
Nanofiber Ion-Exchange Membranes for the Rapid Uptake and Recovery of Heavy Metals from Water



Abstract
:1. Introduction
2. Experimental Section
2.1. Materials
2.2. Methods
2.2.1. Preparation of Poly(glycidyl Methacrylate) (PGMA)
2.2.2. Preparation of Regenerated Cellulose Nanofiber Membrane Support
2.2.3. Preparation of Nanofiber Ion-Exchange Membranes
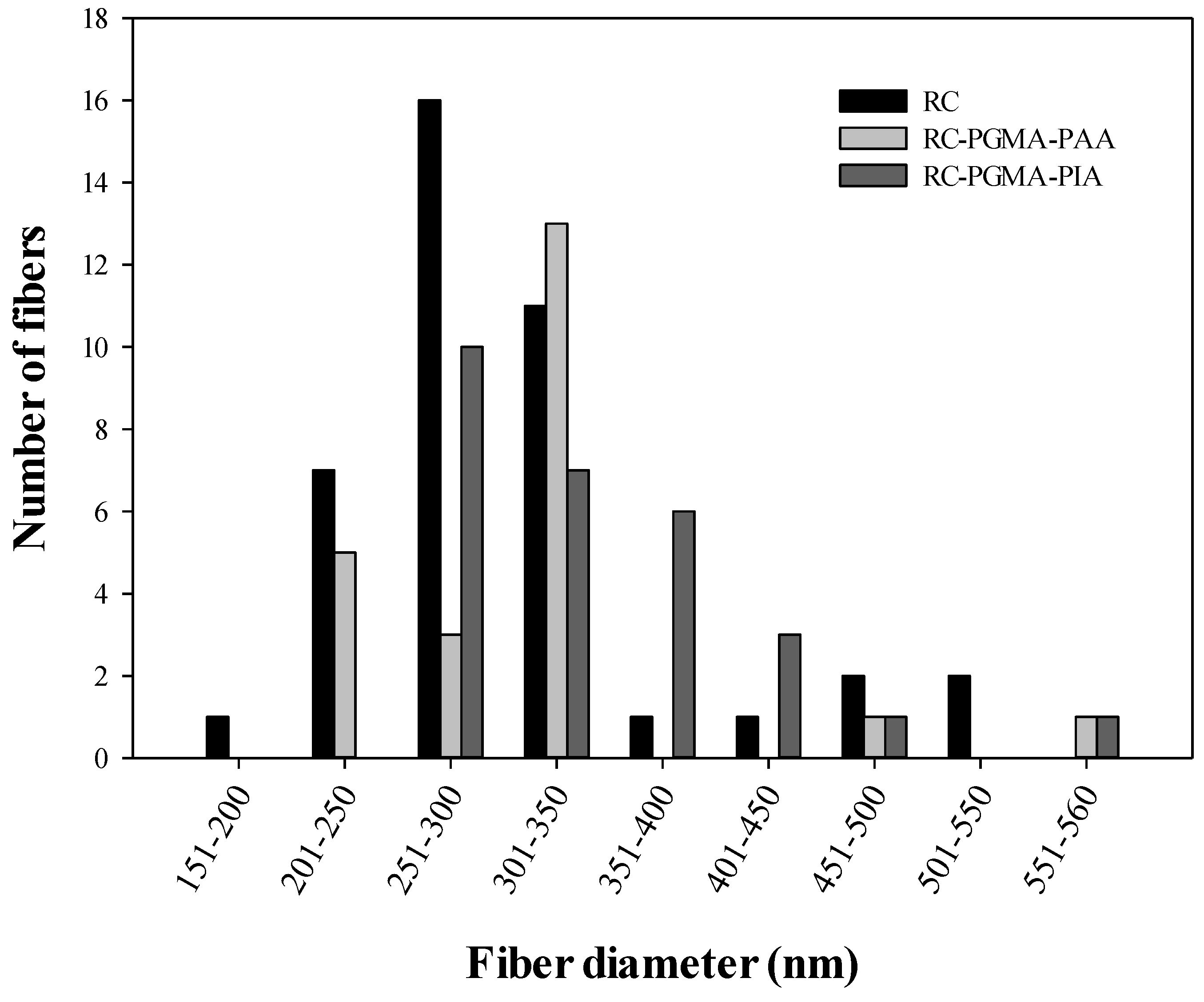
2.3. Membrane Morphology
2.4. Performance Properties of Nanofiber Membranes
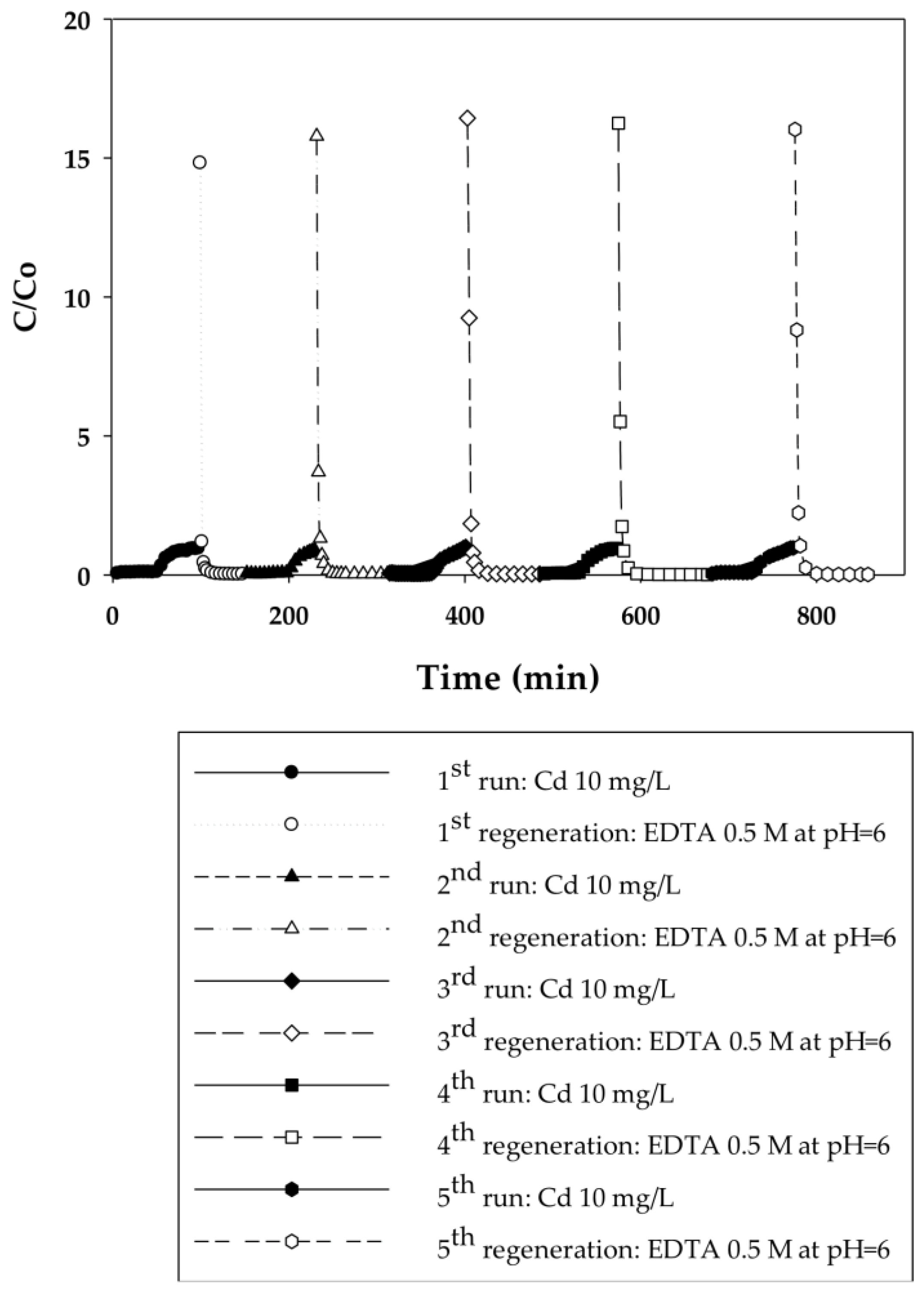
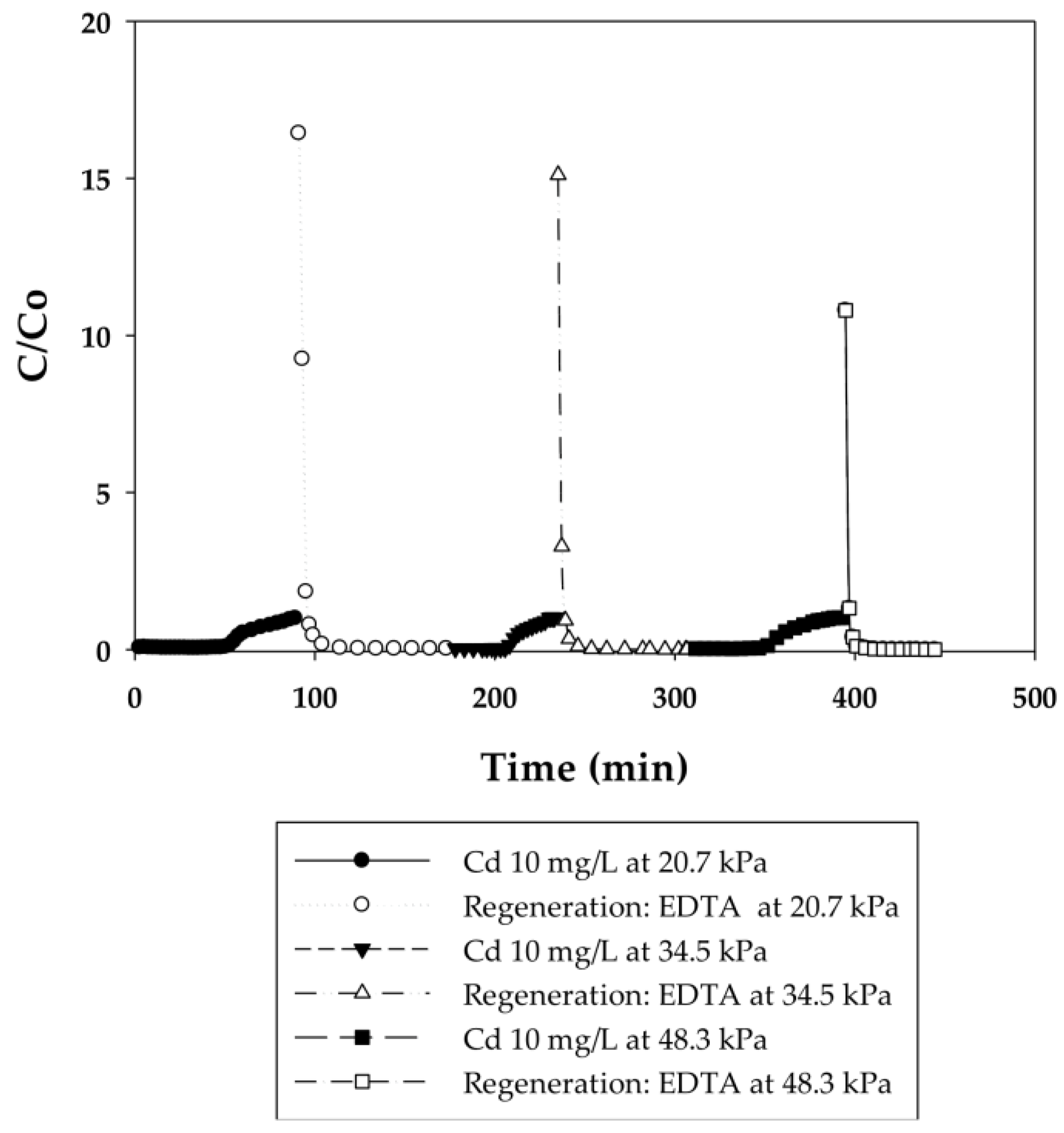
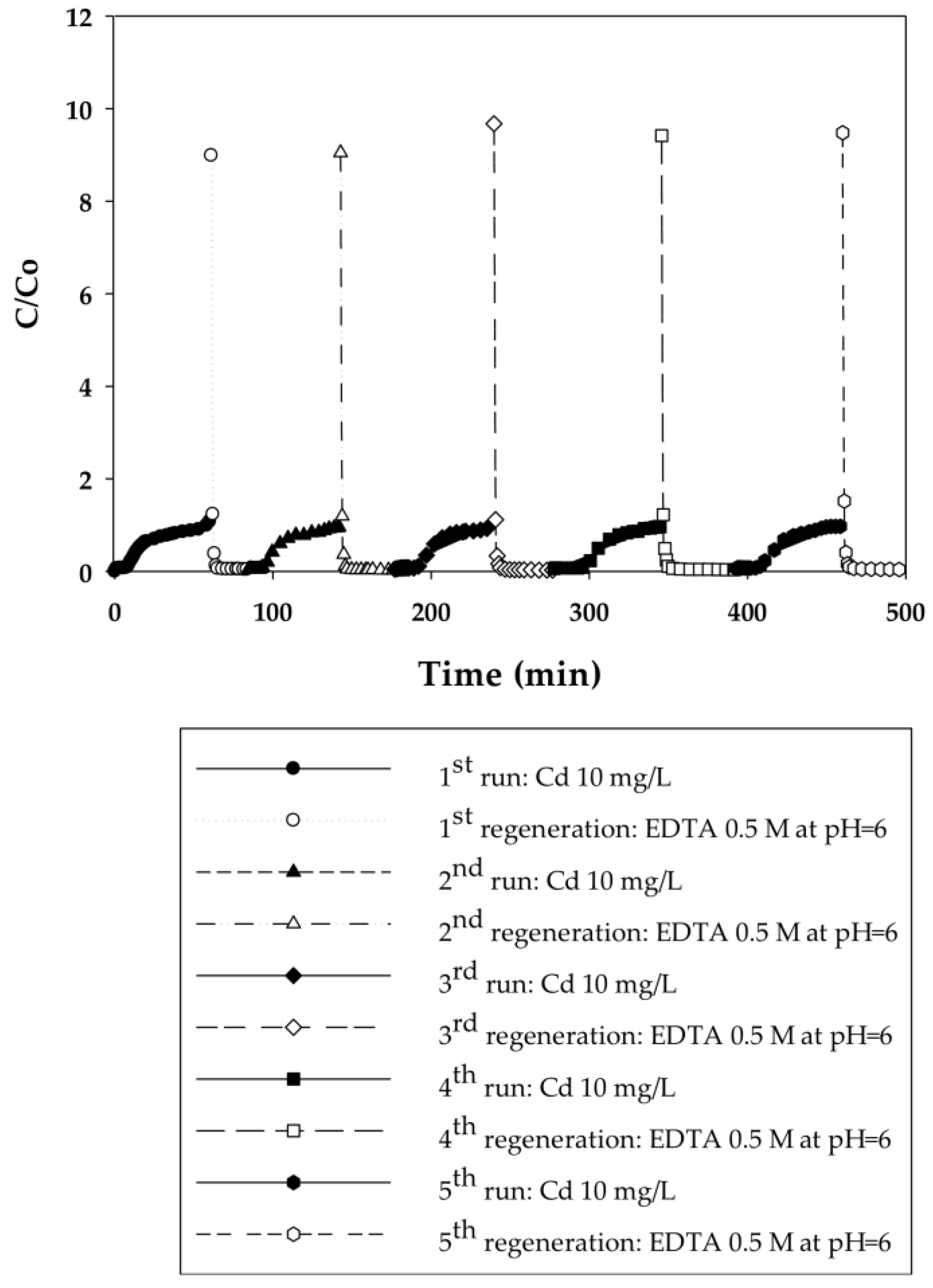
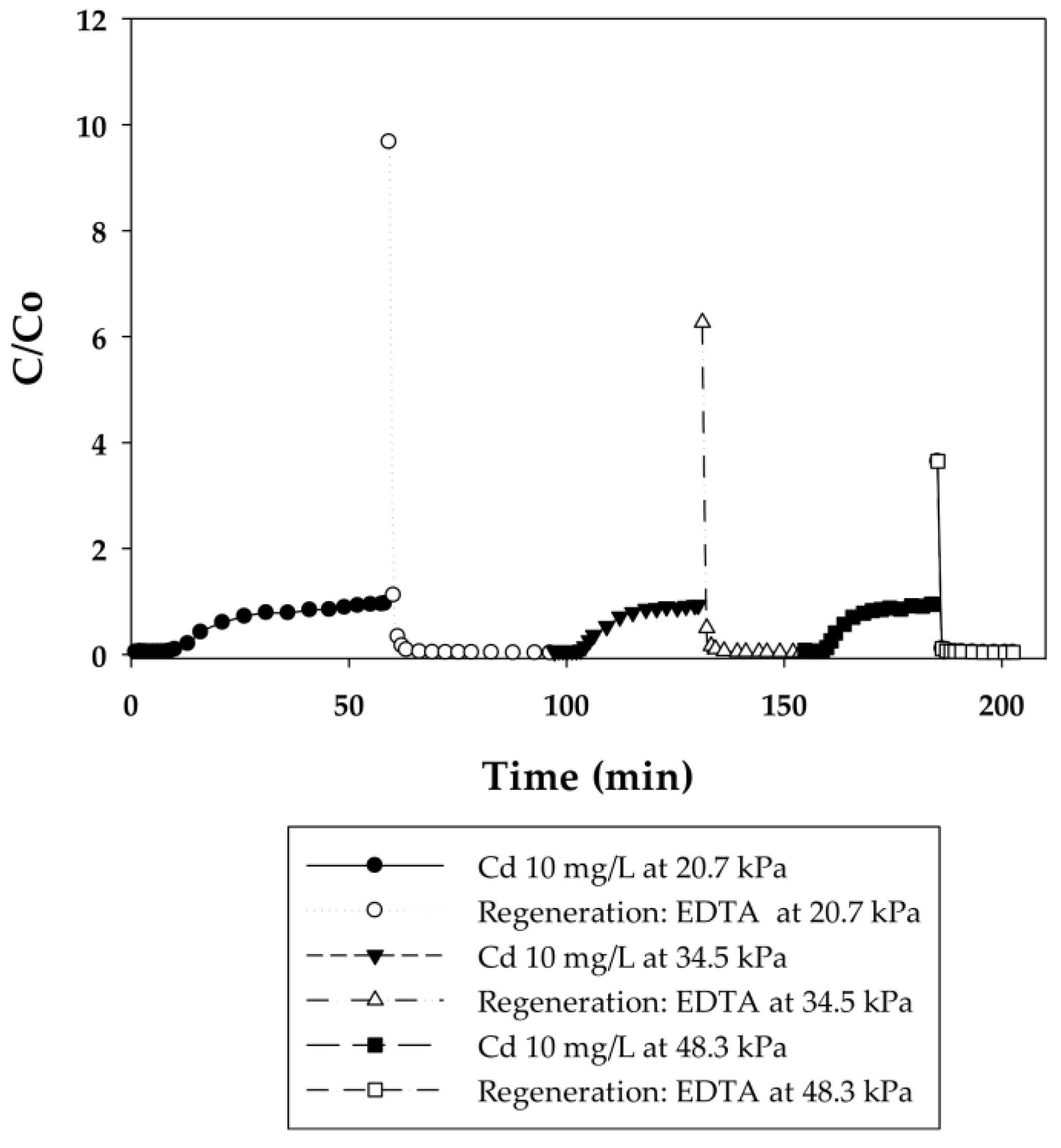
2.4.1. Dynamic Binding Capacity and Regeneration
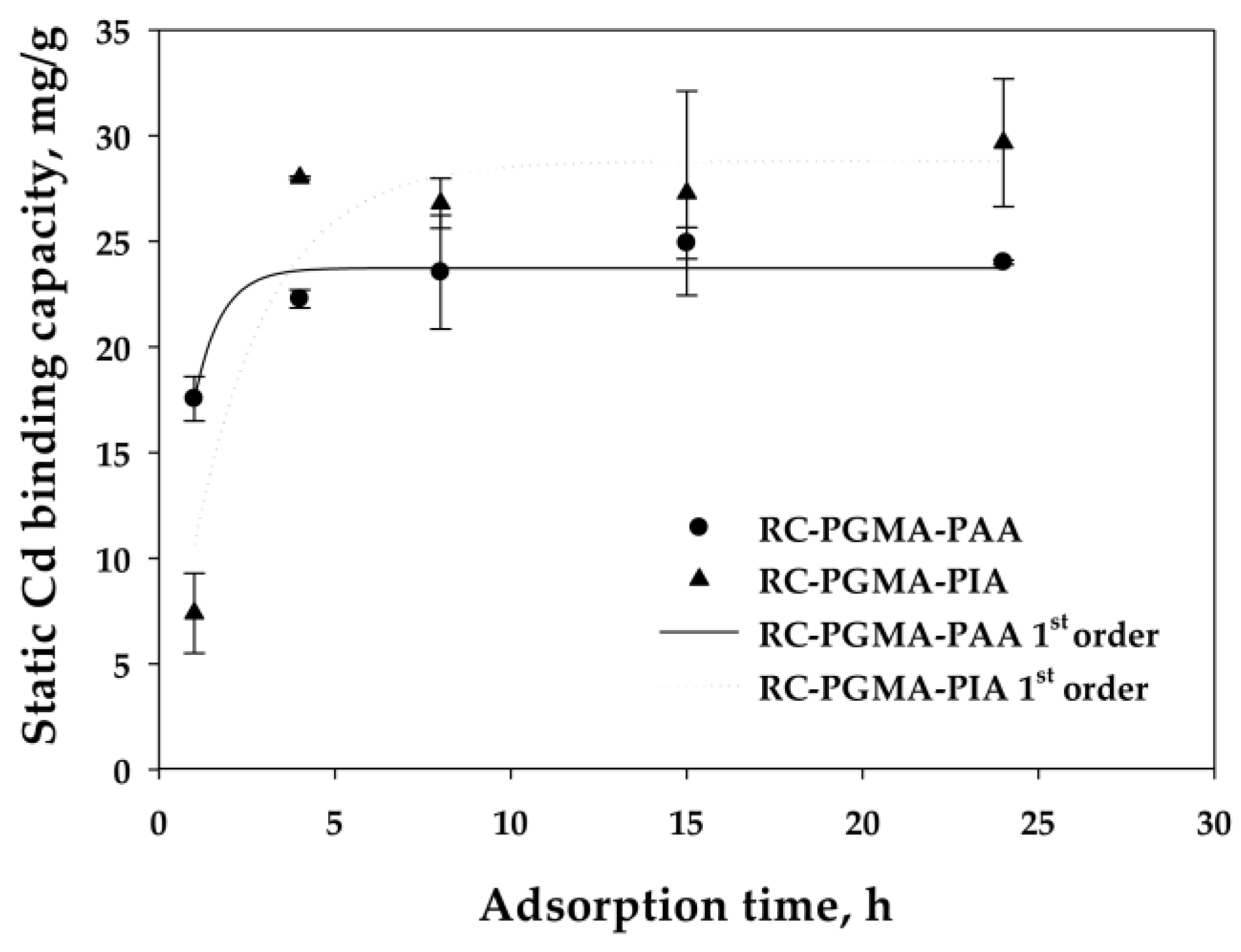
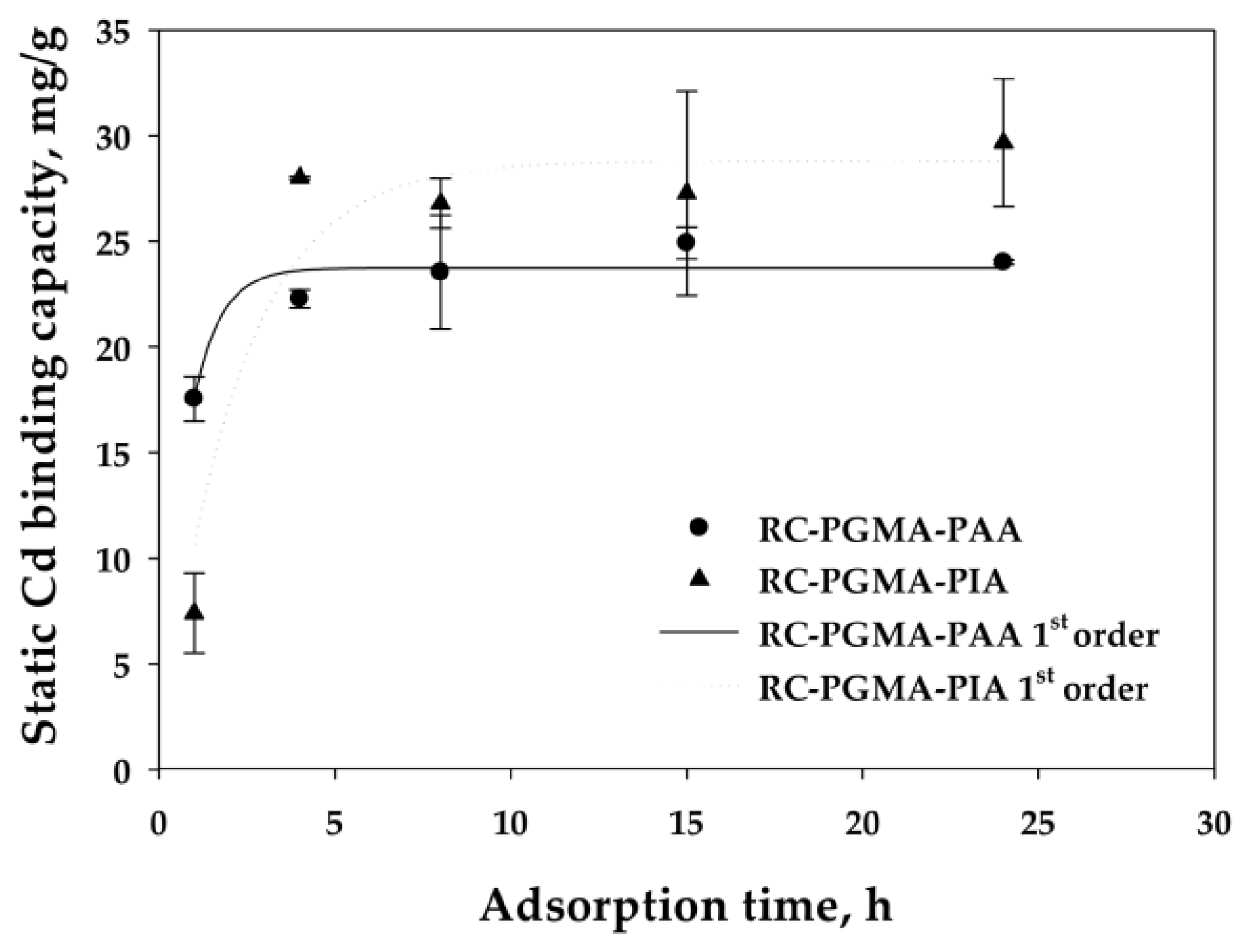
2.4.2. Static Cadmium Binding Kinetic Study
2.4.3. Polymer Characterization in Solution
3. Results and Discussion
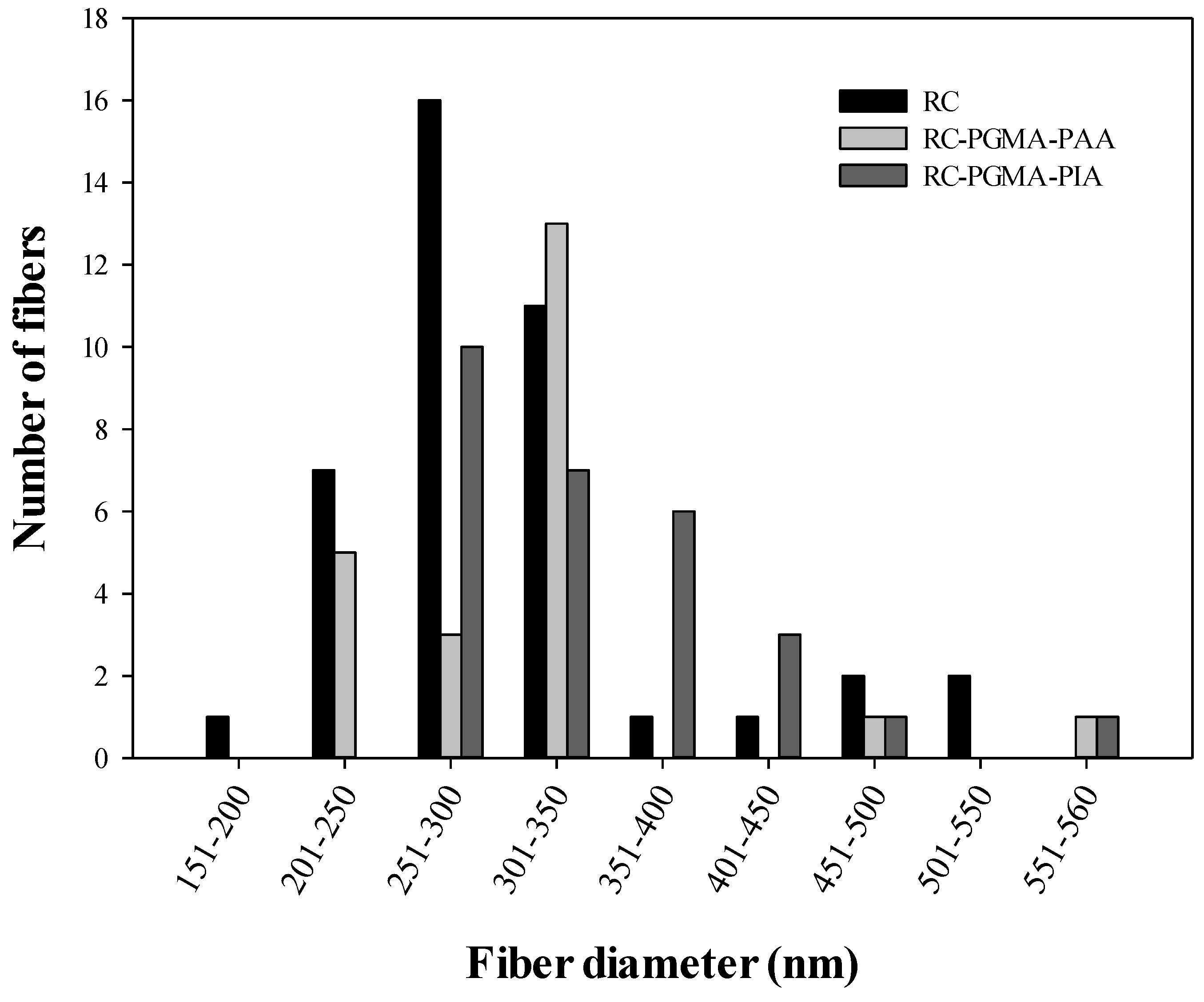
3.1. Membrane Morphology
3.2. Membrane Performance Properties
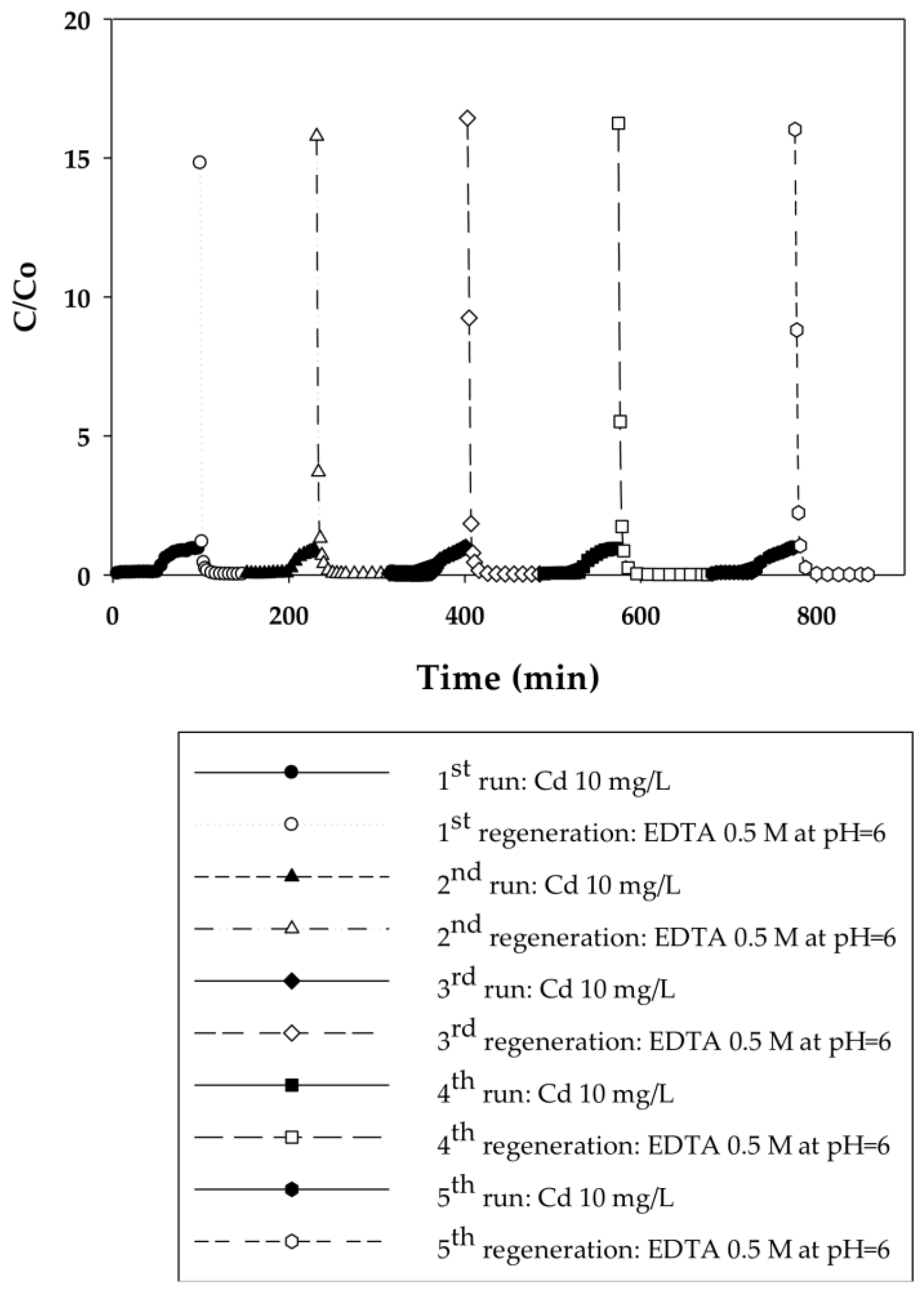
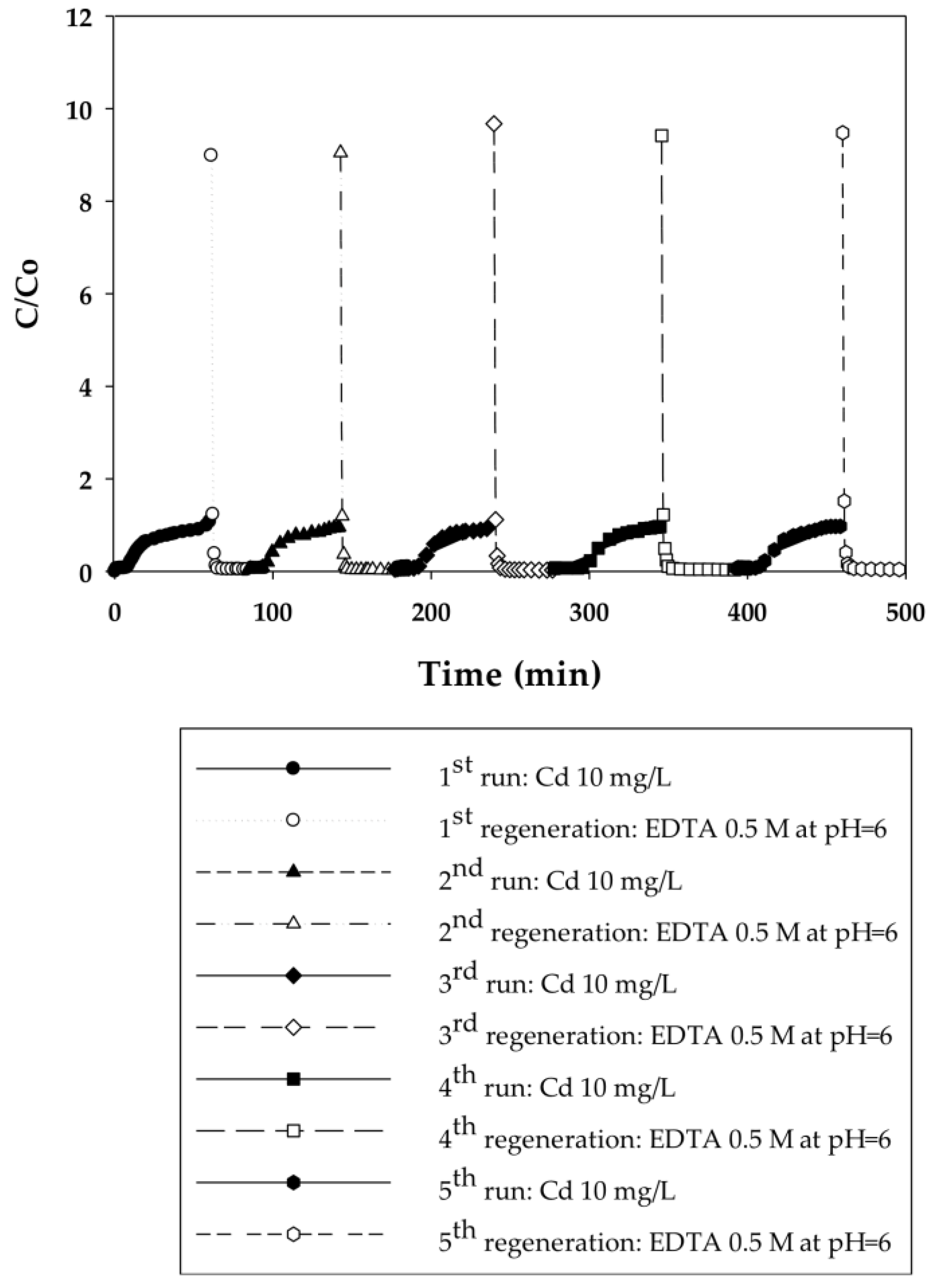
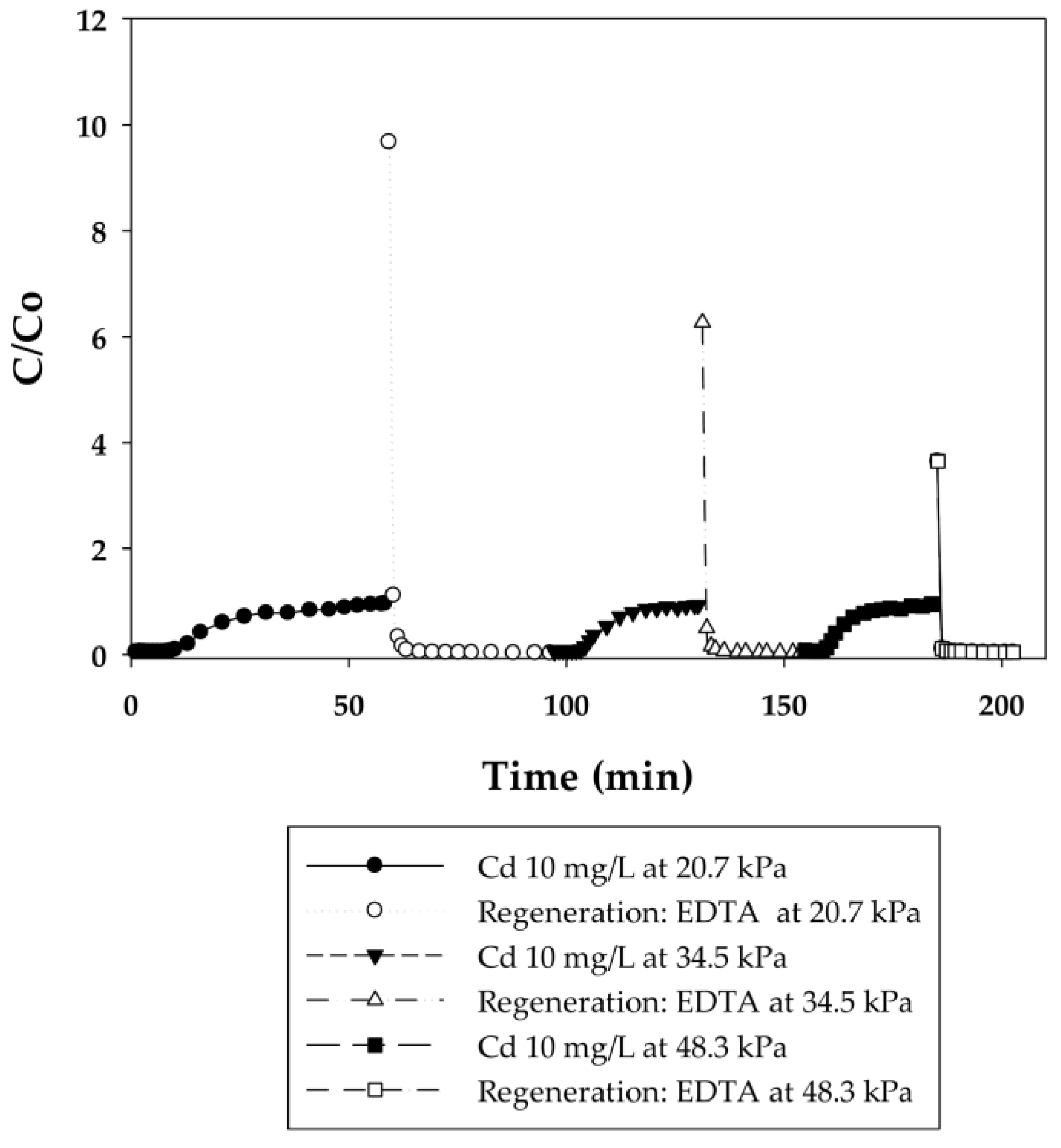
3.2.1. Dynamic Sorption-Regeneration Cycles for Cd(II) Ion Exchange on PAA- and PIA-Modified Nanofiber Membranes
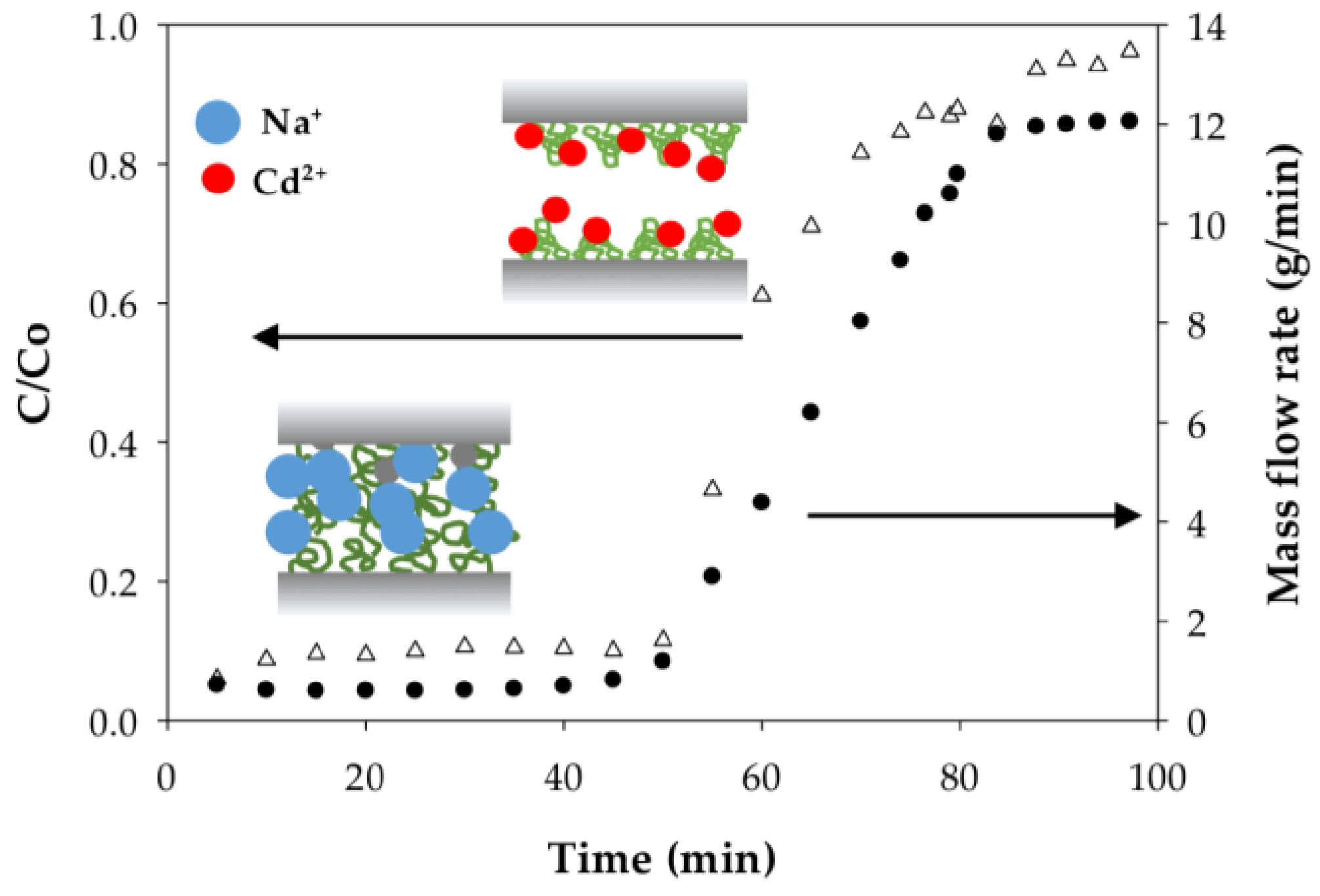
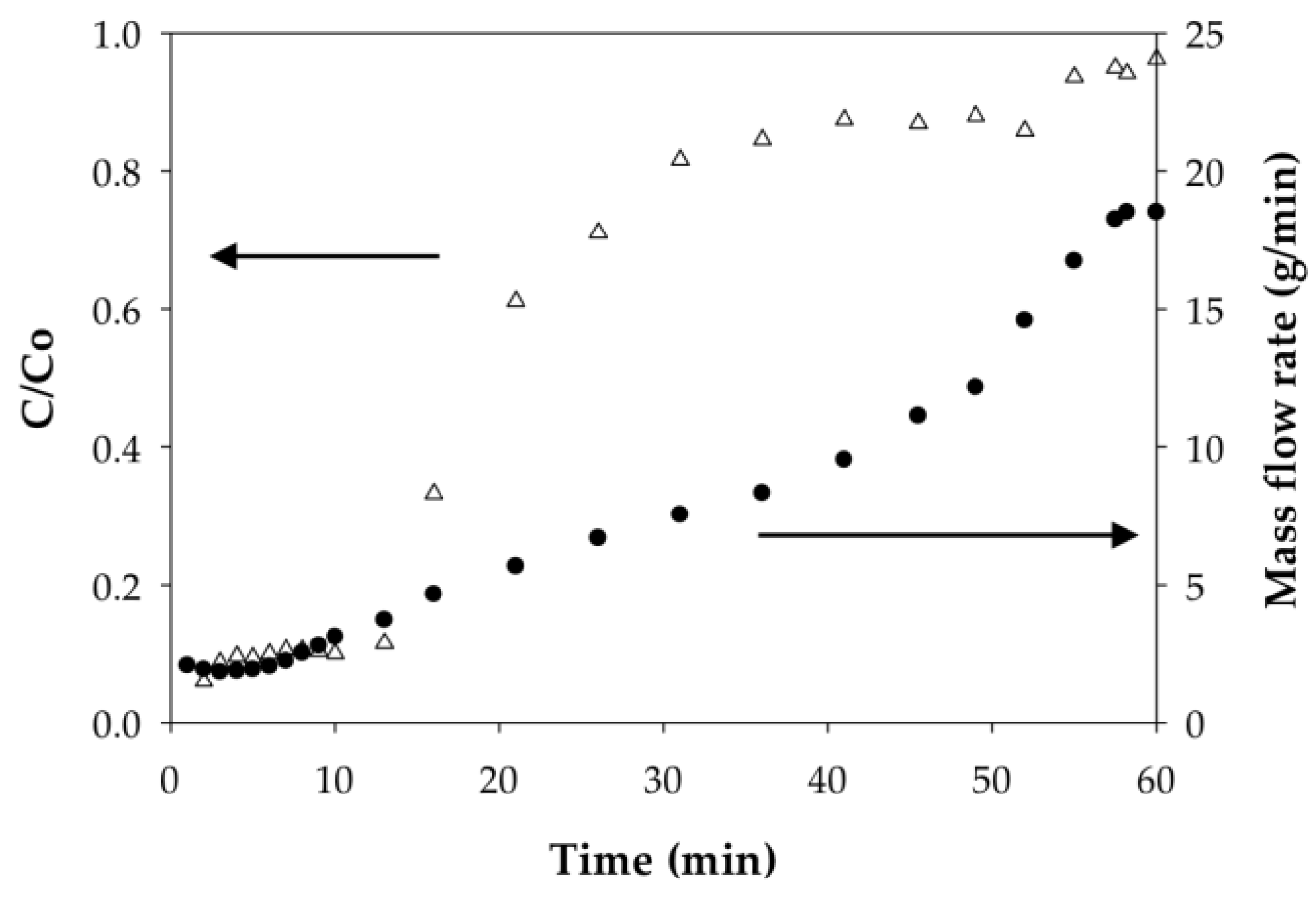
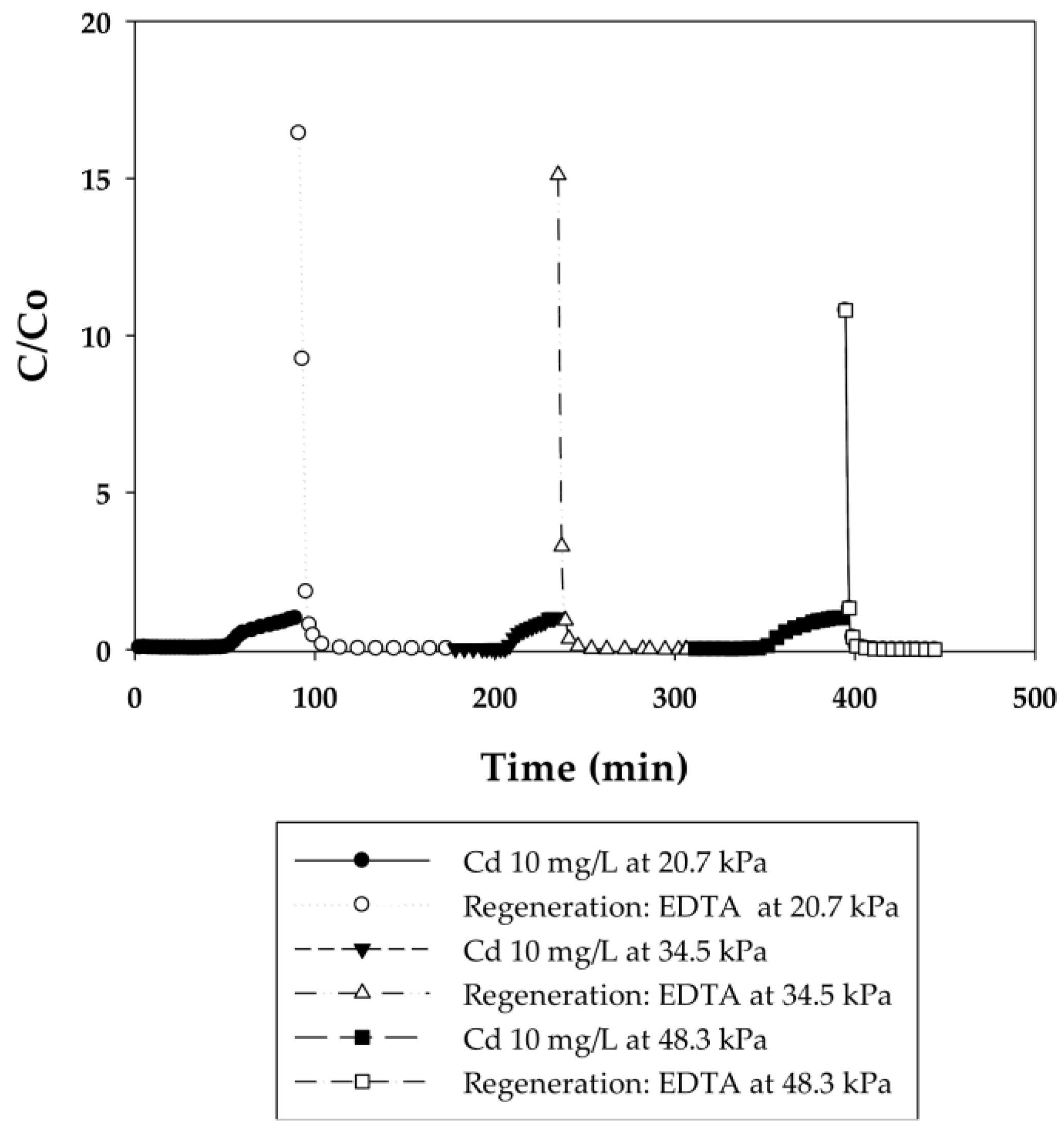
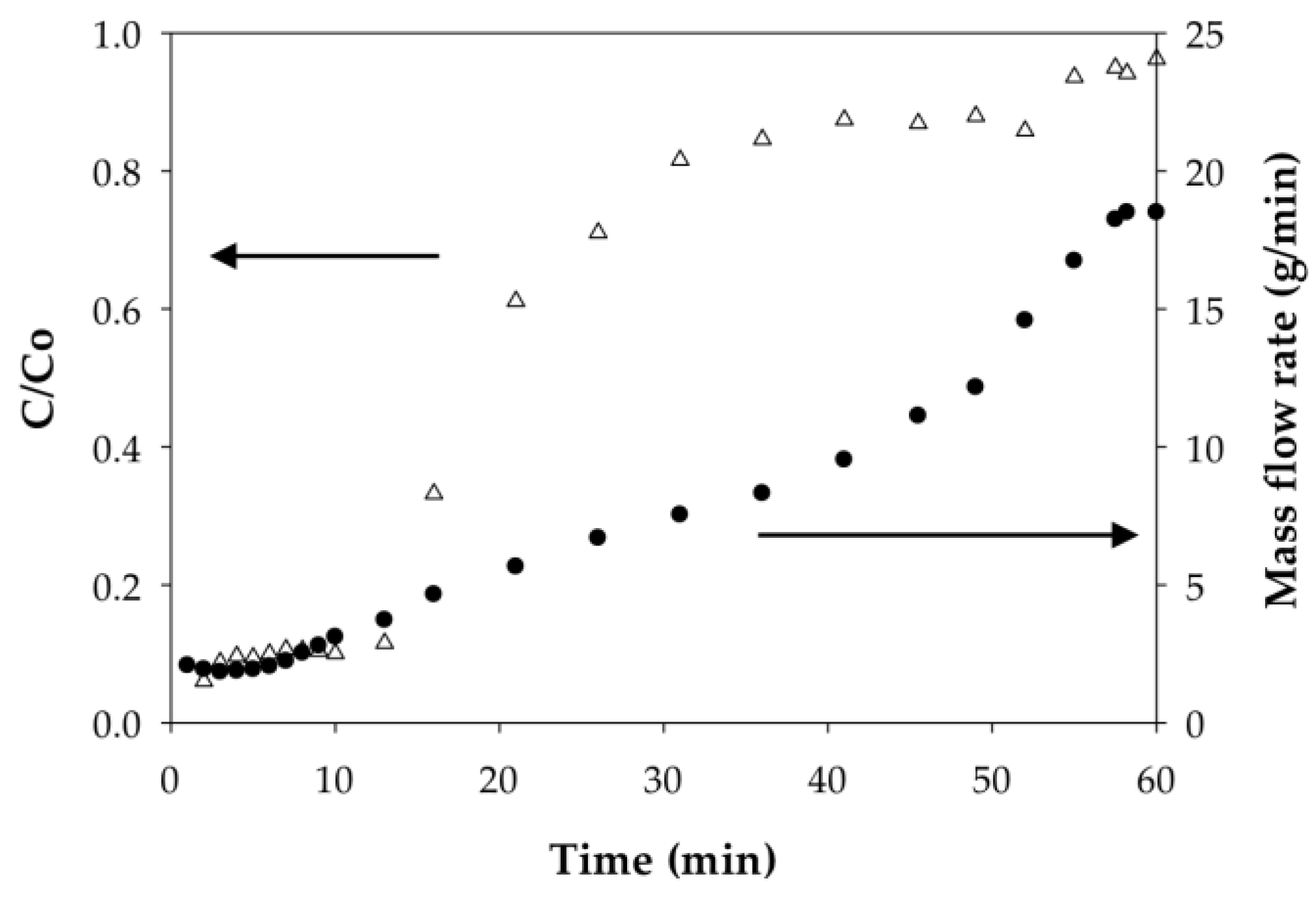
3.2.2. Effect of Flow Rate on the Binding Capacity of Cd(II) on PAA- and PIA-Modified Nanofiber Membranes
3.2.3. Reaction Rate Analysis
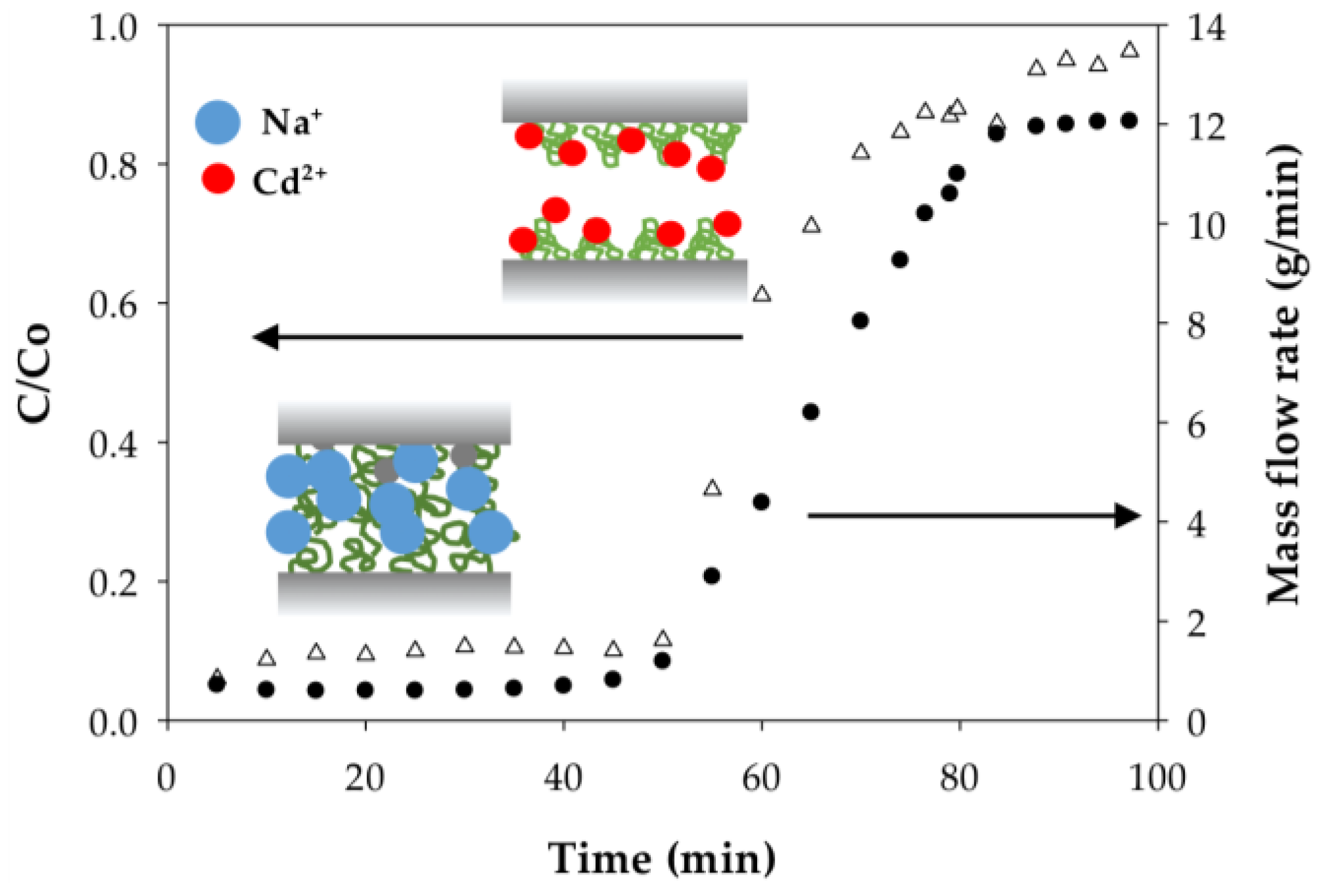
3.2.4. Polymer Behavior before and after Cd Ion Binding
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Srivastava, N.; Majumder, C. Novel biofiltration methods for the treatment of heavy metals from industrial wastewater. J. Hazard. Mater. 2008, 151, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Barakat, M. New trends in removing heavy metals from industrial wastewater. Arab. J. Chem. 2011, 4, 361–377. [Google Scholar] [CrossRef]
- Srivastava, V.C.; Mall, I.D.; Mishra, I.M. Competitive adsorption of cadmium(II) and nickel(II) metal ions from aqueous solution onto rice husk ash. Chem. Eng. Process. Process Intensif. 2009, 48, 370–379. [Google Scholar] [CrossRef]
- Fu, F.; Wang, Q. Removal of heavy metal ions from wastewaters: A review. J. Environ. Manag. 2011, 92, 407–418. [Google Scholar] [CrossRef] [PubMed]
- Nakate, S. Three Major Types of Environmental Pollution. Available online: http://www.buzzle.com/articles/three-major-types-of-environmental-pollution.html (accessed on 1 May 2016).
- Han, B.; Carvalho, W.; Canilha, L.; da Silva, S.S.; e Silva, J.B.A.; McMillan, J.D.; Wickramasinghe, S.R. Adsorptive membranes vs. Resins for acetic acid removal from biomass hydrolysates. Desalination 2006, 193, 361–366. [Google Scholar] [CrossRef]
- Irani, M.; Keshtkar, A.R.; Mousavian, M.A. Removal of Cd(II) and Ni(II) from aqueous solution by PVA/TEOS/TMPTMS hybrid membrane. Chem. Eng. J. 2011, 175, 251–259. [Google Scholar] [CrossRef]
- Horvath, C.G.; Preiss, B.; Lipsky, S.R. Fast liquid chromatography. Investigation of operating parameters and the separation of nucleotides on pellicular ion exchangers. Anal. Chem. 1967, 39, 1422–1428. [Google Scholar] [CrossRef] [PubMed]
- Urmann, M.; Graalfs, H.; Joehnck, M.; Jacob, L.R.; Frech, C. Cation-exchange chromatography of monoclonal antibodies: Characterisation of a novel stationary phase designed for production-scale purification. MAbs 2010, 2, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Gaikwad, R.; Sapkal, V.; Sapkal, R. Ion exchange system design for removal of heavy metals from acid mine drainage wastewater. Acta Montan. Slovaca 2010, 15, 298–304. [Google Scholar]
- Ramakrishna, S.; Ma, Z.; Matsuura, T. Polymer Membranes in Biotechnology: Preparation, Functionalization and Application; Imperial College Press: London, UK, 2011. [Google Scholar]
- Hamdaoui, O. Removal of copper(II) from aqueous phase by purolite C100-MB cation exchange resin in fixed bed columns: Modeling. J. Hazard. Mater. 2009, 161, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Malla, M.E.; Alvarez, M.B.; Batistoni, D.A. Evaluation of sorption and desorption characteristics of cadmium, lead and zinc on amberlite IRC-718 iminodiacetate chelating ion exchanger. Talanta 2002, 57, 277–287. [Google Scholar] [CrossRef]
- Wada, G.; Ishihara, R.; Miyoshi, K.; Umeno, D.; Saito, K.; Asai, S.; Yamada, S.; Hirota, H. Crosslinked-chelating porous sheet with high dynamic binding capacity of metal ions. Solvent Extr. Ion Exch. 2013, 31, 210–220. [Google Scholar] [CrossRef]
- Figoli, A.; Cassano, A.; Basile, A. Membrane Technologies for Biorefining; Woodhead Publishing: Cambridge, UK, 2016; pp. 225–226. [Google Scholar]
- Zhang, L.; Zhao, Y.-H.; Bai, R. Development of a multifunctional membrane for chromatic warning and enhanced adsorptive removal of heavy metal ions: Application to cadmium. J. Membr. Sci. 2011, 379, 69–79. [Google Scholar] [CrossRef]
- Ling, P.; Liu, F.; Li, L.; Jing, X.; Yin, B.; Chen, K.; Li, A. Adsorption of divalent heavy metal ions onto IDA-chelating resins: Simulation of physicochemical structures and elucidation of interaction mechanisms. Talanta 2010, 81, 424–432. [Google Scholar] [CrossRef] [PubMed]
- He, Z.-Y.; Nie, H.-L.; Branford-White, C.; Zhu, L.-M.; Zhou, Y.-T.; Zheng, Y. Removal of Cu2+ from aqueous solution by adsorption onto a novel activated nylon-based membrane. Bioresour. Technol. 2008, 99, 7954–7958. [Google Scholar] [CrossRef] [PubMed]
- Chitpong, N.; Husson, S.M. Polyacid functionalized cellulose nanofiber membranes for removal of heavy metals from impaired waters. J. Membr. Sci. 2017, 523, 418–429. [Google Scholar] [CrossRef]
- World Health Organization. Explosure to Cadmium: A Major Public Health Concern; The WHO Document Production Services: Geneva, Switzerland, 2010. [Google Scholar]
- United Nations Environment Programme. Final Review of Scientific Information on Cadmium; United Nations Environment Programme (UNEP) Chemicals Branch: Nairobi, Kenya, 2010. [Google Scholar]
- Haider, S.; Park, S.-Y. Preparation of the electrospun chitosan nanofibers and their applications to the adsorption of Cu(II) and Pb(II) ions from an aqueous solution. J. Membr. Sci. 2009, 328, 90–96. [Google Scholar] [CrossRef]
- Callegari, G.; Tyomkin, I.; Kornev, K.G.; Neimark, A.V.; Hsieh, Y.-L. Absorption and transport properties of ultra-fine cellulose webs. J. Colloid Interface Sci. 2011, 353, 290–293. [Google Scholar] [CrossRef] [PubMed]
- Saeed, K.; Haider, S.; Oh, T.-J.; Park, S.-Y. Preparation of amidoxime-modified polyacrylonitrile (PAN-oxime) nanofibers and their applications to metal ions adsorption. J. Membr. Sci. 2008, 322, 400–405. [Google Scholar] [CrossRef]
- Ramakrishna, S.; Fujihara, K.; Teo, W.-E.; Yong, T.; Ma, Z.; Ramaseshan, R. Electrospun nanofibers: Solving global issues. Mater. Today 2006, 9, 40–50. [Google Scholar] [CrossRef]
- Chaiyasith, S.; Chaiyasith, P.; Septhum, C. Removal of cadmium and nickel from aqueous solution by adsorption onto treated fly ash from Thailand. Thammasat Int. J. Sci. Technol. 2006, 11, 13–20. [Google Scholar]
- Chenette, H.C.; Robinson, J.R.; Hobley, E.; Husson, S.M. Development of high-productivity, strong cation-exchange adsorbers for protein capture by graft polymerization from membranes with different pore sizes. J. Membr. Sci. 2012, 423, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Njikam, E.; Schiewer, S. Optimization and kinetic modeling of cadmium desorption from citrus peels: A process for biosorbent regeneration. J. Hazard. Mater. 2012, 213, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Hasan, S.; Krishnaiah, A.; Ghosh, T.K.; Viswanath, D.S.; Boddu, V.M.; Smith, E.D. Adsorption of divalent cadmium (Cd(II)) from aqueous solutions onto chitosan-coated perlite beads. Ind. Eng. Chem. Res. 2006, 45, 5066–5077. [Google Scholar] [CrossRef]
- Jha, I.; Iyengar, L.; Rao, A.P. Removal of cadmium using chitosan. J. Environ. Eng. 1988, 114, 962–974. [Google Scholar] [CrossRef]
- Schulte, S. Recovery/recycling Methods for Metal Finishers. Hixson Inc., 2011. Available online: http://www.pfonline.com/articles/recoveryrecycling-methods-for-platers (accessed on 7 December 2016).
- Di Palma, L.; Ferrantelli, P.; Merli, C.; Biancifiori, F. Recovery of EDTA and metal precipitation from soil flushing solutions. J. Hazard. Mater. 2003, 103, 153–168. [Google Scholar] [CrossRef]
- Bhut, B.V.; Husson, S.M. Dramatic performance improvement of weak anion-exchange membranes for chromatographic bioseparations. J. Membr. Sci. 2009, 337, 215–223. [Google Scholar] [CrossRef]
- Bhut, B.V.; Christensen, K.A.; Husson, S.M. Membrane chromatography: Protein purification from E. coli lysate using newly designed and commercial anion-exchange stationary phases. J. Chromatogr. A 2010, 1217, 4946–4957. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhou, J.; Zhou, D.; Tang, Y. Adsorption of cadmium and strontium on cellulose/alginic acid ion-exchange membrane. J. Membr. Sci. 1999, 162, 103–109. [Google Scholar] [CrossRef]
- Pehlivan, E.; Altun, T. The study of various parameters affecting the ion exchange of Cu2+, Zn2+, Ni2+, Cd2+, and Pb2+ from aqueous solution on Dowex 50W synthetic resin. J. Hazard. Mater. 2006, 134, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Kumar, U. Agricultural products and by-products as a low cost adsorbent for heavy metal removal from water and wastewater: A review. Sci. Res. Essays 2006, 1, 033–037. [Google Scholar]
- Hinojosa Reyes, L.; Saucedo Medina, I.; Navarro Mendoza, R.; Revilla Vázquez, J.; Avila Rodriguez, M.; Guibal, E. Extraction of cadmium from phosphoric acid using resins impregnated with organophosphorus extractants. Ind. Eng. Chem. Res. 2001, 40, 1422–1433. [Google Scholar] [CrossRef]
- Ho, Y.-S.; McKay, G. Pseudo-second order model for sorption processes. Process Biochem. 1999, 34, 451–465. [Google Scholar] [CrossRef]
- Chiron, N.; Guilet, R.; Deydier, E. Adsorption of Cu(II) and Pb(II) onto a grafted silica: Isotherms and kinetic models. Water Res. 2003, 37, 3079–3086. [Google Scholar] [CrossRef]
- Aksu, Z. Equilibrium and kinetic modelling of cadmium(II) biosorption by C. Vulgaris in a batch system: Effect of temperature. Sep. Purif. Technol. 2001, 21, 285–294. [Google Scholar] [CrossRef]
- Boparai, H.K.; Joseph, M.; O’Carroll, D.M. Kinetics and thermodynamics of cadmium ion removal by adsorption onto nano zerovalent iron particles. J. Hazard. Mater. 2011, 186, 458–465. [Google Scholar] [CrossRef] [PubMed]
- Fogler, H.S. Elements of Chemical Reaction Engineering; Prentice-Hall International, Inc.: Upper Saddle River, NJ, USA, 1999; Chapter 4; pp. 137–138. [Google Scholar]
- Wang, R.; Liu, Y.; Li, B.; Hsiao, B.S.; Chu, B. Electrospun nanofibrous membranes for high flux microfiltration. J. Membr. Sci. 2012, 392, 167–174. [Google Scholar] [CrossRef]
- Wandera, D.; Wickramasinghe, S.R.; Husson, S.M. Stimuli-responsive membranes. J. Membr. Sci. 2010, 357, 6–35. [Google Scholar] [CrossRef]
- Konradi, R.; Rühe, J. Interaction of poly (methacrylic acid) brushes with metal ions: Swelling properties. Macromolecules 2005, 38, 4345–4354. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cycle | Pressure | Static Binding Capacity | Dynamic Binding Capacity |
|---|---|---|---|
| (kPa) | qmax (mg/g) | qmax (mg/g) | |
| 1 | 20.7 | 25.5 | 10.0 |
| 2 | 20.7 | 24.5 | 9.4 |
| 3 | 20.7 | 24.6 | 9.6 |
| 4 | 20.7 | 26.2 | 9.7 |
| 5 | 20.7 | 26.0 | 9.5 |
| Cycle | Pressure | Static Binding Capacity | Dynamic Binding Capacity |
|---|---|---|---|
| (kPa) | qmax (mg/g) | qmax (mg/g) | |
| 1 | 20.7 | 37.5 | 5.5 |
| 2 | 20.7 | 34.5 | 9.6 |
| 3 | 20.7 | 37.1 | 8.2 |
| 4 | 20.7 | 33.9 | 9.5 |
| 5 | 20.7 | 33.6 | 10.2 |
| Cycle | Pressure | Static Binding Capacity | Dynamic Binding Capacity |
|---|---|---|---|
| (kPa) | qmax (mg/g) | qmax (mg/g) | |
| 1 | 20.7 | 24.6 | 9.6 |
| 2 | 34.5 | 25.9 | 10.6 |
| 3 | 48.3 | 25.7 | 9.7 |
| Cycle | Pressure | Static Binding Capacity | Dynamic Binding Capacity |
|---|---|---|---|
| (kPa) | qmax (mg/g) | qmax (mg/g) | |
| 1 | 20.7 | 34.6 | 9.6 |
| 2 | 34.5 | 32.9 | 7.9 |
| 3 | 48.3 | 33.3 | 8.2 |
| Ion Exchange Materials | qmax | Permeability | Flow Rate | Productivity (Co = 10 mg/L) | |
|---|---|---|---|---|---|
| mg Cd/g | L/m2/h/bar | BV/h | mL/min/g | mg Cd/g/min | |
| Cellulose/alginic acid IEX membrane [35] | 44.4 | 8.0 | - | - | - |
| Chitosan/CA IEX membrane [16] | 43.8 | 7.7 | - | - | - |
| Dowex 50W [36] | 277 | - | 0.5 | - | - |
| Duolite CT-73 [37] | 106 | - | 10 | - | - |
| Amberlite 200 [37] | 225 | - | 40 | - | - |
| Amberlite XAD-7/Cyanex-301 [38] | - | - | - | 0.64 | 3.5 × 10−2 |
| RC-PGMA-PAA [19] | 163 | 400 | 280 | 12 | 2.2 × 10−1 |
| RC-PGMA-PIA | 222 | 500 | 1890 | 80 | 5.5 × 10−1 |
| % Polymer in Na and Cd forms | 100% Na + 0% Cd | 92.5% Na + 7.5% Cd | 85% Na + 15% Cd | 75% Na + 25% Cd | 50% Na + 50% Cd | 0% Na + 100% Cd | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Rh (nm) | SD | Rh (nm) | SD | Rh (nm) | SD | Rh (nm) | SD | Rh (nm) | SD | Rh (nm) | SD | |
| PAA | 67 | 0.67 | 11 | 0.21 | 9.4 | 0.08 | 10 | 0.10 | 9.4 | 0.04 | * | - |
| PIA | 62 | 0.81 | 5.5 | 0.04 | 6.1 | 0.01 | 7.4 | 0.01 | * | - | * | - |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chitpong, N.; Husson, S.M. Nanofiber Ion-Exchange Membranes for the Rapid Uptake and Recovery of Heavy Metals from Water. Membranes 2016, 6, 59. https://doi.org/10.3390/membranes6040059
Chitpong N, Husson SM. Nanofiber Ion-Exchange Membranes for the Rapid Uptake and Recovery of Heavy Metals from Water. Membranes. 2016; 6(4):59. https://doi.org/10.3390/membranes6040059
Chicago/Turabian StyleChitpong, Nithinart, and Scott M. Husson. 2016. "Nanofiber Ion-Exchange Membranes for the Rapid Uptake and Recovery of Heavy Metals from Water" Membranes 6, no. 4: 59. https://doi.org/10.3390/membranes6040059
APA StyleChitpong, N., & Husson, S. M. (2016). Nanofiber Ion-Exchange Membranes for the Rapid Uptake and Recovery of Heavy Metals from Water. Membranes, 6(4), 59. https://doi.org/10.3390/membranes6040059