Plasma Modification and Synthesis of Membrane Materials—A Mechanistic Review

 , , ,
, , ,  and
and 
Abstract
1. Introduction
2. Low-Pressure Plasma Processes
2.1. Plasma Gas Treatments of Membranes
2.1.1. Inert Gas Plasma—Argon and Helium Plasma
2.1.2. Oxidative Gases Plasma Including Oxygen/Carbon Dioxide/Water Vapor
Oxygen Gas Plasma
Carbon Dioxide Gas Plasma
Water Vapor Plasma
2.1.3. Reductive Gas Plasma—Ammonia (NH3)
2.1.4. The Mechanistic Overview of the Plasma Gas Processes
2.2. Low-Pressure Plasma Polymerization Treatments toward Surface Modification
2.2.1. Plasma Polymerization of Amine Monomers onto Membranes
2.2.2. Plasma Polymerization of Carboxyl Functional Groups onto Polymeric Membranes
2.2.3. Plasma Polymerization of Hydroxyl (–OH) Functional Monomers onto Membranes
2.2.4. Plasma Polymerization of Organosilicon and Fluorocarbon Moieties for Membranes Coating
Deposition of Silicon Oxide (SiOx) Thin Films
Deposition of Fluorocarbon (CFx) Thin Films
2.2.5. The Mechanistic Overview of the Plasma Polymerization Processes
3. Atmospheric Pressure Plasma Processes
3.1. Plasma Gas Treatments
3.2. Plasma Polymerization Modification by Thin Film Deposition at Atmospheric Pressure
3.2.1. Atmospheric Plasma Polymerization (APP)
Deposition of Carboxyl-Enriched Films
Deposition of Silicon Oxide (SiOx) Thin Films
3.2.2. Aerosol-Assisted-Atmospheric Plasma Polymerization (AA-APP)
3.2.3. The Mechanistic Overview of the Plasma Gas and Polymerization Processes at Atmospheric Pressure
4. Conclusions and Prospects
Funding
Acknowledgments
Conflicts of Interest
Nomenclature
| A | atoms |
| AA-APP | aerosol-assisted atmospheric plasma polymerization |
| AAc | acrylic acid |
| AC | alternating current |
| AlCeO3 | aluminum–cerium oxide |
| APP | atmospheric plasma polymerization |
| AP-PECVD | atmospheric-pressure plasma enhanced chemical vapor deposition |
| Ar | argon |
| BSA | bovine serum albumin |
| C=O | carbonyl group |
| C6F14 | perfluorohexane |
| C7F16 | heptane |
| CF4 | tetrafluoromethane |
| CFx | fluorocarbon |
| CO2 | carbon dioxide |
| –COO–/–COOH | carboxylic group |
| CW | continuous wave |
| DBD | dielectric barrier discharge |
| DI | deionized |
| DSSC | dye-sensitized solar cells |
| e− | electrons |
| FTIR-ATR | Fourier transform infrared spectroscopy-Attenuated total reflectance |
| H2O | water |
| He | helium |
| HMDSO | hexamethyldisiloxane |
| IEP | isoelectric point |
| LIB | lithium-ion battery |
| M | monomers |
| MA | maleic anhydride |
| MF | microfiltration |
| MTMOS | methyltrimethoxysilane |
| N2 | nitrogen |
| NC | nanocomposite |
| NF | nanofiltration |
| NH3 | ammonia |
| NP | nanoparticles |
| NR | not report |
| O2 | oxygen |
| OES | optical emission spectroscopy |
| -OH | hydroxyl group |
| PA | poly(amide) |
| PC | poly(carbonate) |
| PECVD | plasma enhanced chemical vapor deposition |
| PEG | poly(ethylene glycol) |
| PEO | poly(ethylene oxide) |
| PES | poly(ethersulfone) |
| PET | poly(ethylene terephthalate) |
| PET-TM | poly(ethylene terephthalate) track-etched membranes |
| PFSA | perfluorosulfonic acid |
| PP | poly(propylene) |
| PSf | poly(sulfone) |
| PTFE | poly(tetrafluoroethylene) |
| PVDF | poly(vinylidene fluoride) |
| PVDF-HFP | poly(vinylidene fluoride-co-hexafluoropropylene) |
| RF | radio frequency |
| RMS | roughness |
| RO | reverse osmosis |
| sccm | standard cubic centimeter per minute |
| SEM | scanning electron microscope |
| SiO2 | silica |
| SiO2–ZrO2 | silicon dioxide–zirconium dioxide |
| SiOx | silicon oxide |
| slm | standard liters per minute |
| TFC | thin film composite |
| TMMOS | trimethylmethoxysilane |
| TSEM | transmission scanning electron microscopy |
| UF | ultrafiltration |
| VIM | 1-vinylimidazole |
| WCA | water contact angle |
| XPS | X-ray photoelectron spectroscopy |
| ZnO | zinc oxide |
References
- Li, D.; Yan, Y.; Wang, H. Recent advances in polymer and polymer composite membranes for reverse and forward osmosis processes. Prog. Polym. Sci. 2016, 61, 104–155. [Google Scholar] [CrossRef]
- Kang, G.D.; Cao, Y.M. Application and modification of poly(vinylidene fluoride) (PVDF) membranes—A review. J. Membr. Sci. 2014, 463, 145–165. [Google Scholar] [CrossRef]
- Guo, W.; Ngo, H.-H.; Li, J. A mini-review on membrane fouling. Bioresour. Technol. 2012, 122, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Wang, J. Biofouling Control of Reverse Osmosis Membranes Using Free Nitrous Acid. In Proceedings of the 4th IWA Regional Conference on Membrane Technology, Ho Chi Minh City, Vietnam, 3–6 December 2014. [Google Scholar]
- Filloux, E.; Wang, J.; Pidou, M.; Gernjak, W.; Yuan, Z. Biofouling and scaling control of reverse osmosis membrane using one-step cleaning-potential of acidified nitrite solution as an agent. J. Membr. Sci. 2015, 495, 276–283. [Google Scholar] [CrossRef]
- Dumee, L.F.; He, L.; King, P.C.; Le Moing, M.; Güller, I.; Duke, M.; Hodgson, P.D.; Gray, S.; Poole, A.J.; Kong, L. Towards integrated anti-microbial capabilities: Novel bio-fouling resistant membranes by high velocity embedment of silver particles. J. Membr. Sci. 2015, 475, 552–561. [Google Scholar] [CrossRef]
- Reis, R.; Dumée, L.F.; Tardy, B.L.; Dagastine, R.; Orbell, J.D.; Schutz, J.A.; Duke, M.C. Towards enhanced performance thin-film composite membranes via surface plasma modification. Sci. Rep. 2016, 6. Available online: https://www.nature.com/articles/srep29206 (accessed on 26 July 2018). [CrossRef] [PubMed]
- Reis, R.; Dumée, L.F.; He, L.; She, F.; Orbell, J.D.; Winther-Jensen, B.; Duke, M.C. Amine enrichment of thin-film composite membranes via low pressure plasma polymerization for antimicrobial adhesion. ACS Appl. Mater. Interfaces 2015, 7, 14644–14653. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.J.; Du Plessis, J.; Kyratzis, I.L.; Maurdev, G.; Huson, M.G.; Coombs, C. Controlled amine functionalization and hydrophilicity of a poly(lactic acid) fabric. Plasma Process. Polym. 2009, 6, 489–497. [Google Scholar] [CrossRef]
- Meyyappan, M. Plasma nanotechnology: Past, present and future. J. Phys. D: Appl. Phys. 2011, 44. [Google Scholar] [CrossRef]
- Kravets, L.I.; Gilman, A.B.; Dinescu, G. Modification of polymer membrane properties by low-temperature plasma. Rus. J. Gen. Chem. 2015, 85, 1284–1301. [Google Scholar] [CrossRef]
- Kang, M.S.; Chun, B.; Kim, S.S. Surface modification of polypropylene membrane by low-temperature plasma treatment. J. Appl. Polym. Sci. 2001, 81, 1555–1566. [Google Scholar] [CrossRef]
- Kim, H.I.; Kim, S.S. Fabrication of reverse osmosis membrane via low temperature plasma polymerization. J. Membr. Sci. 2001, 190, 21–33. [Google Scholar] [CrossRef]
- Michelmore, A.; Steele, D.A.; Whittle, J.D.; Bradley, J.W.; Short, R.D. Nanoscale deposition of chemically functionalised films via plasma polymerisation. RSC Adv. 2013, 3, 13540–13557. [Google Scholar] [CrossRef]
- Siow, K.S.; Britcher, L.; Kumar, S.; Griesser, H.J. Plasma methods for the generation of chemically reactive surfaces for biomolecule immobilization and cell colonization—A review. Plasma Process. Polym. 2006, 3, 392–418. [Google Scholar] [CrossRef]
- Khelifa, F.; Ershov, S.; Habibi, Y.; Snyders, R.; Dubois, P. Free-Radical-Induced Grafting from Plasma Polymer Surfaces. Chem. Rev. 2016, 116, 3975–4005. [Google Scholar] [CrossRef] [PubMed]
- Bryjak, M.; Gancarz, I.; Pozniak, G. Plasma-modified porous membranes. Chem. Pap. Chem. Zvesti 2000, 54, 496–501. [Google Scholar]
- Bryjak, M.; Gancarz, I.; Smolinska, K. Plasma nanostructuring of porous polymer membranes. Adv. Colloid Interface Sci. 2010, 161, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Khulbe, K.C.; Feng, C.; Matsuura, T. The art of surface modification of synthetic polymeric membranes. J. Appl. Polym. Sci. 2010, 115, 855–895. [Google Scholar] [CrossRef]
- Kochkodan, V.M.; Sharma, V.K. Graft polymerization and plasma treatment of polymer membranes for fouling reduction: A review. J. Environ. Sci. Health Part A 2012, 47, 1713–1727. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.J.; Dreyer, D.R.; Bielawski, C.W.; Paul, D.R.; Freeman, B.D. Surface modification of water purification membranes. Angew. Chem. Int. Ed. 2017, 56, 4662–4711. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, J. Mechanisms of plasma polymerization—Reviewed from a chemical point of view. Plasma Process. Polym. 2011, 8, 783–802. [Google Scholar] [CrossRef]
- Massines, F.; Sarra-Bournet, C.; Fanelli, F.; Naudé, N.; Gherardi, N. Atmospheric Pressure Low Temperature Direct Plasma Technology: Status and Challenges for Thin Film Deposition. Plasma Process. Polym. 2012, 9, 1041–1073. [Google Scholar] [CrossRef]
- Merche, D.; Vandencasteele, N.; Reniers, F. Atmospheric plasmas for thin film deposition: A critical review. Thin Solid Films 2012, 520, 4219–4236. [Google Scholar] [CrossRef]
- Yu, H.Y.; He, X.C.; Liu, L.Q.; Gu, J.S.; Wei, X.W. Surface modification of poly (propylene) microporous membrane to improve its antifouling characteristics in an SMBR: O2 plasma treatment. Plasma Process. Polym. 2008, 5, 84–90. [Google Scholar] [CrossRef]
- Jaleh, B.; Parvin, P.; Wanichapichart, P.; PourakbarSaffar, A.; Reyhani, A. Induced super hydrophilicity due to surface modification of polypropylene membrane treated by O2 plasma. Appl. Surf. Sci. 2010, 257, 1655–1659. [Google Scholar] [CrossRef]
- Wavhal, D.S.; Fisher, E.R. Modification of polysulfone ultrafiltration membranes by CO2 plasma treatment. Desalination 2005, 172, 189–205. [Google Scholar] [CrossRef]
- Wavhal, D.S.; Fisher, E.R. Modification of porous poly(ether sulfone) membranes by low-temperature CO2-plasma treatment. J. Polym. Sci. Part B: Polym. Phys. 2002, 40, 2473–2488. [Google Scholar] [CrossRef]
- Reis, R.; Dumée, L.F.; Merenda, A.; Orbell, J.D.; Schütz, J.A.; Duke, M.C. Plasma-induced physicochemical effects on a poly (amide) thin-film composite membrane. Desalination 2017, 403, 3–11. [Google Scholar] [CrossRef]
- Yan, M.-G.; Liu, L.Q.; Tang, Z.Q.; Huang, L.; Li, W.; Zhou, J.; Gu, J.S.; Wei, X.W.; Yu, H.Y. Plasma surface modification of polypropylene microfiltration membranes and fouling by BSA dispersion. Chem. Eng. J. 2008, 145, 218–224. [Google Scholar] [CrossRef]
- Fridman, A. Plasma Chemistry; Cambridge University: New York, NY, USA, 2008. [Google Scholar]
- Tompkins, B.D.; Dennison, J.M.; Fisher, E.R. H2O plasma modification of track-etched polymer membranes for increased wettability and improved performance. J. Membr. Sci. 2013, 428, 576–588. [Google Scholar] [CrossRef]
- Dumée, L.F.; Alglave, H.; Chaffraix, T.; Lin, B.; Magniez, K.; Schütz, J. Morphology-properties relationship of gas plasma treated hydrophobic meso-porous membranes and their improved performance for desalination by membrane distillation. Appl. Surf. Sci. 2016, 363, 273–285. [Google Scholar] [CrossRef]
- Kravets, L.; Gilman, A.; Yablokov, M.; Elinson, V.; Mitu, B.; Dinescu, G. Surface and electrochemical properties of plasma-treated polypropylene track membrane. Plasma Process. Polym. 2013, 10, 603–618. [Google Scholar] [CrossRef]
- Kim, H.I.; Kim, S.S. Plasma treatment of polypropylene and polysulfone supports for thin film composite reverse osmosis membrane. J. Membr. Sci. 2006, 286, 193–201. [Google Scholar] [CrossRef]
- Pegalajar-Jurado, A.; Mann, M.N.; Maynard, M.R.; Fisher, E.R. Hydrophilic modification of polysulfone ultrafiltration membranes by low temperature water vapor plasma treatment to enhance performance. Plasma Process. Polym. 2016, 13, 598–610. [Google Scholar] [CrossRef]
- Steen, M.L.; Jordan, A.C.; Fisher, E.R. Hydrophilic modification of polymeric membranes by low temperature H2O plasma treatment. J. Membr. Sci. 2002, 204, 341–357. [Google Scholar] [CrossRef]
- Saxena, N.; Prabhavathy, C.; De, S.; DasGupta, S. Flux enhancement by argon–oxygen plasma treatment of polyethersulfone membranes. Sep. Purif. Technol. 2009, 70, 160–165. [Google Scholar] [CrossRef]
- Juang, R.-S.; Huang, C.; Hsieh, C.-L. Surface modification of PVDF ultrafiltration membranes by remote argon/methane gas mixture plasma for fouling reduction. J. Taiwan Inst. Chem. Eng. 2014, 45, 2176–2186. [Google Scholar] [CrossRef]
- Wu, S.; Xing, J.; Zheng, C.; Xu, G.; Zheng, G.; Xu, J. Plasma modification of aromatic polyamide reverse osmosis composite membrane surface. J. Appl. Polym. Sci. 1997, 64, 1923–1926. [Google Scholar] [CrossRef]
- Bryjak, M.; Gancarz, I.; Poźniak, G.; Tylus, W. Modification of polysulfone membranes 4. Ammonia plasma treatment. Eur. Polym. J. 2002, 38, 717–726. [Google Scholar] [CrossRef]
- Steen, M.L.; Hymas, L.; Havey, E.D.; Capps, N.E.; Castner, D.G.; Fisher, E.R. Low temperature plasma treatment of asymmetric polysulfone membranes for permanent hydrophilic surface modification. J. Membr. Sci. 2001, 188, 97–114. [Google Scholar] [CrossRef]
- Kull, K.R.; Steen, M.L.; Fisher, E.R. Surface modification with nitrogen-containing plasmas to produce hydrophilic, low-fouling membranes. J. Membr. Sci. 2005, 246, 203–215. [Google Scholar] [CrossRef]
- Yu, H.-Y.; He, X.C.; Liu, L.Q.; Gu, J.S.; Wei, X.W. Surface modification of polypropylene microporous membrane to improve its antifouling characteristics in an SMBR: N2 plasma treatment. Water Res. 2007, 41, 4703–4709. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.-Y.; Hu, M.X.; Xu, Z.K.; Wang, J.L.; Wang, S.Y. Surface modification of polypropylene microporous membranes to improve their antifouling property in MBR: NH3 plasma treatment. Sep. Purif. Technol. 2005, 45, 8–15. [Google Scholar] [CrossRef]
- Gancarz, I.; Poźniak, G.; Bryjak, M. Modification of polysulfone membranes 1. CO2 plasma treatment. Eur. Polym. J. 1999, 35, 1419–1428. [Google Scholar] [CrossRef]
- Ulbricht, M.; Belfort, G. Surface modification of ultrafiltration membranes by low temperature plasma II. Graft polymerization onto polyacrylonitrile and polysulfone. J. Membr. Sci. 1996, 111, 193–215. [Google Scholar] [CrossRef]
- Pal, D.; Neogi, S.; De, S. Improved antifouling characteristics of acrylonitrile co-polymer membrane by low temperature pulsed ammonia plasma in the treatment of oil-water emulsion. Vacuum 2016, 131, 293–304. [Google Scholar] [CrossRef]
- Gancarz, I.; Poźniak, G.; Bryjak, M.; Frankiewicz, A. Modification of polysulfone membranes. 2. Plasma grafting and plasma polymerization of acrylic acid. Acta Polym. 1999, 50, 317–326. [Google Scholar] [CrossRef]
- Lee, J.; Hill, A.; Kentish, S. Formation of a thick aromatic polyamide membrane by interfacial polymerisation. Sep. Purif. Technol. 2013, 104, 276–283. [Google Scholar] [CrossRef]
- Wavhal, D.S.; Fisher, E.R. Hydrophilic modification of polyethersulfone membranes by low temperature plasma-induced graft polymerization. J. Membr. Sci. 2002, 209, 255–269. [Google Scholar] [CrossRef]
- Gancarz, I.; Poźniak, G.; Bryjak, M. Modification of polysulfone membranes: 3. Effect of nitrogen plasma. Eur. Polym. J. 2000, 36, 1563–1569. [Google Scholar] [CrossRef]
- Yasuda, H. Glow discharge polymerization. J. Polym. Sci. Macromol. Rev. 1981, 16, 199–293. [Google Scholar] [CrossRef]
- Reis, R.; Duke, M.; Merenda, A.; Winther-Jensen, B.; Puskar, L.; Tobin, M.J.; Orbell, J.D.; Dumée, L.F. Customizing the surface charge of thin-film composite membranes by surface plasma thin film polymerization. J. Membr. Sci. 2017, 537, 1–10. [Google Scholar] [CrossRef]
- Kramer, P.W.; Yeh, Y.S.; Yasuda, H. Low-Temperature Plasma for the Preparation of Separation Membranes. J. Membr. Sci. 1989, 46, 1–28. [Google Scholar] [CrossRef]
- Yasuda, H.; Lamaze, C.E. Preparation of reverse osmosis membranes by plasma polymerization of organic compounds. J. Appl. Polym. Sci. 1973, 17, 201–222. [Google Scholar] [CrossRef]
- Yasuda, H. Composite reverse osmosis membranes prepared by plasma polymerization. In Reverse Osmosis and Synthetic Membranes; Sourirajan, S., Ed.; National Research Council Canada: Ottawa, ON, Canada, 1977; pp. 263–294. [Google Scholar]
- Yasuda, H. Plasma polymerization for protective coatings and composite membranes. J. Membr. Sci. 1984, 18, 273–284. [Google Scholar] [CrossRef]
- Gancarz, I.; Poźniak, G.; Bryjak, M.; Tylus, W. Modification of polysulfone membranes 5. Effect of n-butylamine and allylamine plasma. Eur. Polym. J. 2002, 38, 1937–1946. [Google Scholar] [CrossRef]
- Poźniak, G.; Gancarz, I.; Bryjak, M.; Tylus, W. N-butylamine plasma modifying ultrafiltration polysulfone membranes. Desalination 2002, 146, 293–299. [Google Scholar] [CrossRef]
- Kravets, L.; Dmitriev, S.; Gilman, A.; Drachev, A.; Dinescu, G. Water permeability of poly(ethylene terephthalate) track membranes modified by DC discharge plasma polymerization of dimethylaniline. J. Membr. Sci. 2005, 263, 127–136. [Google Scholar] [CrossRef]
- Reis, R.; Duke, M.C.; Tardy, B.L.; Oldfield, D.; Dagastine, R.R.; Orbell, J.D.; Dumée, L.F. Charge tunable thin-film composite membranes by gamma-ray triggered surface polymerization. Sci. Rep. 2017, 7, 4426. [Google Scholar] [CrossRef] [PubMed]
- Topala, I.; Dumitrascu, N.; Popa, G. Properties of the acrylic acid polymers obtained by atmospheric pressure plasma polymerization. Nucl. Instrum. Methods Phys. Res. Sect. B 2009, 267, 442–445. [Google Scholar] [CrossRef]
- Çökeliler, D. Enhancement of polycarbonate membrane permeability due to plasma polymerization precursors. Appl. Surf. Sci. 2013, 267, 28–36. [Google Scholar] [CrossRef]
- Zou, L.; Vidalis, I.; Steele, D.; Michelmore, A.; Low, S.P.; Verberk, J.Q.J.C. Surface hydrophilic modification of RO membranes by plasma polymerization for low organic fouling. J. Membr. Sci. 2011, 369, 420–428. [Google Scholar] [CrossRef]
- Nagasawa, H.; Minamizawa, T.; Kanezashi, M.; Yoshioka, T.; Tsuru, T. Microporous organosilica membranes for gas separation prepared via PECVD using different O/Si ratio precursors. J. Membr. Sci. 2015, 489, 11–19. [Google Scholar] [CrossRef]
- Nagasawa, H.; Yamamoto, Y.; Tsuda, N.; Kanezashi, M.; Yoshioka, T.; Tsuru, T. Atmospheric-pressure plasma-enhanced chemical vapor deposition of microporous silica membranes for gas separation. J. Membr. Sci. 2017, 524, 644–651. [Google Scholar] [CrossRef]
- Trunec, D.; Navratil, Z.; Stahel, P.; Zajíčková, L.; Buršíková, V.; Cech, J. Deposition of thin organosilicon polymer films in atmospheric pressure glow discharge. J. Phys. D Appl. Phys. 2004, 37, 2112–2120. [Google Scholar] [CrossRef]
- Nagasawa, H.; Shigemoto, H.; Kanezashi, M.; Yoshioka, T.; Tsuru, T. Characterization and gas permeation properties of amorphous silica membranes prepared via plasma enhanced chemical vapor deposition. J. Membr. Sci. 2013, 441, 45–53. [Google Scholar] [CrossRef]
- Ngamou, P.H.T.; Overbeek, J.P.; Kreiter, R.; van Veen, H.M.; Vente, J.F.; Wienk, I.M.; Cuperus, P.F.; Creatore, M. Plasma-deposited hybrid silica membranes with a controlled retention of organic bridges. J. Mater. Chem. A 2013, 1, 5567–5576. [Google Scholar] [CrossRef]
- Bae, B.; Chun, B.H.; Kim, D. Surface characterization of microporous polypropylene membranes modified by plasma treatment. Polymer 2001, 42, 7879–7885. [Google Scholar] [CrossRef]
- Lue, S.J.; Hsiaw, S.Y.; Wei, T.C. Surface modification of perfluorosulfonic acid membranes with perfluoroheptane (C7F16)/argon plasma. J. Membr. Sci. 2007, 305, 226–237. [Google Scholar] [CrossRef]
- Trofimov, D.; Shkinev, V.M.; Spivakov, B.Y.; Schué, F. Improvement of pore geometry and performances of poly (ethylene terephthalate) track membranes by a protective layer method using plasma-induced graft polymerization of 1H, 1H, 2H-perfluoro-1-octene monomer. J. Membr. Sci. 2009, 326, 265–269. [Google Scholar] [CrossRef]
- Gancarz, I.; Bryjak, M.; Kujawski, J.; Wolska, J.; Kujawa, J.; Kujawski, W. Plasma deposited fluorinated films on porous membranes. Mater. Chem. Phys. 2015, 151, 233–242. [Google Scholar] [CrossRef]
- Wei, X.; Zhao, B.; Li, X.M.; Wang, Z.; He, B.Q.; He, T.; Jiang, B. CF4 plasma surface modification of asymmetric hydrophilic polyethersulfone membranes for direct contact membrane distillation. J. Membr. Sci. 2012, 407, 164–175. [Google Scholar] [CrossRef]
- Biloiu, C.; Biloiu, I.A.; Sakai, Y.; Sugawara, H.; Ohta, A. Amorphous fluorocarbon polymer (a-C:F) films obtained by plasma enhanced chemical vapor deposition from perfluoro-octane (C8F18) vapor. II. Dielectric and insulating properties. J. Vac. Sci. Technol. A 2004, 22, 1158–1165. [Google Scholar] [CrossRef]
- Yasuda, H.; Yasuda, T. The competitive ablation and polymerization (CAP) principle and the plasma sensitivity of elements in plasma polymerization and treatment. J. Polym. Sci. Part A Polym. Chem. 2000, 38, 943–953. [Google Scholar] [CrossRef]
- Huang, C.; Lin, P.J.; Tsai, C.Y.; Juang, R.S. Electrospun Microfibrous Membranes with Atmospheric-Pressure Plasma Surface Modification for the Application in Dye-Sensitized Solar Cells. Plasm. Process. Polym. 2013, 10, 938–947. [Google Scholar] [CrossRef]
- Shenton, M.J.; Stevens, G.C. Surface modification of polymer surfaces: Atmospheric plasma versus vacuum plasma treatments. J. Phys. D Appl. Phys. 2001, 34, 2761–2768. [Google Scholar] [CrossRef]
- Juang, R.S.; Chen, K.S.; Wei, T.C.; Liu, C.H.; Tsai, C.Y.; Jheng, H.Y.; Huang, C. Surface Characterization of Argon/Methane Mixture Atmospheric-Pressure Plasma-Treated Filtration Poly(Vinylidene Fluoride) Membrane and Its Flux Enhancement. IEEE Trans. Plasma Sci. 2014, 42, 3698–3702. [Google Scholar] [CrossRef]
- Yin, M.; Huang, J.; Yu, J.; Chen, G.; Qu, S.; Wang, X.; Li, C. The polypropylene membrane modified by an atmospheric pressure plasma jet as a separator for lithium-ion button battery. Electrochimica Acta 2018, 260, 489–497. [Google Scholar] [CrossRef]
- Donegan, M.; Dowling, D.P. Protein adhesion on water stable atmospheric plasma deposited acrylic acid coatings. Surf. Coat. Technol. 2013, 234, 53–59. [Google Scholar] [CrossRef]
- Manakhov, A.; Michlíček, M.; Nečas, D.; Polčák, J.; Makhneva, E.; Eliáš, M.; Zajíčková, L. Carboxyl-rich coatings deposited by atmospheric plasma co-polymerization of maleic anhydride and acetylene. Surf. Coat. Technol. 2016, 295, 37–45. [Google Scholar] [CrossRef]
- Chen, X.; Chen, Z.; Dumée, L.F.; O’Dell, L.A.; du Plessis, J.; d’Agostino, R.; Dai, X.J.; Magniez, K. Grafting of N-moieties onto octa-methyl polyhedral oligomeric silsesquioxane microstructures by sequential continuous wave and pulsed plasma. Plasm. Process. Polym. 2017, 14, 1600244. [Google Scholar] [CrossRef]
- Massines, F.; Gouda, G. A comparison of polypropylene-surface treatment by filamentary, homogeneous and glow discharges in helium at atmospheric pressure. J. Phys. D Appl. Phys. 1998, 31, 3411. [Google Scholar] [CrossRef]
- Massines, F.; Gherardi, N.; Fornelli, A.; Martin, S. Atmospheric pressure plasma deposition of thin films by Townsend dielectric barrier discharge. Surf. Coat. Technol. 2005, 200, 1855–1861. [Google Scholar] [CrossRef]
- Gomathi, N.; Sureshkumar, A.; Neogi, S. RF plasma-treated polymers for biomedical applications. Curr. Sci. 2008, 94, 1478–1486. [Google Scholar]
- Chun, S.J.; Choi, E.S.; Lee, E.H.; Kim, J.H.; Lee, S.Y.; Lee, S.Y. Eco-friendly cellulose nanofiber paper-derived separator membranes featuring tunable nanoporous network channels for lithium-ion batteries. J. Mater. Chem. 2012, 22, 16618–16626. [Google Scholar] [CrossRef]
- Zhang, J.; Xu, Y.; Li, W.; Zhou, J.; Zhao, J.; Qian, G.; Xu, Z.P. Enhanced remediation of Cr(VI)-contaminated soil by incorporating a calcined-hydrotalcite-based permeable reactive barrier with electrokinetics. J. Hazard. Mater. 2012, 239, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Drews, J.; Launay, H.; Hansen, C.M.; West, K.; Hvilsted, S.; Kingshott, P.; Almdal, K. Hydrolysis and stability of thin pulsed plasma polymerised maleic anhydride coatings. Appl. Surf. Sci. 2008, 254, 4720–4725. [Google Scholar] [CrossRef]
- Schiller, S.; Hu, J.; Jenkins, A.T.A.; Timmons, R.B.; Sanchez-Estrada, F.S.; Knoll, W.; Förch, R. Chemical Structure and Properties of Plasma-Polymerized Maleic Anhydride Films. Chem. Mater. 2002, 14, 235–242. [Google Scholar] [CrossRef]
- Friedrich, J.F.; Mix, R.; Schulze, R.D.; Meyer-Plath, A.; Joshi, R.; Wettmarshausen, S. New Plasma Techniques for Polymer Surface Modification with Monotype Functional Groups. Plasm. Process. Polym. 2008, 5, 407–423. [Google Scholar] [CrossRef]
- Fanelli, F.; Mastrangelo, A.M.; Fracassi, F. Aerosol-Assisted Atmospheric Cold Plasma Deposition and Characterization of Superhydrophobic Organic–Inorganic Nanocomposite Thin Films. Langmuir 2014, 30, 857–865. [Google Scholar] [CrossRef] [PubMed]
- Fanelli, F.; Fracassi, F. Aerosol-Assisted Atmospheric Pressure Cold Plasma Deposition of Organic–Inorganic Nanocomposite Coatings. Plasm. Chem. Plasm. Process. 2014, 34, 473–487. [Google Scholar] [CrossRef]
- Da Ponte, G.; Sardella, E.; Fanelli, F.; d’Agostino, R.; Gristina, R.; Favia, P. Plasma Deposition of PEO-Like Coatings with Aerosol-Assisted Dielectric Barrier Discharges. Plasm. Process. Polym. 2012, 9, 1176–1183. [Google Scholar] [CrossRef]
- Nisol, B.; Poleunis, C.; Bertrand, P.; Reniers, F. Poly(ethylene glycol) Films Deposited by Atmospheric Pressure Plasma Liquid Deposition and Atmospheric Pressure Plasma-Enhanced Chemical Vapour Deposition: Process, Chemical Composition Analysis and Biocompatibility. Plasm. Process. Polym. 2010, 7, 715–725. [Google Scholar] [CrossRef]
- Bardon, J.; Bour, J.; Del Frari, D.; Arnoult, C.; Ruch, D. Dispersion of Cerium-Based Nanoparticles in an Organosilicon Plasma Polymerized Coating: Effect on Corrosion Protection. Plasm. Process. Polym. 2009, 6, S655–S659. [Google Scholar] [CrossRef]
- Robert, E.; Barbosa, E.; Dozias, S.; Vandamme, M.; Cachoncinlle, C.; Viladrosa, R.; Pouvesle, J.M. Experimental Study of a Compact Nanosecond Plasma Gun. Plasma Process. Polym. 2009, 6, 795–802. [Google Scholar] [CrossRef]
- Robert, E.; Sarron, V.; Riès, D.; Dozias, S.; Vandamme, M.; Pouvesle, J.M. Characterization of pulsed atmospheric-pressure plasma streams (PAPS) generated by a plasma gun. Plasma Sources Sci. Technol. 2012, 21, 034017. [Google Scholar] [CrossRef]
- Seok, O.J.; Yolanda, A.G.; James, W.B. Time-resolved mass spectroscopic studies of an atmospheric-pressure helium microplasma jet. J. Phys. D Appl. Phys. 2011, 44, 365202. [Google Scholar]
- McKay, K.; Oh, J.S.; Walsh, J.L.; Bradley, J.W. Mass spectrometric diagnosis of an atmospheric pressure helium microplasma jet. J. Phys. D Appl. Phys. 2013, 46, 464018. [Google Scholar] [CrossRef]
- Bhusan, S.B.; Han, J.G.; Shin, K.S.; Hori, M. Nitrogen Radical and Plasma Diagnostics in Dual Frequency Hybrid Plasmas to Investigate N2/SiH4 PECVD Process. Plasma Process. Polym. 2016, 13, 447–458. [Google Scholar]
- Vetter, J.; Barbezat, G.; Crummenauer, J.; Avissar, J. Surface treatment selections for automotive applications. Surf. Coat. Technol. 2005, 200, 1962–1968. [Google Scholar] [CrossRef]
- Andrea, Z.; Ribeiro, O.F.; Pedro, S.A. Plasma Treatment in Textile Industry. Plasma Process. Polym. 2015, 12, 98–131. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Entry | Plasma Treatment | Plasma Conditions | Membrane | Flux (L m−2 h−1) | Salt Rejection (%) | Water Contact Angle(°) | Surface Charge (pH) | Roughness RMS (nm) | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Ar | 10, 50, or 80 W RF power; 0.2 mbar; 1, 5, 15, or 30 min | RO PA Hydrophilic BW30 TFC (Dow Filmtec Corp.) | Raised by 22% (10–50 W) and then dropped by 76% (80 W; 30 min) compared to control 45 | 98 (control) to 97 (10–50 W; 1–15min), ~60% (50 W; 30 min), ~6% (80 W; 30 min) 1 | Declined ~15 with increasing power density and time from 60 | Negative charge from pH 3 to 8 for both control and modified | Declined to ~40 (80 W; 30 min) from 60 (control) | [7] |
| 2 | He | 10 or 80 W RF power; 0.2 mbar; 1, 2 or 5 min | RO PA Hydrophilic BW30 TFC (Dow Filmtec Corp.) | Raised by 66% (10 W, 5 min); by 25% (80 W, 5 min) compared to pristine RO PA 30 | Maintained at 98% 1 | 47 (PA control) to 10 (5 min 10 W) | NR | 63 (PA control) to 58 (10W, 2 min); to ~40 (80 W, 5 min) | [29] |
| 3 | O2 | 30 W RF power; 10 cm3/min O2 vapor flow rate; 0.1 mbar; 0–10 min. | UF PP Hydrophobic Laboratory synthesized | Increased 30% after 1 min, and 15% after 4 min, compared to its control 350 | NR 2 | 128 (control) to 72 after 9 min treatment | NR | NR | [25] |
| 4 | O2 | 25 W RF power; 0.1 mbar; 1–5 min. | MF PP Hydrophobic Osmonics, Germany | Increased >50% after 5 min, compared to its control 243 | NR | 135 to 20 after 5 min treatment | NR | NR | [26] |
| 5 | O2 or Ar | 100 W; 20 kHz frequency; 0.13 mbar; 0–6 min. | RO PA Hydrophilic Laboratory synthesized | Increased more than 2.5 times its control (20) after 3 min O2 plasma; whilst only 4% higher than its control after 3 min Ar plasma | NR | 77 (Control, laboratory synthesized) to 70 after 2 min, and to 44 after 6 min O2 plasma; to 69 after 6 min Ar plasma | NR | NR | [40] |
| 6 | CO2 | 5, 10, and 20 W RF power; 0.2 mbar; 10–300 s | UF PSf Hydrophobic US Filter, Inc. | Increased 2.3-fold compared to control (175) modified at 10 W | NR | 94 (control) declined to 47 (10 s), to 15 (30 s), and to 0 (60 s and 180 s) at 10 W 3 | NR | NR | [27] |
| 7 | CO2 | 20 and 35 W RF power; 0.2 mbar; 0.5–15 min | UF PES Hydrophobic Millipore Corporation | NR | NR | 66 (control) to 0, with the water drop, disappears within 25 s (35 W, 30 s) and 75 s (20 W, 30 s) | NR | NR | [28] |
| 8 | H2O | 25 W RF power; 0.5 mbar; 2 min | UF PSf Hydrophobic US Filter, Inc. | NR | NR | 86 (control) to 0 | NR | NR | [42] |
| 9 | H2O | 25 W RF power; 0.5 mbar; 2–4 min | UF PES and PE Hydrophobic Millipore Corporation | Increased 28.3% for PES (compared to its control 4856) and 28.4% for PE (compared to its control 421) | NR | 63 (control) to 0 for PES, 123 to 0 for PE 3 | NR | NR | [37] |
| 10 | H2O | 25 W RF power; 0.7 mbar; 2 min | MF PC and PET Hydrophobic Sterlitech Corporation | Increased from 25 (control) to 68 for PC, and raised from 20 to 45 for PET | NR | 97 (control) to 38 for PC, 59 (control) to 27 for PET | NR | NR | [32] |
| 11 | H2O | 10 and 80 W RF power; 0.2 mbar; 1, 2, and 5 min | RO PA Hydrophilic BW30 TFC Dow Filmtec Corporation | Declined by >50% compared to pristine RO PA 30 | 98–84% (80W) 1 | Declined to ~11 (modified –10 W) ~20 (modified –80 W) from 47 (control) | Negative charged from pH 3 to 8 for both control and modified | Declined to 58 (10 W), ~36 (80 W) from 63 (control) after 2 min | [29] |
| 12 | NH3 | 30 W RF power; 0.1 mbar; 0–8 min. | UF PP Hydrophobic Laboratory synthesized | Two times higher than control (350) for 1 min-treated sample, 20% higher 8 min treated samples | NR | 128 (control) to 54 after 8 min | NR | NR | [30] |
| 13 | NH3 | 30 W RF power; 0.1 mbar; 4 min. | UF PP Hydrophobic Laboratory synthesized | NR | NR | 128 (control) to 71 under 10 Pa; to 90 under 104 Pa | NR | NR | [45] |
| 14 | NH3 | 450 V Pulsed DC power supply; 20 kHz; 0.12 mbar; 9.6 cm3/min; three duty cycles (Dt), 30%, 50%, and 70%; 0–8 min | UF PAN Hydrophobic Laboratory synthesized | 32% higher than PAN (control ca. 55) after 1 h oil-water filtration test 4 | NR | 89 (control) to 29 (8 min, 30% Dt), to 13 (8 min, 70% Dt) | NR | NR | [48] |
| 15 | NH3, NH3/Ar | 60 W microwave power; 125 Hz frequency and 25% of duty cycle; 1 mbar; 10 cm3/min Ar flow rate; 0–10 min. | UF PSf Hydrophobic (Amoco, CO., US) | NR | NR | 87 (control) to 46 (not specified in the study) | NR | NR | [41] |
| 16 | NH3, NH3/O2 | 15–120 (25) W RF power; 0.07–0.53 mbar; 2–25 (3) min | UF PES Hydrophobic Millipore Inc. | 70% (25 W, 3 min, 3:5 NH3/O2) higher than PES (control ca. 260) after 30 min PW filtration | NR | 66 (control) to 0 (25 W, 3 min, 3:5 NH3/O2) | NR | NR | [43] |
| Constant Conditions | Variable Conditions | PP Membranes | PSf Membranes | ||
|---|---|---|---|---|---|
| Water Flux (L m−2 h−1) | Salt Rejection (%) | Water Flux (L m−2 h−1) | Salt Rejection (%) | ||
| 10 W/0.8 sccm | pristine | 15.5 | 0 | 15.2 | 0 |
| After 60 min | 0.1 | 92 | 0.5 | 95 | |
| 0.8 sccm/30 min | 10 W | ~0.7 | ~88 | ~1.5 | 85 |
| 50 W | 0.2 | ~88 | ~1.55 | 88 | |
| 10 W/60 min | 0.8 sccm | 0.1 | 92 | 0.5 | 95 |
| 1.8 sccm | 5.5 | 20 | ~2.75 | 30 | |
| Entry | Plasma Polymerization Treatment | Plasma Conditions | Application | Flux (L m−2 h−1) d | Salt/Solute Rejection (%) | Water Contact Angle(°) | Surface Charge - IEP (pH) | Roughness RMS (nm) | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Allylamine  | 10–50 W RF power; monomer flow rate = 0.6–1.8 sccm (standard cm3/min); 1–60 min; | MF PP (Hoechst-Celanese Co.) and UF PSf Laboratory synthesized Hydrophobic | Declined 91% for PP, and 96% for PSf (10 W, 0.8 sccm, 50 min), compared to its control 15.5 | Salt rejection of PP and PSf increased 90% and 86% from 0%, respectively 1 | NR 2 | NR | NR | [13] |
| 2 | Allylamine | 10–50 W RF power; reactor pressure at 0.053, 0.093 and 0.133 mbar; 10–30 min | MF PP Hydrophobic (Hoechst-Celanese Co.) | Increased by ~38.5% (5 W, 5.332 Pa, and 10 min), compared to its control 48 | 89.8% of BSA rejection at pH 7 (5 W, 5.332 Pa, and 10 min) 3 | 108 (PP control) declined 38 (5 W, 5.332 Pa, and 30 min) | Negative charged at pH 7 | NR | [12] |
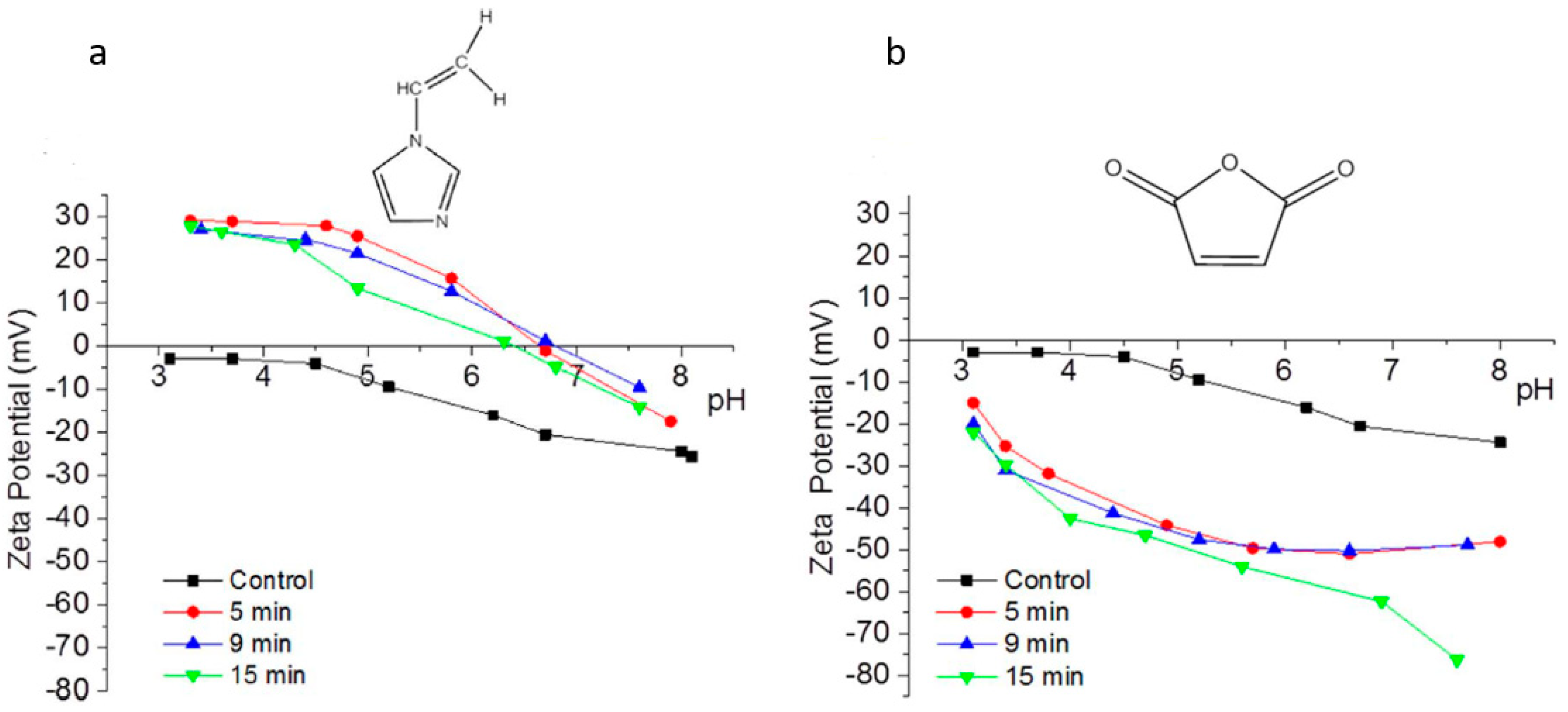
| 3 | 1-vinyl(imidazole) with Ar  | 1 W/L AC power; 0.07 mbar; 1.60 mL/min; 5, 9, and 15 min; | RO PA Hydrophilic BW30 TFC (Dow Filmtec Corp.) | Statistically stable compared to its control 44.2 | 96 to 97 4 | NR | Positively charged from pH 3 to 7 and IEP changed from pH 3.5 to ~7 | Reduced by 30% from 24 (control) to 17 (15 min) | [8,54] |
| 4 | Acrylic acid  | 10–50 W RF power; reactor pressure at 0.053, 0.093, and 0.133 mbar; 10–30 min; | MF PP Hydrophobic (Hoechst-Celanese Co.) | Increased by ~50.0% (5 W, 5.332 Pa, and 10 min), compared to its control 48 | 96.2% of BSA rejection at pH 7 (5 W, 5.332 Pa, and 10 min) 3 | 108 (PP control) declined 25 (5 W, 5.332 Pa, and 30 min) | Negatively charged at pH 7 | NR | [12] |
| 5 | Acrylic acid | 20 W RF power; 25 mL/min (monomer vapor flow rate); 10 min | UF PC(TE) Hydrophobic (Poretics, USA) | NR | NR | Decreased from 71.8 to 36.4 (10 min) | NR | NR | [64] |
| 6 | MA with Ar  | 1 W/L AC power; 0.07 mbar; 1.60 mL/min; 5, 9, and 15 min; | RO PA Hydrophilic BW30 TFC (Dow Filmtec Corp.) | Declined by 33% and 18% after 9 and 15 min, compared to its control 44.2 | 96.8 to 97.5 4 | NR | Negative from pH 3 to 8 | NR | [54] |
| 7 | Triglyme Polyethylene glycol (PEG)—like monomers  | 1 W RF power; monomer flow rate: 0.4 sscm at 80–90 °C; 10, 15, 30, 60, and 120 s; | RO PA Hydrophilic SW30HR TFC (Dow Filmtec Corp.) | 10–15% decline compared to control, compared to its control 44.2 5 | Maintained ~98 5 | 32 (control) to 7 (modified 120 s) | _ | 62 (control) to 89 (modified 60 s) | [65] |
| 8 | HMDSO, TMMOS, and MTMOS with Ar | 30 W RF power; Ar flow rate 10 sscm; 1.5 mbar; 0–20 min; | SiO2–ZrO2 intermediate layer Laboratory synthesized | High H2 permeance of 6.5 × 107 mol/(m2 s Pa) with an H2/SF6 selectivity of 410 at 200 °C | NR | NR | NR | NR | [66] |
| 9 | Heptane (C7F16) and Ar | 30, 50, 70 W RF power; 0.03 mbar; monomer flow rate: 5 sccm; heated at 30 °C; 0.03 mbar; 30, 60, 90 s; | PFSA used in proton exchange membrane fuel cell (PEMFC) | Methanol permeability: decreased from 2.42 to 0.033 (10−6 cm2/s) 6 | NR | 86.9 increased to 117.3 (70 W, 90 s, 400 mTorr) | NR | 11.8 increased to 80.2 (70 W, 90 s, 400 mTorr) | [72] |
| 10 | Perfluorohexane (C6F14) and Ar | 0.018–0.064 W, 75 kHz discharged; 0.13–0.53 mbar; 5 min; distance between the electrodes is 39 mm; | MF PET-TM (0.4 µm, Sterlitech) | Pure water flux: 3.5–3.6 (its control ~3); Apple juice flux: 2.8–2.9 (its control ~2–2.2) | Sugar rejection: 98–100% | Increased from ca. 48 to 105 | NR | Decreased from ca.33 to 14 nm as the degree of deposition increased from 30.3 to 102 µg/cm2 | [74] |
| 11 | Tetrafluoromethane (CF4) | 50–400 W RF power; monomer flow rate = 18 sccm (standard cm3/min); 1–60 min; | UF PES Hydrophilic (Nanjing, China Altrateck Co., Ltd.) | 66.7 (control is not given) | 100% | Increased from 60 to 125 (modified at 200 W for 40 min) | NR | NR | [75] |
| Experience Series | Variables | Constants | WCA (Dropped from 137°) | ||
|---|---|---|---|---|---|
| Series 1 | Duration | RF Power | Argon Plow Rate | Gap Between Pubstrates and Glow | |
| 0–150, 30 s interval | 100 W | 10 slm | 5 mm | 19° after 150 s | |
| Series 2 | Argon Flow Rate | Plasma Power | Duration | Gap between Substrates and Glow | |
| 0–10, 1 slm interval | 100 W | 150 s | 5 mm | 22° at 10 slm | |
| Series 3 | Gap Between Substrates and Glow | Duration | Plasma Power | Argon Flow Rate | |
| 5.0, 7.5, 10, and 12.5 mm | 150 s | 100 W | 10 slm | 23° at 10 mm | |
| Control | Working Gas Component | Elemental Composition (%) | Elemental Ratio | ||||
|---|---|---|---|---|---|---|---|
| Si 2p | C 1s | O 1s | N 1s | C/Si | O/Si | ||
| HMDSO | - | - | - | - | 3.0 | 0.5 | |
| SiO2 | - | - | - | - | - | 2 | |
| Pure Ar | 32.3 | 3.6 | 64.1 | - | 0.11 | 1.98 | |
| O2/Ar (5.0 vol.%) | 31.9 | 3.2 | 64.9 | - | 0.10 | 2.04 | |
| N2/Ar (5.0 vol.%) | 29.3 | 20.2 | 46.0 | 4.5 | 0.69 | 1.57 | |
| Entry | Plasma Treatment | Plasma Conditions | Application | Membrane Performance | Water Contact Angle (°) | Pore Size/Porosity | Roughness RMS | Ref. |
|---|---|---|---|---|---|---|---|---|
| 1 | Ar Gas | 100 W RF powered two-rotating double-pipe type plasma jets; Ar flow rate = 0–10 slm; 0–150 s; gap between substrates and discharge nozzle: 5.0, 7.5, 10, and 12.5 mm | PVDF-HFP Laboratory synthesized Hydrophobic | For DSSC, the electrolyte update rate is 26.9% higher than the pristine PVDF, 10.8 ± 0.8 g/g | 137 declined to 21.3 ± 2.1 at 100 W, 10 slm, 5 mm gap, after 150 s | Increased from 0.6 to 0.7 µm; the porosity increased from 73.6 to 86.0%, compared to pristine control | NR 1 | [78] |
| 2 | AA with Ar/O2 or He/O2 | 30 kV AC powered plasma jet; Ar or He flow rate: 0.7 m3 h−1; O2 flow rate 0.1 m3 h−1; 1–20 min; AA heating temperature: 60 °C | MF PP Hydrophobic (Celgard 2500) | As a separator in the lithium-ion battery (LIB), the columbic efficiency maintained at about 99.0% and 99.5% upon 20 min treatment, respectively, compared to the pristine PP (97.5%) | 112 (PP control) declined to 63 and 39 upon 20-min Ar/O2/AA and He/O2/AA, respectively | Increased from 57.8 (control) to 180 nm upon 20-min Ar/O2/AA, but decreased to 10 nm upon 20 min He/O2/AA | Decreased from 68.91 (control)to 52.73 nm and 46.16 nm after 10 min Ar/O2/AA and He/O2/AA plasma, respectively | [81] |
| 3 | MA with Ar and C2H2 | 8 W plasma DBD; 5.0–6.6 kHz; 95kPa; MA flow rate: 0.06–0.33; C2H2 flow rate: 2–3 sccm; 5 or 10 min; cap between top and bottom electrodes is 1.6 mm | Silicon (c-Si) wafers | The carboxyl-enriched films were stable when deposited at MA:C2H2 = 0.037, with a thickness of 544 nm | NR | NR | NR | [83] |
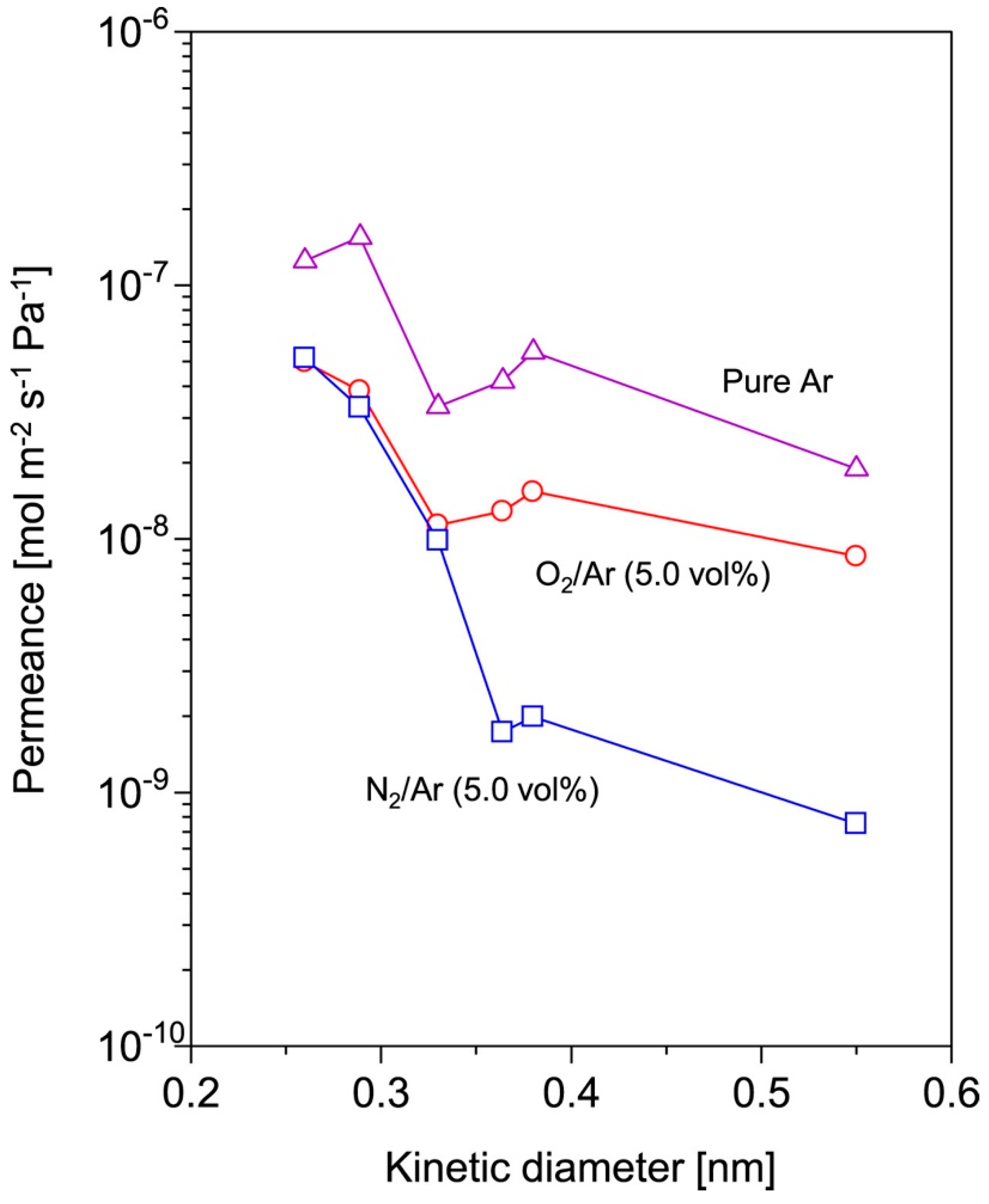
| 4 | HMDSO with pure Ar or O2/Ar or N2/Ar | 6 kV DBD plasma jet; 50 kHz; Ar or He flow rate: 0.7 m3 h−1; O2 flow rate 0.1 m3 h−1; Flow rate of pure Ar or a mixture of Ar with O2 (5.0 vol.%) or N2 (0.25–10.0 vol.%): 5.0 L min−1; 20 min; gap between substrates and discharge nozzle: 2.0 mm; HMDSO heating temperature: 40 °C | Tubular porous α-alumina substrates (SiO2-ZrO2) Laboratory synthesized | The He permeance of the HMDSO/N2/Ar deposited films was 0.52 × 10−7, which is lower than that of 1.50 × 10−7 mol m−2 s−1 Pa−1, achieved by HMDSO/O2/Ar prepared films; HMDSO/N2/Ar films also provided highest permeance ratio of He/H2 as 1.6 | NR | NR | NR | [67] |
| 5 | TEGDME with He  | 8–13 W AC powered plasma DBD; 15–50 kHz frequency; the total flow of TEGDME/He aerosol and He carrier gas: 8–10 slm; flow rate of He via aerosol: 3.15 slm; 5 min; 4 mm interelectrode gap | Glass substrates | NR | Static WCA is 52 for the film deposited at 27 kHz and 57 at 36 kHz; static WCA of control is not given | NR | NR | [95] |
| 6 | Oleate-capped ZnO NPs in n-octane (0.5–5 wt.%.) | 0.28 ± 0.02 W cm−2 AC powered plasma DBD; 105 Pa; total flow of He: 8000 sccm; flow rate of He via aerosol: 2800 sccm; 10 min; 4 mm interelectrode gap | Borosilicate glass slides, CaF2 substrates, carbon-coated Cu grids for TEM | NR | Advancing WCA and receding WCA increased from 113 to 170 and from 64 to 168, respectively | NR | The roughness of the films prepared from pure n-octane and 3 wt.% NPs dispersion in n-octane was 0.345 ± 0.007, and 574 ± 11 nm, respectively. | [93] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Chen, X.; Reis, R.; Chen, Z.; Milne, N.; Winther-Jensen, B.; Kong, L.; Dumée, L.F. Plasma Modification and Synthesis of Membrane Materials—A Mechanistic Review. Membranes 2018, 8, 56. https://doi.org/10.3390/membranes8030056
Wang J, Chen X, Reis R, Chen Z, Milne N, Winther-Jensen B, Kong L, Dumée LF. Plasma Modification and Synthesis of Membrane Materials—A Mechanistic Review. Membranes. 2018; 8(3):56. https://doi.org/10.3390/membranes8030056
Chicago/Turabian StyleWang, Jingshi, Xiao Chen, Rackel Reis, Zhiqiang Chen, Nick Milne, Bjorn Winther-Jensen, Lingxue Kong, and Ludovic F. Dumée. 2018. "Plasma Modification and Synthesis of Membrane Materials—A Mechanistic Review" Membranes 8, no. 3: 56. https://doi.org/10.3390/membranes8030056
APA StyleWang, J., Chen, X., Reis, R., Chen, Z., Milne, N., Winther-Jensen, B., Kong, L., & Dumée, L. F. (2018). Plasma Modification and Synthesis of Membrane Materials—A Mechanistic Review. Membranes, 8(3), 56. https://doi.org/10.3390/membranes8030056