CrossRef]
- Gralinski, L.E.; Sheahan, T.P.; Morrison, T.E.; Menachery, V.D.; Jensen, K.; Leist, S.R.; Whitmore, A.; Heise, M.T.; Baric, R.S. Complement Activation Contributes to Severe Acute Respiratory Syndrome Coronavirus Pathogenesis. mBio 2018, 9, e01753-18. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Zhao, G.; Song, N.; Li, P.; Chen, Y.; Guo, Y.; Li, J.; Du, L.; Jiang, S.; Guo, R.; et al. Blockade of the C5a-C5aR axis alleviates lung damage in hDPP4-transgenic mice infected with MERS-CoV. Emerg. Microbes Infect. 2018, 7, 77. [Google Scholar] [CrossRef] [Green Version]
- Jaimes, J.A.; André, N.M.; Chappie, J.S.; Millet, J.K.; Whittaker, G.R. Phylogenetic Analysis and Structural Modeling of SARS-CoV-2 Spike Protein Reveals an Evolutionary Distinct and Proteolytically Sensitive Activation Loop. J. Mol. Biol. 2020, 432, 3309–3325. [Google Scholar] [CrossRef]
- WHO. Consensus Document on the Epidemiology of Severe Acute Respiratory Syndrome (SARS). Available online: https://www.who.int/csr/sars/en/WHOconsensus.pdf (accessed on 14 April 2021).
- WHO. Middle East Respiratory Syndrome Coronavirus (MERS-CoV). Available online: https://www.who.int/emergencies/mers-cov/en/ (accessed on 14 April 2021).
- WHO. Coronavirus Disease (COVID-19) Dashboard. Available online: https://covid19.who.int/?gclid=EAIaIQobChMIxemjxKPw6gIV1-DICh2EEwenEAAYASAAEgJvPPD_BwE (accessed on 14 April 2021).
- Poor, H.D.; Ventetuolo, C.E.; Tolbert, T.; Chun, G.; Serrao, G.; Zeidman, A.; Dangayach, N.S.; Olin, J.; Kohli-Seth, R.; Powell, C.A. COVID-19 critical illness pathophysiology driven by diffuse pulmonary thrombi and pulmonary endothelial dysfunction responsive to thrombolysis. Clin. Transl. Med. 2020, 10, 44. [Google Scholar] [CrossRef]
- Reynolds, A.S.; Lee, A.G.; Renz, J.; DeSantis, K.; Liang, J.; Powell, C.A.; Ventetuolo, C.E.; Poor, H.D. Pulmonary Vascular Dilatation Detected by Automated Transcranial Doppler in COVID-19 Pneumonia. Am. J. Respir. Crit. Care Med. 2020, 202, 1037–1039. [Google Scholar] [CrossRef]
- Risitano, A.M.; Mastellos, D.C.; Huber-Lang, M.; Yancopoulou, D.; Garlanda, C.; Ciceri, F.; Lambris, J.D. Complement as a target in COVID-19? Nat. Rev. Immunol. 2020, 20, 343–344. [Google Scholar] [CrossRef] [Green Version]
- Magro, C.; Mulvey, J.J.; Berlin, D.; Nuovo, G.; Salvatore, S.; Harp, J.; Baxter-Stoltzfus, A.; Laurence, J. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: A report of five cases. Transl. Res. 2020, 220, 1–13. [Google Scholar] [CrossRef]
- Chauhan, A.J.; Wiffen, L.J.; Brown, T.P. COVID-19: A collision of complement, coagulation and inflammatory pathways. J. Thromb. Haemost. 2020, 18, 2110–2117. [Google Scholar] [CrossRef]
- Ritis, K.; Doumas, M.; Mastellos, D.; Micheli, A.; Giaglis, S.; Magotti, P.; Rafail, S.; Kartalis, G.; Sideras, P.; Lambris, J.D. A Novel C5a Receptor-Tissue Factor Cross-Talk in Neutrophils Links Innate Immunity to Coagulation Pathways. J. Immunol. 2006, 177, 4794–4802. [Google Scholar] [CrossRef]
- Hill, A.; Kelly, R.J.; Hillmen, P. Thrombosis in paroxysmal nocturnal hemoglobinuria. Blood 2013, 121, 4985–4996. [Google Scholar] [CrossRef] [Green Version]
- Dobó, J.; Schroeder, V.; Jenny, L.; Cervenak, L.; Závodszky, P.; Gál, P. Multiple roles of complement MASP-1 at the interface of innate immune response and coagulation. Mol. Immunol. 2014, 61, 69–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobó, J.; Pál, G.; Cervenak, L.; Gál, P. The emerging roles of mannose-binding lectin-associated serine proteases (MASPs) in the lectin pathway of complement and beyond. Immunol. Rev. 2016, 274, 98–111. [Google Scholar] [CrossRef] [Green Version]
- Elhadad, S.; Chapin, J.; Copertino, D.; Van Besien, K.; Ahamed, J.; Laurence, J. MASP2 levels are elevated in thrombotic microangiopathies: Association with microvascular endothelial cell injury and suppression by anti-MASP2 antibody narsoplimab. Clin. Exp. Immunol. 2021, 203, 96–104. [Google Scholar] [CrossRef]
- Yu, J.; Yuan, X.; Chen, H.; Chaturvedi, S.; Braunstein, E.M.; Brodsky, R.A. Direct activation of the alternative complement pathway by SARS-CoV-2 spike proteins is blocked by factor D inhibition. Blood 2020, 136, 2080–2089. [Google Scholar] [CrossRef]
- Gao, T.; Hu, M.; Zhang, X.; Li, H.; Zhu, L.; Liu, H.; Dong, Q.; Zhang, Z.; Wang, Z.; Hu, Y.; et al. Highly pathogenic coronavirus N protein aggravates lung injury by MASP-2-mediated complement over-activation. medRxiv 2020. [Google Scholar] [CrossRef]
- Carvelli, J.; Demaria, O.; Vély, F.; Batista, L.; Chouaki Benmansour, N.; Fares, J.; Carpentier, S.; Thibult, M.-L.; Morel, A.; Remark, R.; et al. Association of COVID-19 inflammation with activation of the C5a–C5aR1 axis. Nature 2020, 588, 146–150. [Google Scholar] [CrossRef]
- Shen, B.; Yi, X.; Sun, Y.; Bi, X.; Du, J.; Zhang, C.; Quan, S.; Zhang, F.; Sun, R.; Qian, L.; et al. Proteomic and Metabolomic Characterization of COVID-19 Patient Sera. SSRN Electron. J. 2020, 182, 59–75. [Google Scholar]
- Messner, C.B.; Demichev, V.; Wendisch, D.; Michalick, L.; White, M.; Freiwald, A.; Textoris-Taube, K.; Vernardis, S.I.; Egger, A.-S.; Kreidl, M.; et al. Ultra-High-Throughput Clinical Proteomics Reveals Classifiers of COVID-19 Infection. Cell Syst. 2020, 11, 11–24. [Google Scholar] [CrossRef]
- D’Alessandro, A.; Thomas, T.; Dzieciatkowska, M.; Hill, R.C.; Francis, R.O.; Hudson, K.E.; Zimring, J.C.; Hod, E.A.; Spitalnik, S.L.; Hansen, K.C. Serum Proteomics in COVID-19 Patients: Altered Coagulation and Complement Status as a Function of IL-6 Level. J. Proteome Res. 2020, 19, 4417–4427. [Google Scholar] [CrossRef]
- Pfister, F.; Vonbrunn, E.; Ries, T.; Jäck, H.-M.; Überla, K.; Lochnit, G.; Sheriff, A.; Herrmann, M.; Büttner-Herold, M.; Amann, K.; et al. Complement Activation in Kidneys of Patients With COVID-19. Front. Immunol. 2021, 11, 594849. [Google Scholar] [CrossRef] [PubMed]
- Rendeiro, A.F.; Ravichandran, H.; Bram, Y.; Chandar, V.; Kim, J.; Meydan, C.; Park, J.; Foox, J.; Hether, T.; Warren, S.; et al. The spatial landscape of lung pathology during COVID-19 progression. Nature 2021. [Google Scholar] [CrossRef] [PubMed]
- Holter, J.C.; Pischke, S.E.; de Boer, E.; Lind, A.; Jenum, S.; Holten, A.R.; Tonby, K.; Barratt-Due, A.; Sokolova, M.; Schjalm, C.; et al. Systemic complement activation is associated with respiratory failure in COVID-19 hospitalized patients. Proc. Nat. Acad. Sci. USA 2020, 117, 25018–25025. [Google Scholar] [CrossRef]
- Cugno, M.; Meroni, P.L.; Gualtierotti, R.; Griffini, S.; Grovetti, E.; Torri, A.; Lonati, P.; Grossi, C.; Borghi, M.O.; Novembrino, C.; et al. Complement activation and endothelial perturbation parallel COVID-19 severity and activity. J. Autoimmun. 2020, 116, 102560. [Google Scholar] [CrossRef] [PubMed]
- De Nooijer, A.H.; Grondman, I.; Janssen, N.A.F.; Netea, M.G.; Willems, L.; van de Veerdonk, F.L.; Giamarellos-Bourboulis, E.J.; Toonen, E.J.M.; Joosten, L.A.B.; RCI-COVID-19 study group. Complement Activation in the Disease Course of Coronavirus Disease 2019 and Its Effects on Clinical Outcomes. J. Infect. Dis. 2021, 223, 214–224. [Google Scholar] [CrossRef] [PubMed]
- Peffault de Latour, R.; Bergeron, A.; Lengline, E.; Dupont, T.; Marchal, A.; Galicier, L.; de Castro, N.; Bondeelle, L.; Darmon, M.; Dupin, C.; et al. Complement C5 inhibition in patients with COVID-19—A promising target? Haematologica 2020, 105, 2847–2850. [Google Scholar] [CrossRef]
- Java, A.; Apicelli, A.J.; Liszewski, M.K.; Coler-Reilly, A.; Atkinson, J.P.; Kim, A.H.; Kulkarni, H.S. The complement system in COVID-19: Friend and foe? JCI Insight 2020, 5. [Google Scholar] [CrossRef]
- Allegra, A.; Innao, V.; Allegra, A.G.; Musolino, C. Coagulopathy and thromboembolic events in patients with SARS-CoV-2 infection: Pathogenesis and management strategies. Ann. Hematol. 2020, 99, 1953–1965. [Google Scholar] [CrossRef]
- Hanff, T.C.; Mohareb, A.M.; Giri, J.; Cohen, J.B.; Chirinos, J.A. Thrombosis in COVID-19. Am. J. Hematol. 2020, 95, 1578–1589. [Google Scholar] [CrossRef]
- Fletcher-Sandersjöö, A.; Bellander, B.-M. Is COVID-19 associated thrombosis caused by overactivation of the complement cascade? A literature review. Thromb. Res. 2020, 194, 36–41. [Google Scholar] [CrossRef]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef]
- Bryce, C.; Grimes, Z.; Pujadas, E.; Ahuja, S.; Beasley, M.B.; Albrecht, R.; Hernandez, T.; Stock, A.; Zhao, Z.; Al Rasheed, M.; et al. Pathophysiology of SARS-CoV-2: Targeting of endothelial cells renders a complex disease with thrombotic microangiopathy and aberrant immune response. The Mount Sinai COVID-19 autopsy experience. medRxiv 2020. [Google Scholar] [CrossRef]
- Jayarangaiah, A.; Kariyanna, P.T.; Chen, X.; Jayarangaiah, A.; Kumar, A. COVID-19-Associated Coagulopathy: An Exacerbated Immunothrombosis Response. Clin. Appl. Thromb. 2020, 26. [Google Scholar] [CrossRef] [PubMed]
- Perico, L.; Benigni, A.; Casiraghi, F.; Ng, L.F.P.; Renia, L.; Remuzzi, G. Immunity, endothelial injury and complement-induced coagulopathy in COVID-19. Nat. Rev. Nephrol. 2021, 17, 46–64. [Google Scholar] [CrossRef]
- Conway, E.M.; Pryzdial, E.L.G. Is the COVID-19 thrombotic catastrophe complement-connected? J. Thromb. Hemost. 2020, 18, 2812–2822. [Google Scholar] [CrossRef] [PubMed]
- Manolis, A.S.; Manolis, T.A.; Manolis, A.A.; Papatheou, D.; Melita, H. COVID-19 Infection: Viral Macro- and Micro-Vascular Coagulopathy and Thromboembolism/Prophylactic and Therapeutic Management. J. Cardiovasc. Pharmacol. Ther. 2021, 26, 12–24. [Google Scholar] [CrossRef]
- DiNicolantonio, J.J.; Mccarty, M. Thrombotic complications of COVID-19 may reflect an upregulation of endothelial tissue factor expression that is contingent on activation of endosomal NADPH oxidase. Open Heart 2020, 7, e001337. [Google Scholar] [CrossRef]
- Rosell, A.; Havervall, S.; von Meijenfeldt, F.; Hisada, Y.; Aguilera, K.; Grover, S.P.; Lisman, T.; Mackman, N.; Thålin, C. Patients with COVID-19 Have Elevated Levels of Circulating Extracellular Vesicle Tissue Factor Activity That is Associated with Severity and Mortality—Brief Report. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 878–882. [Google Scholar] [CrossRef] [PubMed]
- Gavriilaki, E.; Anyfanti, P.; Gavriilaki, M.; Lazaridis, A.; Douma, S.; Gkaliagkousi, E. Endothelial Dysfunction in COVID-19: Lessons Learned from Coronaviruses. Curr. Hypertens. Rep. 2020, 22, 1–12. [Google Scholar] [CrossRef] [PubMed]
- McFadyen, J.D.; Stevens, H.; Peter, K. The Emerging Threat of (Micro)Thrombosis in COVID-19 and Its Therapeutic Implications. Circ. Res. 2020, 127, 571–587. [Google Scholar] [CrossRef]
- Varghese, P.M.; Tsolaki, A.G.; Yasmin, H.; Shastri, A.; Ferluga, J.; Vatish, M.; Madan, T.; Kishore, U. Host-pathogen interaction in COVID-19: Pathogenesis, potential therapeutics and vaccination strategies. Immunobiology 2020, 225, 152008. [Google Scholar] [CrossRef]
- Chau, A.S.; Weber, A.G.; Maria, N.I.; Narain, S.; Liu, A.; Hajizadeh, N.; Malhotra, P.; Bloom, O.; Marder, G.; Kaplan, B. The Longitudinal Immune Response to Coronavirus Disease 2019: Chasing the Cytokine Storm. Arthritis Rheumatol. 2021, 73, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Lippi, G.; Sanchis-Gomar, F.; Henry, B.M. COVID-19: Unravelling the clinical progression of nature’s virtually perfect biological weapon. Ann. Transl. Med. 2020, 8, 693. [Google Scholar] [CrossRef]
- Urwyler, P.; Moser, S.; Charitos, P.; Heijnen, I.A.F.M.; Rudin, M.; Sommer, G.; Giannetti, B.M.; Bassetti, S.; Sendi, P.; Trendelenburg, M.; et al. Treatment of COVID-19 With Conestat Alfa, a Regulator of the Complement, Contact Activation and Kallikrein-Kinin System. Front. Immunol. 2020, 11, 2072. [Google Scholar] [CrossRef]
- Conestat Alfa in the Prevention of Severe SARS-CoV-2 Infection in Hospitalized Patients with COVID-19. Available online: https://clinicaltrials.gov/ct2/show/NCT04414631 (accessed on 14 April 2021).
- Prevention of Severe SARS-CoV-2 Infection in Hospitalized Patients with COVID-19. Available online: https://clinicaltrials.gov/ct2/show/NCT04530136 (accessed on 14 April 2021).
- Mastaglio, S.; Ruggeri, A.; Risitano, A.M.; Angelillo, P.; Yancopoulou, D.; Mastellos, D.C.; Huber-Lang, M.; Piemontese, S.; Assanelli, A.; Garlanda, C.; et al. The first case of COVID-19 treated with the complement C3 inhibitor AMY-101. Clin. Immunol. 2020, 215, 108450. [Google Scholar] [CrossRef] [PubMed]
- A Study of the C3 Inhibitor AMY-101 in Patients with ARDS Due to COVID-19 (SAVE). Available online: https://clinicaltrials.gov/ct2/show/NCT04395456 (accessed on 14 April 2021).
- A Study of APL-9 in Adults with Mild to Moderate ARDS Due to COVID-19. Available online: https://clinicaltrials.gov/ct2/show/NCT04402060 (accessed on 14 April 2021).
- Efficacy and Safety Study of BDB-001 in Severe COVID-19 with ALI/ARDS. Available online: https://clinicaltrials.gov/ct2/show/NCT04449588 (accessed on 14 April 2021).
- Vlaar, A.P.J.; de Bruin, S.; Busch, M.; Timmermans, S.A.M.E.G.; van Zeggeren, I.E.; Koning, R.; ter Horst, L.; Bulle, E.B.; van Baarle, F.E.H.P.; van de Poll, M.C.G.; et al. Anti-C5a antibody IFX-1 (vilobelimab) treatment versus best supportive care for patients with severe COVID-19 (PANAMO): An exploratory, open-label, phase 2 randomised controlled trial. Lancet Rheumatol. 2020, 2, e764–e773. [Google Scholar] [CrossRef]
- Randomized, Controlled Study of IFX-1 in Patients with Severe COVID-19 Pneumonia (PANAMO). Available online: https://clinicaltrials.gov/ct2/show/NCT04333420 (accessed on 14 April 2021).
- Avdoralimab an Anti-C5aR Antibody, in Patients with COVID-19 Severe Pneumonia (FORCE). Available online: https://www.clinicaltrials.gov/ct2/show/NCT04371367 (accessed on 14 April 2021).
- Annane, D.; Heming, N.; Grimaldi-Bensouda, L.; Frémeaux-Bacchi, V.; Vigan, M.; Roux, A.-L.; Marchal, A.; Michelon, H.; Rottman, M.; Moine, P. Eculizumab as an emergency treatment for adult patients with severe COVID-19 in the intensive care unit: A proof-of-concept study. EClinicalMedicine 2020, 28, 100590. [Google Scholar] [CrossRef]
- Diurno, F.; Numis, F.G.; Porta, G.; Cirillo, F.; Maddaluno, S.; Ragozzino, A.; De Negri, P.; Di Gennaro, C.; Pagano, A.; Allegorico, E.; et al. Eculizumab treatment in patients with COVID-19: Preliminary results from real life ASL Napoli 2 Nord experience. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 4040–4047. [Google Scholar]
- Laurence, J.; Mulvey, J.J.; Seshadri, M.; Racanelli, A.; Harp, J.; Schenck, E.J.; Zappetti, D.; Horn, E.M.; Magro, C.M. Anti-complement C5 therapy with eculizumab in three cases of critical COVID-19. Clin. Immunol. 2020, 219, 108555. [Google Scholar] [CrossRef] [PubMed]
- CORIMUNO19-ECU: Trial Evaluating Efficacy and Safety of Eculizumab (Soliris) in Patients with COVID-19 Infection, Nested in the CORIMUNO-19 Cohort (CORIMUNO19-ECU). Available online: https://www.clinicaltrials.gov/ct2/show/NCT04346797?term=complement&cond=Covid19&draw=3 (accessed on 14 April 2021).
- Eculizumab (Soliris) in Covid-19 Infected Patients (SOLID-C19). Available online: https://www.clinicaltrials.gov/ct2/show/NCT04288713?term=complement&cond=Covid19&draw=2&rank=7 (accessed on 14 April 2021).
- Soliris (Eculizumab) Treatment of Patients with COVID-19. Available online: https://clinicaltrials.gov/ct2/show/NCT04355494 (accessed on 14 April 2021).
- Efficacy and Safety Study of IV Ravulizumab in Patients with COVID-19 Severe Pneumonia. Available online: https://clinicaltrials.gov/ct2/show/NCT04369469 (accessed on 14 April 2021).
- Ravulizumab and COVID-19. Available online: https://clinicaltrials.gov/ct2/show/NCT04570397 (accessed on 14 April 2021).
- Multi-Arm Therapeutic Study in Pre-ICU Patients Admitted with COVID-19—Repurposed Drugs (TACTIC-R). Available online: https://clinicaltrials.gov/ct2/show/NCT04390464 (accessed on 14 April 2021).
- Zelek, W.M.; Cole, J.; Ponsford, M.J.; Harrison, R.A.; Schroeder, B.E.; Webb, N.; Jolles, S.; Fegan, C.; Morgan, M.; Wise, M.P.; et al. Complement Inhibition with the C5 Blocker LFG316 in Severe COVID-19. Am. J. Respir. Crit. Care Med. 2020, 202, 1304–1308. [Google Scholar] [CrossRef]
- Zilucoplan in Improving Oxygenation and Short- and Long-Term Outcome of COVID-19 Patients with Acute Hypoxic Respiratory Failure. Available online: https://clinicaltrials.gov/ct2/show/NCT04382755 (accessed on 14 April 2021).
- Rambaldi, A.; Gritti, G.; Micò, M.C.; Frigeni, M.; Borleri, G.; Salvi, A.; Landi, F.; Pavoni, C.; Sonzogni, A.; Gianatti, A.; et al. Endothelial injury and thrombotic microangiopathy in COVID-19: Treatment with the lectin-pathway inhibitor narsoplimab. Immunobiology 2020, 225, 152001. [Google Scholar] [CrossRef]
- I-SPY COVID-19 TRIAL: An Adaptive Platform Trial for Critically Ill Patients (I-SPY_COVID). Available online: https://www.clinicaltrials.gov/ct2/show/NCT04488081?term=complement&cond=Covid19&draw=3&rank=44#contacts (accessed on 14 April 2021).
- Atkinson, J.P.; Du Clos, T.W.; Mold, C.; Kulkarni, H.; Hourcade, D.; Wu, X. 21—The Human Complement System: Basic Concepts and Clinical Relevance. In Clinical Immunology; Elsevier: London, UK, 2019; pp. 299–317. [Google Scholar]
- Skidgel, R.A.; Erdös, E.G.; Deddish, P.A. Kininases. In Encyclopedia of Endocrine Diseases; Elsevier BV: New York, NY, USA, 2004; pp. 146–153. [Google Scholar]
- Van de Veerdonk, F.L.; Netea, M.G.; van Deuren, M.; van der Meer, J.W.; de Mast, Q.; Brüggemann, R.J.; van der Hoeven, H. Kallikrein-kinin blockade in patients with COVID-19 to prevent acute respiratory distress syndrome. eLife 2020, 9, e57555. [Google Scholar] [CrossRef]
- Mastellos, D.C.; Pires da Silva, B.G.P.; Fonseca, B.A.L.; Fonseca, N.P.; Auxiliadora-Martins, M.; Mastaglio, S.; Ruggeri, A.; Sironi, M.; Radermacher, P.; Chrysanthopoulou, A.; et al. Complement C3 vs C5 inhibition in severe COVID-19: Early clinical findings reveal differential biological efficacy. Clin. Immunol. 2020, 220, 108598. [Google Scholar] [CrossRef]
- Mishra, K.P.; Singh, A.K.; Singh, S.B. Hyperinflammation and Immune Response Generation in COVID-19. Neuroimmunomodulation 2020, 27, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Kajikawa, T.; Briones, R.A.; Resuello, R.R.G.; Tuplano, J.V.; Reis, E.S.; Hajishengallis, E.; Garcia, C.A.G.; Yancopoulou, D.; Lambris, J.D.; Hajishengallis, G. Safety and Efficacy of the Complement Inhibitor AMY-101 in a Natural Model of Periodontitis in Non-human Primates. Mol. Ther. Methods Clin. Dev. 2017, 6, 207–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amyndas Pharmaceutical: Our Pipeline. Available online: https://www.amyndas.com/our-pipeline/ (accessed on 14 April 2021).
- Woodruff, T.M.; Nandakumar, K.S.; Tedesco, F. Inhibiting the C5–C5a receptor axis. Mol. Immunol. 2011, 48, 1631–1642. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Liu, B. The roles and potential therapeutic implications of C5a in the pathogenesis of COVID-19-associated coagulopathy. Cytokine Growth Factor Rev. 2020, 58, 75–81. [Google Scholar] [CrossRef] [PubMed]
- InflaRx: Press Release. Available online: https://www.inflarx.de/Home/Investors/Press-Releases/07-2020-InflaRx-Announces-Decision-to-Enter-Phase-III-Development-of-IFX-1-in-Severe-COVID-19-Induced-Pneumonia.html (accessed on 14 April 2021).
- Kulasekararaj, A.G.; Lazana, I.; Large, J.; Posadas, K.; Eagleton, H.; Villajin, J.L.; Zuckerman, M.; Gandhi, S.; Marsh, J.C.W. Terminal complement inhibition dampens the inflammation during COVID-19. Br. J. Haematol. 2020, 190, 84. [Google Scholar] [CrossRef] [PubMed]
- SOLIRIS® (Eculizumab) Injection, for Intravenous Use. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/125166s422lbl.pdf (accessed on 14 April 2021).
- Zelek, W.M.; Xie, L.; Morgan, B.P.; Harris, C.L. Compendium of current complement therapeutics. Mol. Immunol. 2019, 114, 341–352. [Google Scholar] [CrossRef]
- Safety and Efficacy Study of RA101495 in Subjects with Generalized Myasthenia Gravis. Available online: https://clinicaltrials.gov/ct2/show/NCT03315130 (accessed on 14 April 2021).
- Eriksson, O.; Hultström, M.; Persson, B.; Lipcsey, M.; Ekdahl, K.N.; Nilsson, B.; Frithiof, R. Mannose-Binding Lectin is Associated with Thrombosis and Coagulopathy in Critically Ill COVID-19 Patients. Thromb. Haemost. 2020, 120, 1720–1724. [Google Scholar]
- Narsoplimab (OMS721). Available online: https://www.omeros.com/narsoplimab (accessed on 14 April 2021).
- Alexion Provides Update on Phase 3 Study of Ultomiris (Ravulizumab-cwvz) in Hospitalized Patients with Severe COVID-19. Available online: https://ir.alexion.com/news-releases/news-release-details/alexion-provides-update-phase-3-study-ultomirisr-ravulizumab (accessed on 14 April 2021).
- Guan, W.J.; Ni, Z.Y.; Hu, Y.; Liang, W.H.; Ou, C.Q.; He, J.X.; Liu, L.; Shan, H.; Lei, C.-L.; Hui, D.S.C.; et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Horiuchi, T.; Tsukamoto, H. Complement-targeted therapy: Development of C5- and C5a-targeted inhibition. Inflamm. Regen. 2016, 36, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Sponsor | Study Type | Status | Location | NCT | Publications |
|---|---|---|---|---|---|---|
| C1 esterase inhibitor | ||||||
| Conestat-alfa | Pharming | Case series (n = 5) | Not available | CH | Urwyler [56] | |
| Phase 2 (n = 120) | Recruiting | BR, CH, MX | 04414631 [57] | |||
| Ruconest® | Pharming | Phase 2 (n = 120) | Recruiting | US | 04530136 [58] | |
| C3 inhibitor | ||||||
| AMY-101 | Amyndas | Case report (n = 1) | Not available | IT | Mastaglio [59] | |
| Phase 2 (n = 144) | Not yet recruiting | Not listed | 04395456 [60] | |||
| APL-9 | Apellis | Phase 1,2 (n = 66) | Recruiting | US, BR | 04402060 [61] | |
| C5a inhibitor | ||||||
| BDB-001 | Staidson | Case series (n = 2) | Not available | CN | Gao [28] | |
| Phase 2,3 (n = 368) | Recruiting | BD, CN, ES, ID, IN | 04449588 [62] | |||
| Vilobelimab | InflaRx | Phase 2 (n = 30) | Not available | NL | Vlaar [63] | |
| Phase 2,3 (n = 390) | Recruiting | BE, BR, FR, DE, NL, PE, RU | 04333420 [64] | |||
| C5aR inhibitor | ||||||
| Avdoralimab | Innate | Phase 2 (n = 208) | Active, not recruiting | FR | 04371367 [65] | |
| C5 inhibitor | ||||||
| Eculizumab | Alexion | Cohort * (n = 80) | Not available | FR | Annane [66] | |
| Case series (n = 4) | Not available | IT | Diurno [67] | |||
| Case series (n = 3) | Not available | US | Laurence [68] | |||
| Phase 2 (n = 120) | Recruiting | FR | 04346797 [69] | |||
| Expanded Access | Available | Not listed | 04288713 [70] | |||
| Expanded Access | Not available | FR, US | 04355494 [71] | |||
| Ravulizumab | Alexion | Phase 3 (n = 270) | Active, not recruiting | ES, FR, GB, JP, US | 04369469 [72] | |
| Phase 3 (n = 32) | Recruiting | US | 04570397 [73] | |||
| Phase 4 (n = 1167) | Recruiting | GB | 04390464 [74] | |||
| Tesidolumab | Novartis | Case series (n = 5) | Not available | GB | Zelek [75] | |
| Zilucoplan | UCB | Phase 2 (n = 81) | Active, not recruiting | BE | 04382755 [76] | |
| MASP-2 inhibitor | ||||||
| Narsoplimab | Omeros | Case series (n = 6) | Not available | IT | Rambaldi [77] | |
| Phase 2, Adaptive (n = 1500) | Recruiting | US | 04488081 [78] | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ng, N.; Powell, C.A. Targeting the Complement Cascade in the Pathophysiology of COVID-19 Disease. J. Clin. Med. 2021, 10, 2188. https://doi.org/10.3390/jcm10102188
Ng N, Powell CA. Targeting the Complement Cascade in the Pathophysiology of COVID-19 Disease. Journal of Clinical Medicine. 2021; 10(10):2188. https://doi.org/10.3390/jcm10102188
Chicago/Turabian StyleNg, Nicole, and Charles A. Powell. 2021. "Targeting the Complement Cascade in the Pathophysiology of COVID-19 Disease" Journal of Clinical Medicine 10, no. 10: 2188. https://doi.org/10.3390/jcm10102188
APA StyleNg, N., & Powell, C. A. (2021). Targeting the Complement Cascade in the Pathophysiology of COVID-19 Disease. Journal of Clinical Medicine, 10(10), 2188. https://doi.org/10.3390/jcm10102188