The Importance of Non-Coding RNAs in Neurodegenerative Processes of Diabetes-Related Molecular Pathways

 , ,
, ,  , , and
, , and
Abstract
1. Introduction
2. MiRNAs and Their Link to Neurodegenerative Changes in Metabolic Pathways Related to Diabetes
2.1. MicroRNAs Involved in Neurodegeneration and Regeneration
2.1.1. MiRNAs in Diabetic Neuropathies
2.1.2. MiRNAs Involved in Insulin Signaling Pathways in Neurodegeneration
2.2. Dicer
3. LncRNAs and Their Links to Neurogenesis and Nerve Regeneration in Diabetes
3.1. LncRNAs Involved in Neurodegeneration and Regeneration
3.1.1. LncRNAs in Diabetic Retinopathy
3.1.2. LncRNA Involved in Neuronal Apoptosis, Autophagy and Oxidative Stress
4. Concluding Remarks and Limitations
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AGEs | Advanced glycation end products |
| AgRP | Agouti-related protein |
| AD | Alzheimer’s disease |
| Aβ | Amyloid-β |
| Ang-1 | Angiopoietin 1 |
| ARC | Arcuate nucleus of the hypothalamus |
| Atg4B | Autophagy related gene 4B |
| Bcl-2 | B-cell lymphoma 2 |
| BDNF | Brain-derived neurotrophic factor |
| CNS | Central nervous system |
| CSVD | Cerebral small vessel disease |
| CART | Cocaine and amphetamine-related transcript |
| DM | Diabetes Mellitus |
| DCN | Diabetic corneal neuropathy |
| DPN | Diabetic peripheral neuropathy |
| Dicer | Endoribonuclease |
| DIO | Diet-induced obesity |
| DRG | Dorsal root ganglia |
| ECs | Endothelial cells |
| GAS5 | Growth arrest-specific transcript 5 |
| GFAP | Glial fibrillary acidic protein |
| GFP | Green fluorescent protein |
| HO-1 | Heme oxygenase-1 |
| HG | High glucose |
| hCMEC/D3 | Human cerebral endothelial cell model |
| HD | Huntington’s Disease |
| IRS-1 | Increased insulin receptor substrate 1 |
| iPSC | Induced pluripotent stem cells |
| IRS-1 | Insulin receptor substrate 1 |
| IRAK1 | Interleukin 1 Receptor Associated Kinase 1 |
| IL-1β | Interleukin-1β |
| IL-2 | Interleukin 2 |
| IL-6 | Interleukin 6 |
| IP | Intraperitoneal |
| LARP7 | La ribonucleoprotein domain family member 7 |
| lncRNA | Long noncoding RNA, |
| MSC | Mesenchymal stromal cells, |
| MALAT1 | Metastasis-associated lung adenocarcinoma transcript 1 |
| miRs | MicroRNAs, miRNAs |
| LC3-II | Microtubule-associated protein 1 light chain 3 |
| miRISC | MiRNA induced silencing complex |
| MCP-1 | Monocyte chemotactic protein-1 |
| NOX4 | NADPH oxidase 4 |
| NTC | Negative Treated Control |
| NPY | Neuropeptide Y |
| NEAT1 | Nuclear paraspeckle assembly transcript 1 |
| Nrf2 | Nuclear factor erythroid 2-related factor 2 |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| PD | Parkinson’s Disease, |
| PTIP 1 | Pax transactivation domain-interacting protein 1 |
| p-NFkB | Phospho NFkB |
| PDE3A | Phosphodiesterase 3a |
| POMC | Pro-opiomelanocortin |
| PDCD4 | Programmed cell death protein 4 |
| Ago-2 | Protein argonaute-2 |
| qPCR | Quantitative real-time PCR |
| ROS | Reactive oxygen species |
| RAGE | Receptor for advanced glycation end |
| RGCs | Retinal ganglion cells |
| Sirt1 | Silent mating type information regulation 2 homolog 1 |
| Sox2OT | Sox2 overlapping transcript |
| STZ | Streptozocin |
| Tβ4 | Thymosin beta 4 |
| TRAF6 | TNF Receptor Associated Factor 6 |
| TLR4 | Toll-like receptor 4 |
| TRPM7 | Transient receptor potential melastatin 7 |
| TRAF6 | Tumor necrosis factor (TNFR)-associated factor 6 |
| TNF-α | Tumor necrosis factor α |
| T1DM | Type 1 Diabetes Mellitus |
| UTR | 30 untranslated region |
| VCAM-1 | Vascular cell adhesion molecule-1 |
| VEGF | Vascular endothelial growth factor |
| YFP | Yellow fluorescent protein |
References
- American Diabetes Association Diagnosis and classification of diabetes mellitus. Diabetes Care 2014, 37 (Suppl. 1), S81–S90. [CrossRef] [PubMed]
- Saeedi, P.; Petersohn, I.; Salpea, P.; Malanda, B.; Karuranga, S.; Unwin, N.; Colagiuri, S.; Guariguata, L.; Motala, A.A.; Ogurtsova, K.; et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas, 9th edition. Diabetes Res. Clin. Pract. 2019, 157, 107843. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Ley, S.H.; Hu, F.B. Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nat. Rev. Endocrinol. 2018, 14, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Forbes, J.M.; Cooper, M.E. Mechanisms of diabetic complications. Physiol. Rev. 2013, 93, 137–188. [Google Scholar] [CrossRef]
- Ramos-Rodriguez, J.J.; Ortiz, O.; Jimenez-Palomares, M.; Kay, K.R.; Berrocoso, E.; Murillo-Carretero, M.I.; Perdomo, G.; Spires-Jones, T.; Cozar-Castellano, I.; Lechuga-Sancho, A.M.; et al. Differential central pathology and cognitive impairment in pre-diabetic and diabetic mice. Psychoneuroendocrinology 2013, 38, 2462–2475. [Google Scholar] [CrossRef]
- Moran, C.; Beare, R.; Wang, W.; Callisaya, M.; Srikanth, V. For the Alzheimer’s Disease Neuroimaging Initiative (ADNI) Type 2 diabetes mellitus, brain atrophy, and cognitive decline. Neurology 2019, 92, 823–830. [Google Scholar] [CrossRef]
- Oikawa, Y.; Shimada, A. [Type 1 diabetes]. Nihon Rinsho 2015, 73, 1997–2002. [Google Scholar]
- Thevis, M.; Thomas, A.; Schänzer, W. Insulin. Handb. Exp. Pharm. 2010, 209–226. [Google Scholar] [CrossRef]
- Bluestone, J.A.; Herold, K.; Eisenbarth, G. Genetics, pathogenesis and clinical interventions in type 1 diabetes. Nature 2010, 464, 1293–1300. [Google Scholar] [CrossRef]
- Pemp, B.; Palkovits, S.; Howorka, K.; Pumprla, J.; Sacu, S.; Garhöfer, G.; Bayerle-Eder, M.; Schmetterer, L.; Schmidt-Erfurth, U. Correlation of retinal neurodegeneration with measures of peripheral autonomic neuropathy in type 1 diabetes. Acta Ophthalmol. 2018, 96, e804–e810. [Google Scholar] [CrossRef]
- Gundogan, F.C.; Akay, F.; Uzun, S.; Yolcu, U.; Çağıltay, E.; Toyran, S. Early Neurodegeneration of the Inner Retinal Layers in Type 1 Diabetes Mellitus. Ophthalmologica 2016, 235, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R. Type 2 diabetes: Etiology and reversibility. Diabetes Care 2013, 36, 1047–1055. [Google Scholar] [CrossRef]
- Bharadwaj, P.; Wijesekara, N.; Liyanapathirana, M.; Newsholme, P.; Ittner, L.; Fraser, P.; Verdile, G. The Link between Type 2 Diabetes and Neurodegeneration: Roles for Amyloid-β, Amylin, and Tau Proteins. J. Alzheimer’s Dis. 2017, 59, 421–432. [Google Scholar] [CrossRef]
- Yoneda, R.; Ueda, N.; Uranishi, K.; Hirasaki, M.; Kurokawa, R. Long noncoding RNA reduces cyclin D1 gene expression and arrests cell cycle through RNA mA modification. J. Biol. Chem. 2020, 295, 5626–5639. [Google Scholar] [CrossRef] [PubMed]
- Wolska, M.; Jarosz-Popek, J.; Junger, E.; Wicik, Z.; Porshoor, T.; Sharif, L.; Czajka, P.; Postula, M.; Mirowska-Guzel, D.; Czlonkowska, A.; et al. Long Non-coding RNAs as Promising Therapeutic Approach in Ischemic Stroke: A Comprehensive Review. Mol. Neurobiol. 2020, 1–9. [Google Scholar] [CrossRef]
- Eyileten, C.; Wicik, Z.; De Rosa, S.; Mirowska-Guzel, D.; Soplinska, A.; Indolfi, C.; Jastrzebska-Kurkowska, I.; Czlonkowska, A.; Postula, M. MicroRNAs as Diagnostic and Prognostic Biomarkers in Ischemic Stroke-A Comprehensive Review and Bioinformatic Analysis. Cells 2018, 7, 249. [Google Scholar] [CrossRef] [PubMed]
- Pordzik, J.; Pisarz, K.; De Rosa, S.; Jones, A.D.; Eyileten, C.; Indolfi, C.; Malek, L.; Postula, M. The Potential Role of Platelet-Related microRNAs in the Development of Cardiovascular Events in High-Risk Populations, Including Diabetic Patients: A Review. Front. Endocrinol. 2018, 9, 74. [Google Scholar] [CrossRef]
- Pordzik, J.; Jakubik, D.; Jarosz-Popek, J.; Wicik, Z.; Eyileten, C.; De Rosa, S.; Indolfi, C.; Siller-Matula, J.M.; Czajka, P.; Postula, M. Significance of circulating microRNAs in diabetes mellitus type 2 and platelet reactivity: Bioinformatic analysis and review. Cardiovasc. Diabetol. 2019, 18, 113. [Google Scholar] [CrossRef]
- Jiang, Q.; Shan, K.; Qun-Wang, X.; Zhou, R.-M.; Yang, H.; Liu, C.; Li, Y.-J.; Yao, J.; Li, X.-M.; Shen, Y.; et al. Long non-coding RNA-MIAT promotes neurovascular remodeling in the eye and brain. Oncotarget 2016, 7, 49688–49698. [Google Scholar] [CrossRef]
- Raut, S.K.; Khullar, M. The Big Entity of New RNA World: Long Non-Coding RNAs in Microvascular Complications of Diabetes. Front. Endocrinol. 2018, 9, 300. [Google Scholar] [CrossRef]
- Biswas, S.; Sarabusky, M.; Chakrabarti, S. Diabetic Retinopathy, lncRNAs, and Inflammation: A Dynamic, Interconnected Network. J. Clin. Med. Res. 2019, 8, 1033. [Google Scholar] [CrossRef] [PubMed]
- Ciccacci, C.; Latini, A.; Colantuono, A.; Politi, C.; D’Amato, C.; Greco, C.; Rinaldi, M.E.; Lauro, D.; Novelli, G.; Spallone, V.; et al. Expression study of candidate miRNAs and evaluation of their potential use as biomarkers of diabetic neuropathy. Epigenomics 2020, 12, 575–585. [Google Scholar] [CrossRef] [PubMed]
- Robles-Rivera, R.R.; Castellanos-González, J.A.; Olvera-Montaño, C.; Flores-Martin, R.A.; López-Contreras, A.K.; Arevalo-Simental, D.E.; Cardona-Muñoz, E.G.; Roman-Pintos, L.M.; Rodríguez-Carrizalez, A.D. Adjuvant Therapies in Diabetic Retinopathy as an Early Approach to Delay Its Progression: The Importance of Oxidative Stress and Inflammation. Oxid. Med. Cell. Longev. 2020, 2020, 3096470. [Google Scholar] [CrossRef] [PubMed]
- Yildirim, S.S.; Akman, D.; Catalucci, D.; Turan, B. Relationship between downregulation of miRNAs and increase of oxidative stress in the development of diabetic cardiac dysfunction: Junctin as a target protein of miR-1. Cell Biochem. Biophys. 2013, 67, 1397–1408. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, J.C.R.; Acuña, S.M.; Aoki, J.I.; Floeter-Winter, L.M.; Muxel, S.M. Long Non-Coding RNAs in the Regulation of Gene Expression: Physiology and Disease. Noncoding Rna 2019, 5, 17. [Google Scholar] [CrossRef]
- Iqbal, Z.; Azmi, S.; Yadav, R.; Ferdousi, M.; Kumar, M.; Cuthbertson, D.J.; Lim, J.; Malik, R.A.; Alam, U. Diabetic Peripheral Neuropathy: Epidemiology, Diagnosis, and Pharmacotherapy. Clin. Ther. 2018, 40, 828–849. [Google Scholar] [CrossRef]
- Singh, R.; Kishore, L.; Kaur, N. Diabetic peripheral neuropathy: Current perspective and future directions. Pharm. Res. 2014, 80, 21–35. [Google Scholar] [CrossRef]
- Juster-Switlyk, K.; Smith, A.G. Updates in diabetic peripheral neuropathy. F1000Research 2016, 5, F1000 Faculty Rev-738. [Google Scholar] [CrossRef]
- Rask-Madsen, C.; King, G.L. Vascular complications of diabetes: Mechanisms of injury and protective factors. Cell Metab. 2013, 17, 20–33. [Google Scholar] [CrossRef]
- Roustit, M.; Loader, J.; Deusenbery, C.; Baltzis, D.; Veves, A. Endothelial Dysfunction as a Link Between Cardiovascular Risk Factors and Peripheral Neuropathy in Diabetes. J. Clin. Endocrinol. Metab. 2016, 101, 3401–3408. [Google Scholar] [CrossRef]
- Poggesi, A.; Pasi, M.; Pescini, F.; Pantoni, L.; Inzitari, D. Circulating biologic markers of endothelial dysfunction in cerebral small vessel disease: A review. J. Cereb. Blood Flow Metab. 2016, 36, 72–94. [Google Scholar] [CrossRef] [PubMed]
- Yasmeen, S.; Kaur, S.; Mirza, A.H.; Brodin, B.; Pociot, F.; Kruuse, C. miRNA-27a-3p and miRNA-222-3p as Novel Modulators of Phosphodiesterase 3a (PDE3A) in Cerebral Microvascular Endothelial Cells. Mol. Neurobiol. 2019, 56, 5304–5314. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Chopp, M.; Szalad, A.; Zhang, Y.; Wang, X.; Zhang, R.L.; Liu, X.S.; Jia, L.; Zhang, Z.G. The role of miR-146a in dorsal root ganglia neurons of experimental diabetic peripheral neuropathy. Neuroscience 2014, 259, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Chopp, M.; Lu, X.; Szalad, A.; Jia, L.; Liu, X.S.; Wu, K.-H.; Lu, M.; Zhang, Z.G. miR-146a mediates thymosin β4 induced neurovascular remodeling of diabetic peripheral neuropathy in type-II diabetic mice. Brain Res. 2019, 1707, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chopp, M.; Liu, X.S.; Kassis, H.; Wang, X.; Li, C.; An, G.; Zhang, Z.G. MicroRNAs in the axon locally mediate the effects of chondroitin sulfate proteoglycans and cGMP on axonal growth. Dev. Neurobiol. 2015, 75, 1402–1419. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Wang, L.; Chopp, M.; Zhang, Y.; Szalad, A.; Zhang, Z.G. MicroRNA 146a locally mediates distal axonal growth of dorsal root ganglia neurons under high glucose and sildenafil conditions. Neuroscience 2016, 329, 43–53. [Google Scholar] [CrossRef]
- Ziegler, D.; Papanas, N.; Zhivov, A.; Allgeier, S.; Winter, K.; Ziegler, I.; Brüggemann, J.; Strom, A.; Peschel, S.; Köhler, B.; et al. Early detection of nerve fiber loss by corneal confocal microscopy and skin biopsy in recently diagnosed type 2 diabetes. Diabetes 2014, 63, 2454–2463. [Google Scholar] [CrossRef]
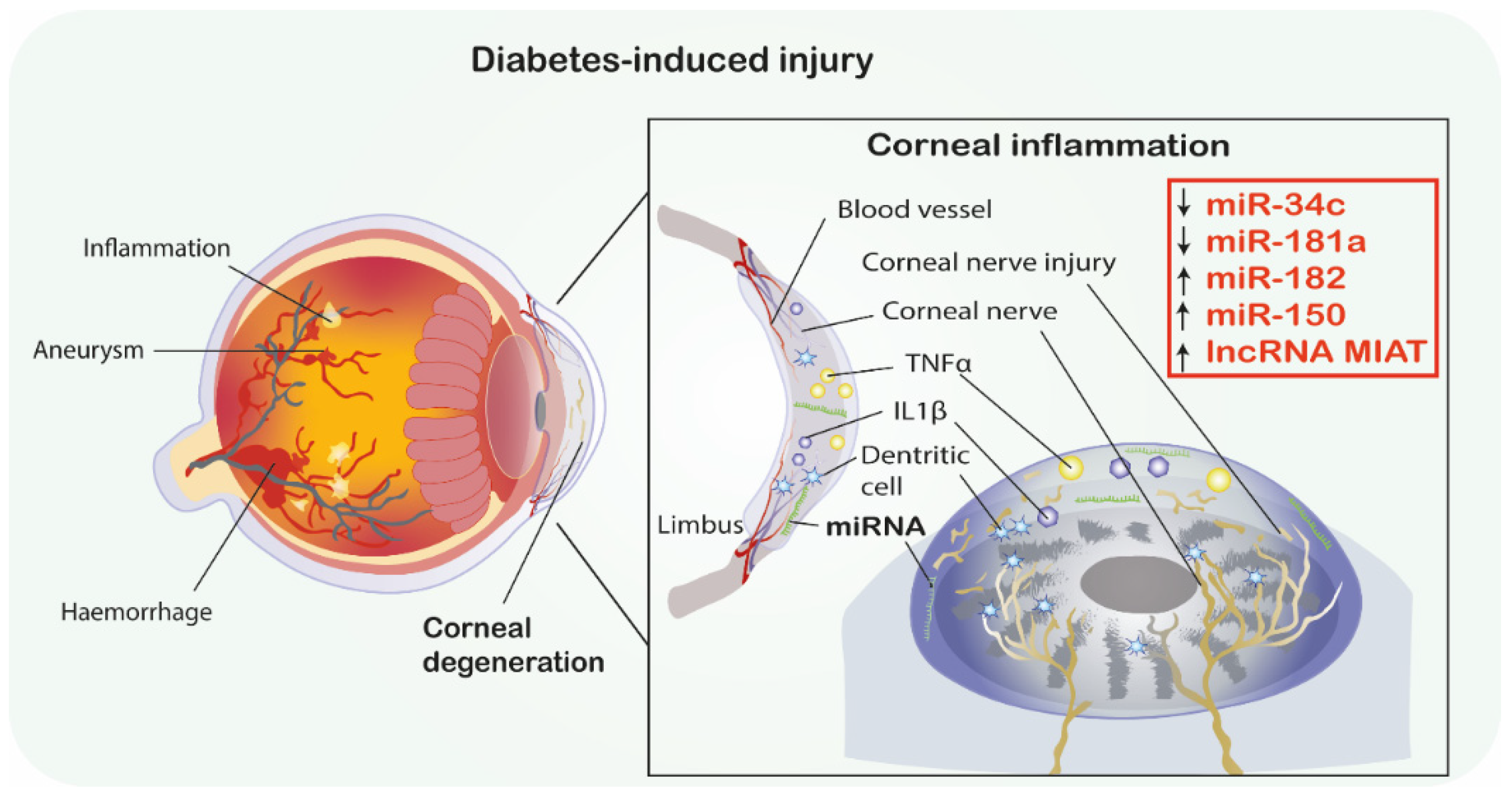
- Hu, J.; Hu, X.; Kan, T. MiR-34c Participates in Diabetic Corneal Neuropathy Via Regulation of Autophagy. Investig. Ophthalmol. Vis. Sci. 2019, 60, 16–25. [Google Scholar] [CrossRef]
- Hu, J.; Huang, Y.; Lin, Y.; Lin, J. Protective effect inhibiting the expression of miR-181a on the diabetic corneal nerve in a mouse model. Exp. Eye Res. 2020, 192, 107925. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, X.; Wu, X.; Dai, Y.; Chen, P.; Xie, L. microRNA-182 Mediates Sirt1-Induced Diabetic Corneal Nerve Regeneration. Diabetes 2016, 65, 2020–2031. [Google Scholar] [CrossRef]
- Wu, J.-H.; Gao, Y.; Ren, A.-J.; Zhao, S.-H.; Zhong, M.; Peng, Y.-J.; Shen, W.; Jing, M.; Liu, L. Altered microRNA expression profiles in retinas with diabetic retinopathy. Ophthalmic Res. 2012, 47, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Schneeberger, M.; Altirriba, J.; García, A.; Esteban, Y.; Castaño, C.; García-Lavandeira, M.; Alvarez, C.V.; Gomis, R.; Claret, M. Deletion of miRNA processing enzyme Dicer in POMC-expressing cells leads to pituitary dysfunction, neurodegeneration and development of obesity. Mol. Metab. 2012, 2, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.-J.; Li, J.; Zhang, S.-Y. Effects of TRPM7/miR-34a Gene Silencing on Spatial Cognitive Function and Hippocampal Neurogenesis in Mice with Type 1 Diabetes Mellitus. Mol. Neurobiol. 2018, 55, 1568–1579. [Google Scholar] [CrossRef] [PubMed]
- Li, H.-H.; Lin, S.-L.; Huang, C.-N.; Lu, F.-J.; Chiu, P.-Y.; Huang, W.-N.; Lai, T.-J.; Lin, C.-L. miR-302 Attenuates Amyloid-β-Induced Neurotoxicity through Activation of Akt Signaling. J. Alzheimers. Dis. 2016, 50, 1083–1098. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.-B.; Ji, T.-F.; Zhou, H.-W.; Yu, J.-L. Effects of microRNA-21 on Nerve Cell Regeneration and Neural Function Recovery in Diabetes Mellitus Combined with Cerebral Infarction Rats by Targeting PDCD4. Mol. Neurobiol. 2018, 55, 2494–2505. [Google Scholar] [CrossRef] [PubMed]
- Venkat, P.; Cui, C.; Chopp, M.; Zacharek, A.; Wang, F.; Landschoot-Ward, J.; Shen, Y.; Chen, J. MiR-126 Mediates Brain Endothelial Cell Exosome Treatment-Induced Neurorestorative Effects After Stroke in Type 2 Diabetes Mellitus Mice. Stroke 2019, 50, 2865–2874. [Google Scholar] [CrossRef]
- Li, X.-J. Long non-coding RNA nuclear paraspeckle assembly transcript 1 inhibits the apoptosis of retina Müller cells after diabetic retinopathy through regulating miR-497/brain-derived neurotrophic factor axis. Diab. Vasc. Dis. Res. 2018, 15, 204–213. [Google Scholar] [CrossRef]
- Mattsson, N.; Andreasson, U.; Zetterberg, H.; Blennow, K. Alzheimer’s Disease Neuroimaging Initiative Association of Plasma Neurofilament Light With Neurodegeneration in Patients with Alzheimer Disease. JAMA Neurol. 2017, 74, 557–566. [Google Scholar] [CrossRef]
- Spielman, L.J.; Little, J.P.; Klegeris, A. Inflammation and insulin/IGF-1 resistance as the possible link between obesity and neurodegeneration. J. Neuroimmunol. 2014, 273, 8–21. [Google Scholar] [CrossRef]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharm. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef]
- Sarlak, G.; Vincent, B. The Roles of the Stem Cell-Controlling Sox2 Transcription Factor: From Neuroectoderm Development to Alzheimer’s Disease? Mol. Neurobiol. 2016, 53, 1679–1698. [Google Scholar] [CrossRef] [PubMed]
- Bayfield, M.A.; Yang, R.; Maraia, R.J. Conserved and divergent features of the structure and function of La and La-related proteins (LARPs). Biochim. Biophys. Acta 2010, 1799, 365–378. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Li, X.; Li, Q. Curcumin accelerates the repair of sciatic nerve injury in rats through reducing Schwann cells apoptosis and promoting myelinization. Biomed. Pharm. 2017, 92, 1103–1110. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Zhu, P.; Yang, J.; Liu, X.; Dong, S.; Wang, X.; Chun, B.; Zhuang, J.; Zhang, C. Ischaemic preconditioning-regulated miR-21 protects heart against ischaemia/reperfusion injury via anti-apoptosis through its target PDCD4. Cardiovasc. Res. 2010, 87, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Gusel’nikova, V.V.; Korzhevskiy, D.E. NeuN As a Neuronal Nuclear Antigen and Neuron Differentiation Marker. Acta Nat. 2015, 7, 42–47. [Google Scholar] [CrossRef]
- Person, F.; Wilczak, W.; Hube-Magg, C.; Burdelski, C.; Möller-Koop, C.; Simon, R.; Noriega, M.; Sauter, G.; Steurer, S.; Burdak-Rothkamm, S.; et al. Prevalence of βIII-tubulin (TUBB3) expression in human normal tissues and cancers. Tumour Biol. 2017, 39, 1010428317712166. [Google Scholar] [CrossRef]
- Schlingmann, K.P.; Waldegger, S.; Konrad, M.; Chubanov, V.; Gudermann, T. TRPM6 and TRPM7--Gatekeepers of human magnesium metabolism. Biochim. Biophys. Acta 2007, 1772, 813–821. [Google Scholar] [CrossRef]
- Lin, X.; Guan, H.; Huang, Z.; Liu, J.; Li, H.; Wei, G.; Cao, X.; Li, Y. Downregulation of Bcl-2 expression by miR-34a mediates palmitate-induced Min6 cells apoptosis. J. Diabetes Res. 2014, 2014, 258695. [Google Scholar] [CrossRef]
- Song, M.-S.; Rossi, J.J. Molecular mechanisms of Dicer: Endonuclease and enzymatic activity. Biochem. J. 2017, 474, 1603–1618. [Google Scholar] [CrossRef]
- Koscianska, E.; Starega-Roslan, J.; Krzyzosiak, W.J. The role of Dicer protein partners in the processing of microRNA precursors. PLoS ONE 2011, 6, e28548. [Google Scholar] [CrossRef]
- Shan, K.; Jiang, Q.; Wang, X.-Q.; Wang, Y.-N.-Z.; Yang, H.; Yao, M.-D.; Liu, C.; Li, X.-M.; Yao, J.; Liu, B.; et al. Role of long non-coding RNA-RNCR3 in atherosclerosis-related vascular dysfunction. Cell Death Dis. 2016, 7, e2248. [Google Scholar] [CrossRef] [PubMed]
- Leung, A.; Natarajan, R. Long Noncoding RNAs in Diabetes and Diabetic Complications. Antioxid. Redox Signal. 2018, 29, 1064–1073. [Google Scholar] [CrossRef] [PubMed]
- Gasecka, A.; Siwik, D.; Gajewska, M.; Jaguszewski, M.J.; Mazurek, T.; Filipiak, K.J.; Postuła, M.; Eyileten, C. Early Biomarkers of Neurodegenerative and Neurovascular Disorders in Diabetes. J. Clin. Med. Res. 2020, 9, 2807. [Google Scholar] [CrossRef] [PubMed]
- Adamska, A.; Pilacinski, S.; Zozulinska-Ziolkiewicz, D.; Gandecka, A.; Grzelka, A.; Konwerska, A.; Malinska, A.; Nowicki, M.; Araszkiewicz, A. An increased skin microvessel density is associated with neurovascular complications in type 1 diabetes mellitus. Diab. Vasc. Dis. Res. 2019, 16, 513–522. [Google Scholar] [CrossRef]
- Zhang, Q.; Fang, W.; Ma, L.; Wang, Z.-D.; Yang, Y.-M.; Lu, Y.-Q. VEGF levels in plasma in relation to metabolic control, inflammation, and microvascular complications in type-2 diabetes: A cohort study. Medicine 2018, 97, e0415. [Google Scholar] [CrossRef]
- Eyileten, C.; Mirowska-Guzel, D.; Milanowski, L.; Zaremba, M.; Rosiak, M.; Cudna, A.; Kaplon-Cieslicka, A.; Opolski, G.; Filipiak, K.J.; Malek, L.; et al. Serum Brain-Derived Neurotrophic Factor is Related to Platelet Reactivity and Metformin Treatment in Adult Patients with Type 2 Diabetes Mellitus. Can. J. Diabetes 2019, 43, 19–26. [Google Scholar] [CrossRef]
- Eyileten, C.; Kaplon-Cieslicka, A.; Mirowska-Guzel, D.; Malek, L.; Postula, M. Antidiabetic Effect of Brain-Derived Neurotrophic Factor and Its Association with Inflammation in Type 2 Diabetes Mellitus. J. Diabetes Res. 2017, 2017, 2823671. [Google Scholar] [CrossRef]
- Eyileten, C.; Zaremba, M.; Janicki, P.K.; Rosiak, M.; Cudna, A.; Kapłon-Cieślicka, A.; Opolski, G.; Filipiak, K.J.; Kosior, D.A.; Mirowska-Guzel, D.; et al. Serum Brain-Derived Neurotrophic Factor is Related to Platelet Reactivity but not to Genetic Polymorphisms within BDNF Encoding Gene in Patients with Type 2 Diabetes. Med. Sci. Monit. 2016, 22, 69–76. [Google Scholar] [CrossRef]
- Mirowska-Guzel, D. The role of neurotrophic factors in the pathology and treatment of multiple sclerosis. Immunopharmacol. Immunotoxicol. 2009, 31, 32–38. [Google Scholar] [CrossRef]
- Eyileten, C.; Sharif, L.; Wicik, Z.; Jakubik, D.; Jarosz-Popek, J.; Soplinska, A.; Postula, M.; Czlonkowska, A.; Kaplon-Cieslicka, A.; Mirowska-Guzel, D. The Relation of the Brain-Derived Neurotrophic Factor with MicroRNAs in Neurodegenerative Diseases and Ischemic Stroke. Mol. Neurobiol. 2020, 58, 329–347. [Google Scholar] [CrossRef]
- Li, Y.; Xu, F.; Xiao, H.; Han, F. Long noncoding RNA BDNF-AS inversely regulated BDNF and modulated high-glucose induced apoptosis in human retinal pigment epithelial cells. J. Cell. Biochem. 2018, 119, 817–823. [Google Scholar] [CrossRef]
- Luo, L.; Ji, L.-D.; Cai, J.-J.; Feng, M.; Zhou, M.; Hu, S.-P.; Xu, J.; Zhou, W.-H. Microarray Analysis of Long Noncoding RNAs in Female Diabetic Peripheral Neuropathy Patients. Cell. Physiol. Biochem. 2018, 46, 1209–1217. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Song, C.; Chen, K.; Zhang, X. Inhibition of long non-coding RNA IGF2AS protects apoptosis and neuronal loss in anesthetic-damaged mouse neural stem cell derived neurons. Biomed. Pharm. 2017, 85, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Gong, H.-Y.; Xu, L. PVT1 protects diabetic peripheral neuropathy via PI3K/AKT pathway. Eur. Rev. Med. Pharm. Sci. 2018, 22, 6905–6911. [Google Scholar] [CrossRef]
- Liu, C.; Li, C.-P.; Wang, J.-J.; Shan, K.; Liu, X.; Yan, B. RNCR3 knockdown inhibits diabetes mellitus-induced retinal reactive gliosis. Biochem. Biophys. Res. Commun. 2016, 479, 198–203. [Google Scholar] [CrossRef]
- Yu, J.-L.; Li, C.; Che, L.-H.; Zhao, Y.-H.; Guo, Y.-B. Downregulation of long noncoding RNA H19 rescues hippocampal neurons from apoptosis and oxidative stress by inhibiting IGF2 methylation in mice with streptozotocin-induced diabetes mellitus. J. Cell. Physiol. 2019, 234, 10655–10670. [Google Scholar] [CrossRef]
- Li, C.-P.; Wang, S.-H.; Wang, W.-Q.; Song, S.-G.; Liu, X.-M. Long Noncoding RNA-Sox2OT Knockdown Alleviates Diabetes Mellitus-Induced Retinal Ganglion Cell (RGC) injury. Cell. Mol. Neurobiol. 2017, 37, 361–369. [Google Scholar] [CrossRef]
- Zhang, Y.-L.; Hu, H.-Y.; You, Z.-P.; Li, B.-Y.; Shi, K. Targeting long non-coding RNA MALAT1 alleviates retinal neurodegeneration in diabetic mice. Int. J. Ophthalmol. 2020, 13, 213–219. [Google Scholar] [CrossRef]
- Jiang, L.; Wang, C.; Shen, X. LncRNA GAS5 suppresses ER stress‑induced apoptosis and inflammation by regulating SERCA2b in HG‑treated retinal epithelial cell. Mol. Med. Rep. 2020, 22, 1072–1080. [Google Scholar] [CrossRef]
- Tu, W.; Wang, H.; Li, S.; Liu, Q.; Sha, H. The Anti-Inflammatory and Anti-Oxidant Mechanisms of the Keap1/Nrf2/ARE Signaling Pathway in Chronic Diseases. Aging Dis. 2019, 10, 637–651. [Google Scholar] [CrossRef]
- Li, H.; Liu, Y.; Huang, J.; Liu, Y.; Zhu, Y. Association of genetic variants in lncRNA GAS5/miR-21/mTOR axis with risk and prognosis of coronary artery disease among a Chinese population. J. Clin. Lab. Anal. 2020, 34, 521. [Google Scholar] [CrossRef]
- Chen, L.; Ren, P.; Zhang, Y.; Gong, B.; Yu, D.; Sun, X. Long non‑coding RNA GAS5 increases the radiosensitivity of A549 cells through interaction with the miR‑21/PTEN/Akt axis. Oncol. Rep. 2020, 43, 897–907. [Google Scholar] [CrossRef]
- Zhou, Y.; Chen, B. GAS5‑mediated regulation of cell signaling (Review). Mol. Med. Rep. 2020, 22, 3049–3056. [Google Scholar] [CrossRef]
- Khoshnoodi, M.; Truelove, S.; Polydefkis, M. Effect of diabetes type on long-term outcome of epidermal axon regeneration. Ann. Clin. Transl. Neurol. 2019, 6, 2088–2096. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Ref | Analyzed miRNAs and Their Deregulation | Analyzed mRNAs | Pathophysiological Mechanism and Axis | Methodology | Conclusion |
|---|---|---|---|---|---|
| [19] | ↑miR-150 | VEGF BDNF, NGF, NT-3, Ang-1 | Oxidative stress, Apoptosis Hypoxia, Angiogenesis MIAT/miR-150-5p/VEGF | STZ-induced diabetes model *, in vitro, in vivo, Mice, cornea and retina * Study used single injection of STZ to mice, 60 mg/kg used to induce diabetes. Seven days after STZ injection, animals with blood glucose levels > 16.7 mmol/L were included in diabetic group. | MIAT regulates neural and vascular cell function via MIAT/miR-150-5p/VEGF network. MIAT knockdown leads to cerebral microvascular degeneration, progressive neuronal loss and neurodegeneration, behavioral deficits in a CNS neurovascular disorder, Alzheimer’s disease. MIAT may represent a pharmacological target for treating neurovascular-related disorders |
| [42] | No miRNA, Dicer | Nhlrc1, Park2, Rps24, Rps9UN Agrp, Cart, Pomc, Npy, Crh, Mc3r, Mc4r, Tpit, Crhr1, Ntrk2, Myc, Naglu, Acp2, UN Dicer UN Drosha, UN Dgcr8, UN Ago2, UN Pit-1, UN Gh, UN Tshβ, | Apoptosis, Inflammation, Autophagy | The role of Dicer in pituitary dysfunction and neurodegeneration, In vivo, In vitro, POMCDicerKO mice, pituitary gland, hypothalamus, | The absence of Dicer protein in pituitary neurons in mice leads to impaired neuronal function, development of obesity and neurodegenerative processes |
| [40] | ↑miR-182 | NOX4 | Oxidative stress | T2DM model In silico, In vivo, BKS.Cg-m+/+Leprdb/J (db/db) mice as the model of T2DM, cornea | Sirt1 binds to miR-182 promoter and modulates its transcription. MiR-182 plays a key role in DM-induced corneal nerve regeneration via targeting NOX4. Administration of miR-182 lead to corneal healing effect in diabetic mice. |
| [43] | ↑miR-34a | TRPM7 ICA IAA GAD-Ab Bax Cyt-c Caspase-3 Bcl-2 | Apoptosis TRPM7/miR-34a | T1DM model STZ-induced T1DM, In vitro, In vivo, mice, hippocamp | Inhibition of TRPM7/miR-34a axis decreased apoptosis markers, improved spatial cognitive function and promoted hippocampal neurogenesis in mice with DM. |
| [32] | ↑miR-27a-3p↑ miR-222-3p | PDE3A | CSVD | Endothelial dysfunction In silico, In vitro, hCMEC/D3 cell line model of cerebral endothelial micro-vessel cells | Increased levels of miR-222-3p and miR-27-3p diminished expression of PDE3A in cerebral endothelial cells via decreasing ischemic penumbra and diminishing damage caused by CSVD, therefore may play a role in endothelial function and improve cerebral circulation. |
| [33] | ↓miR-146a | IRAK1, TRAF6 Caspase-3 | Apoptosis, Inflammation | T2DM In vitro, In vivo, BKS.Cg-m+/+Leprdb/J (db/db) mice as the model of T2DM, DRGs neurons | DM increased IRAK1, TRAF6, caspase-3 and decreased miR-146a expression in DRG neurons. Sildenafil treatment reversed those effects. Mir-146a may play a crucial role in DM-induced DRG apoptosis. |
| [44] | ↑ miR-302 | Nrf2 HO-1 Nanog PTEN | Oxidative stress, Akt/Nfr2/HO-1 Apoptosis, Akt/GSK3, PI3K/Akt | AD and neurodegeneration in insulin signaling pathways, In silico, In vitro, In vivo, Human AD patients’ blood samples, human neuroblastoma SK-N-MC cells | Overexpression of miR-302 reduces Aβ-induced oxidative stress via activating Akt/Nfr2/HO-1, reduces mitochondrial dysfunction via Nrf2, reduces neurotoxicity and apoptosis via PI3K/Akt pathway. MiR-302 upregulation results with activating Akt/GSK2B by inhibition of PTEN and therefore targets Nanong which is a proliferation factor.MiR-302 can protect neuronal cells by restoring impaired insulin signaling via the prevention of Aβ-induced neurotoxicity. MiR-302 plays a neuroprotective role. |
| [45] | ↑ miR-21 | NeuN, GFAP, β-III-Tubulin, PDCD4 HNA, Nestine, PTEN, FasL, PDCD4 | Apoptosis | Alloxan-induced diabetes *, In vitro, In vivo, Diabetic rats, brain frontal pole * Rats were intraperitoneally injected with 100 mg/kg alloxan for two consecutive days. The fasting blood glucose of the rats in 72 h after the last injection was detected. The DM model rats were successfully established when fasting blood glucose value reached 16.7 mmol/L or more. After being fed with a high-fat diet for five consecutive weeks, the DM rats were used for the experiments. | Overexpression miR-21 may stimulate nerve regeneration and the recovery of neural function by inhibition of apoptosis via PDCD4 downregulation. |
| [46] | ↓miR-126 | NM | Angiogenesis | T2DM In vitro, In vivo, BKS.Cg-m+/+Leprdb/J(db/db-T2DM), bone marrow stromal cells (BMCs), smooth muscle cells (SMCs), mouse brain (ECs), astrocytes cells culture | Ischemic brain tissue and serum present diminished expression of miR-126. EC-Exo contains elevated levels of miR-126 and induces neurorestorative effects in post-stroke DM mice. EC-Exo treatment force angiogenesis, growth of PCN, increased myelin and vascular density. EC-Exo treatment improves cognitive and neurological function. |
| [36] | ↓miR-146a | IRAK1 TRAF6 Ago2 | Inflammation | The direct effect of HG on distal axonal growth In vitro, rats DRG neurons, cultured under HG and sildenafil conditions | HG-induced downregulation of miR-146a leads to reduced axonal growth in DRG neurons. Sildenafil treatment reverses the HG-induced effect of axonal growth reduction. Downregulation of miR-146a exacerbates diabetes wound-healing. |
| [39] | ↑miR-181a | ATG5, LC3b-II Bcl-2 | Apoptosis, Autophagy, | T1DM STZ-induced T1DM model, In vitro, In vivo, mice, trigeminal ganglion, corneas, | Inhibition of miR-181a upregulates ATG5 and Bcl-2 levels and therefore enhances autophagy and antagonizes apoptosis and promotes neurodegeneration by neuronal axon growth of corneal epithelium in HG condition and therefore play a neuroprotective role in DM mice. |
| [38] | ↑miR-34c | Atg4B LC3-II | Autophagy, | T1DM STZ-induced T1DM model In silico, In vitro, In vivo, mice, trigeminal ganglion, corneas, | MiR-34c inhibition promotes growth of trigeminal ganglion cells. Additionally, miR-34c inhibition restores corneal nerve function via directly targeting both Atg4B and LC3-II. Inhibition of miR-34c accelerates epithelial wound healing of cornea in DM mice by autophagy. |
| [41] | ↑miR-182, ↑miR-96, ↑miR-183, ↑miR-211, ↑miR-204 ↑miR-124 ↓miR-199a-3p, ↓miR-10b, ↓miR-10a, ↓miR-219-2-3p, ↓miR-144 ↓miR-338 | NM | Capillary endothelial function | STZ-induced diabetes model *, In vitro, In vivo, rats, retinas *Male Sprague-Dawley rats were fed with standard rat chow and water ad libitum. DM was induced by a single intraperitoneal injection of STZ at a dose of 60 mg/kg body weight, and was defined as a blood glucose level above 16.7 mmol/l determined at 3 days after injection. | The study shows that altered miRNA expression profiles are associated with diabetic retinopathy development. Thus, modulation of those miRNAs may be potentially useful in diabetic retinopathy treatment. |
| [47] | ↑miR-497 | BDNF | Apoptosis NEAT1/BDNF/miR-497 | STZ-induced diabetes * In silico, In vitro, rats, retinas, primary Muller glial cells * The Male Sprague Dawley rats in the diabetes group received STZ (65 mg/kg) by intravenous injection once. The rat with blood glucose levels>16.7 mmol/L after 5 days of injection was considered successful. After 4 weeks of induction, all rats were sacrificed to isolate the retinal tissues. | Decreased levels of NEAT1 increased the HG-induced apoptosis ratio of Müller cells via regulating miR-497/BDNF axis and promoted retinopathy progression. Injections of pcDNA-NEAT1 enhanced retinal NEAT1 expression and decelerated retina thickening in DM rats. |
| Ref | Analyzed lncRNAs and Their Deregulation | Analyzed mRNAs | Pathophysiological Mechanism and Axis | Methodology | Conclusion |
|---|---|---|---|---|---|
| [74] | ↑PVT1 | PI3K/ AKT | Apoptosis, PI3K/AKT signaling pathway | STZ-induced diabetes * In vitro, In vivo, rats, DRG * Male Sprague Dawley rats were injected 55 mg/kg of STZ intraperitoneally. The diabetes rat model was considered to be successfully established when the fasting blood-glucose level in rats was detected to be higher than 16.67 mmol/L. | PVT1 can significantly inhibit the apoptosis of DRG cells in diabetic rats. PVT1 protects DPN via inhibiting the PI3K/AKT pathway |
| [19] | ↓MIAT | VEGF, BDNF, NGF, NT-3, Ang-1 | Angiogenesis, MIAT/miR-150-5p/VEGF axis | STZ-induced diabetes *, In vivo, In vitro, mice, corneas, retinas * Study used single injection of STZ to mice, 60 mg/kg used to induce diabetes. Seven days after STZ injection, animals with blood glucose levels > 16.7 mmol/L were included in diabetic group. | MIAT regulates neural and vascular cell function via MIAT/miR-150-5p/VEGF network. MIAT knockdown leads to microvascular degeneration, progressive neuronal loss, neurodegeneration and behavioral deficits in a CNS. MIAT may represent a pharmacological target for treating neurovascular-related disorders. |
| [47] | ↓NEAT1 | BDNF | Apoptosis NEAT1/BDNF/miR-497 | STZ-induced diabetes * In silico, In vitro, rats, retinas, primary Muller glial cells * The Male Sprague Dawley rats in the diabetes group received STZ (65 mg/kg) by intravenous injection once. The rat with blood glucose levels>16.7 mmol/L after 5 days of injection was considered successful. After 4 weeks of induction, all rats were sacrificed to isolate the retinal tissues. | Decreased levels of NEAT1 increased the HG-induced apoptosis ratio of Müller cells via regulating miR-497/BDNF axis and promoted retinopathy progression. Injections of pcDNA-NEAT1 enhanced retinal NEAT1 expression and decelerated retina thickening in DM rats. |
| [75] | ↑RNCR3 | GFAP Vimentin | Apoptosis, Inflammation, | STZ-induced diabetes * In vitro, In vivo, mice, primary Muller glial cells, retinas * Male C57BL/6 mice (4-month old) were used for the induction of diabetes. Diabetes was induced by the intraperitoneal injection of STZ (70 mg/kg). Blood glucose level was measured, in non-fasted animals, in venous blood using a glucometer. Blood glucose level ≥16.7 mmol/L was considered to be hyperglycemia | RNCR3 knockdown treatment reduces IL-2, IL-3, IL-4, IL-5, IL-9, IL-13, IL-17, MCP-1, VEGF and TNF-α. RNCR 3 knockdown could inhibit retinal glial reactivity and prevent HG-induced retinal neurodegeneration and may be used as a treatment. |
| [76] | ↑H19 | Bax, Caspase-3, IGF2, Bcl-2, CAT, SOD2, GPx4 | Apoptosis, Oxidative stress, | STZ-induced diabetes * In vitro, In vivo, mice, hippocampal cells * Male 6 weeks old Kunming mice were injected intraperitoneally with STZ (70 mg/kg) to induce diabetes. After the STZ injections, blood from the tail vein was collected accordingly. The mice that were confirmed to have a blood glucose concentration greater than 18 mmol/L were deemed to have been successfully modeled. | Downregulation of H19 results with downregulation of Bax, Caspase-3 and upregulation of Bcl-2 therefore inhibits apoptosis of hippocampal neurons in DM mice, and could be a potential treatment target. H19 knockdown diminishes oxidative stress by decrease of LPO and enhances GSH, CAT, SOD and GSH-Px. |
| [77] | ↓Sox2OT | NRF2, HO1 | Oxidative stress, Apoptosis | STZ-induced diabetes * In vitro, In vivo, mice retinal ganglion cells (RGCs) * Adult male C57BL/6 J mice were used in the study. Diabetes was induced by intraperitoneal injection of STZ (70 mg/kg). Animals with blood glucose levels higher than 16.7 mmol/L were deemed as having diabetes. | Sox2OT knockdown decreases apoptosis ratio via downregulation of Bax, Bxl-2 and Caspase-3. Sox2OT knockdown protects against HG condition RGC damage through the NRF2/HO1 signaling pathway, and may become a therapeutic target of DM-induced neural retinal neurodegeneration. |
| [78] | ↑MALAT1 | - | Morphological and functional changes in retinal photoreceptor cells | STZ-induced diabetes *, In vivo, mice * Eight weeks old male C57BL/6 mice with no ocular diseases were used. Except fasting for diabetes establishment, food and water available at all times. Mice were intraperitoneally injected with STZ (50mg/kg) for 5 consecutive days after an 8h fast to induce diabetes. On the 7th day after first injection, blood samples were collected to measure blood glucose concentration. Effective induction of the diabetes was defined as glucose levels greater than 300 mg/dL. | Retinal MALAT1 knockdown presents mitigative effects on rod and cone cells, thus MALAT1 silencing can be potentially used in diabetic neurodegeneration therapy |
| [79] | ↓GAS5 | TNF-α, IL-1β, IL-6 | Inflammation, Apoptosis, Endoplasmic reticulum stress | In vitro, HG condition, ARPE-19 human adult retinal pigment epithelial cells | The treatment of GAS5 suppressed endoplasmic reticulum stress-induced apoptosis and inflammation in HG-treated cells. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jarosz-Popek, J.; Wolska, M.; Gasecka, A.; Czajka, P.; Jakubik, D.; Sharif, L.; Adem, T.; Liu, W.-L.; Mirowska-Guzel, D.; Postula, M.; et al. The Importance of Non-Coding RNAs in Neurodegenerative Processes of Diabetes-Related Molecular Pathways. J. Clin. Med. 2021, 10, 9. https://doi.org/10.3390/jcm10010009
Jarosz-Popek J, Wolska M, Gasecka A, Czajka P, Jakubik D, Sharif L, Adem T, Liu W-L, Mirowska-Guzel D, Postula M, et al. The Importance of Non-Coding RNAs in Neurodegenerative Processes of Diabetes-Related Molecular Pathways. Journal of Clinical Medicine. 2021; 10(1):9. https://doi.org/10.3390/jcm10010009
Chicago/Turabian StyleJarosz-Popek, Joanna, Marta Wolska, Aleksandra Gasecka, Pamela Czajka, Daniel Jakubik, Lucia Sharif, Taqwa Adem, Wei-Ling Liu, Dagmara Mirowska-Guzel, Marek Postula, and et al. 2021. "The Importance of Non-Coding RNAs in Neurodegenerative Processes of Diabetes-Related Molecular Pathways" Journal of Clinical Medicine 10, no. 1: 9. https://doi.org/10.3390/jcm10010009
APA StyleJarosz-Popek, J., Wolska, M., Gasecka, A., Czajka, P., Jakubik, D., Sharif, L., Adem, T., Liu, W.-L., Mirowska-Guzel, D., Postula, M., & Eyileten, C. (2021). The Importance of Non-Coding RNAs in Neurodegenerative Processes of Diabetes-Related Molecular Pathways. Journal of Clinical Medicine, 10(1), 9. https://doi.org/10.3390/jcm10010009