

The Utility of Fluorescence Recovery after Photobleaching (FRAP) to Study the Plasma Membrane


{kind=link}
{kind=link}
Abstract
1. Introduction
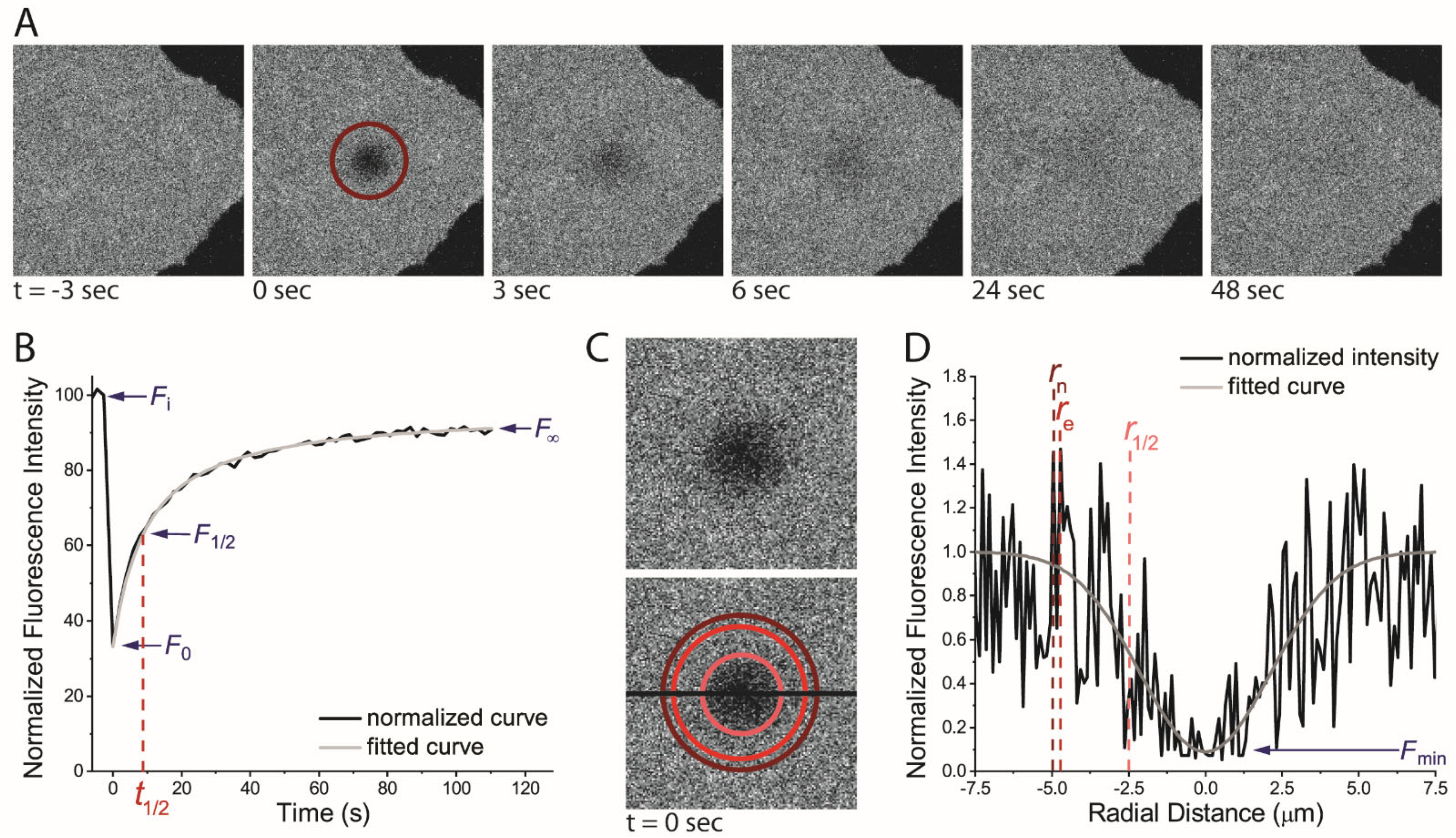
2. Basic FRAP Methodology and Data Analysis
2.1. FRAP Methodology
2.2. Calculating the Mobile Fraction
2.3. Deriving a Diffusion Coefficient from FRAP Data
2.3.1. Deriving a Diffusion Coefficient Using the Half-Time of Recovery from a Static Bleaching Beamline FRAP
2.3.2. Deriving a Diffusion Coefficient by Modeling Recovery from a Static Bleaching Beamline
2.3.3. Deriving a Diffusion Coefficient by Modeling Recovery from Line-Scanning Confocal FRAP Data
2.4. Variations on FRAP
3. Alternative Methods for Measuring Diffusion
3.1. Single-Particle Tracking
3.2. Fluorescence Correlation Spectroscopy
4. What Diffusion Measurements in Live Cells Have Taught Us about the Plasma Membrane
4.1. The Lipid Raft Hypothesis
4.2. Particle Trapping in Caveolae
4.3. Association with Clathrin-Coated Pits
4.4. Diffusion Confined by Cortical Actin
4.5. Effects of Membrane Line Tension
4.6. Effects of Protein Density on Diffusion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Frye, L.D.; Edidin, M. The rapid intermixing of cell surface antigens after formation of mouse-human heterokaryons. J. Cell. Sci. 1970, 7, 319–335. [Google Scholar] [CrossRef]
- Singer, S.J.; Nicolson, G.L. The fluid mosaic model of the structure of cell membranes. Science 1972, 175, 720–731. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, W. A Dynamical Theory of Diffusion for Non-Electrolytes and the Molecular Mass of Albumin. Lond. Edinb. Dublin Philos. Mag. J. Sci. 1905, 9, 781–785. [Google Scholar] [CrossRef]
- Einstein, A. Über die von der molekularkinetischen Theorie der Wärme geforderte Bewegung von in ruhenden Flüssigkeiten suspendierten Teilchen. Ann. Phys. 1905, 322, 549–560. [Google Scholar] [CrossRef]
- Saffman, P.G.; Delbrück, M. Brownian motion in biological membranes. Proc. Natl. Acad. Sci. USA 1975, 72, 3111–3113. [Google Scholar] [CrossRef]
- Jacobson, K.; Sheets, E.D.; Simson, R. Revisiting the fluid mosaic model of membranes. Science 1995, 268, 1441–1442. [Google Scholar] [CrossRef]
- Day, C.A.; Kraft, L.J.; Kang, M.; Kenworthy, A.K. Analysis of protein and lipid dynamics using confocal fluorescence recovery after photobleaching (FRAP). Curr. Protoc. Cytom. 2012, 62, 2.19.1–2.19.29. [Google Scholar] [CrossRef]
- Fritzsche, M.; Charras, G. Dissecting protein reaction dynamics in living cells by fluorescence recovery after photobleaching. Nat. Protoc. 2015, 10, 660–680. [Google Scholar] [CrossRef]
- Carnell, M.; Macmillan, A.; Whan, R. Fluorescence recovery after photobleaching (FRAP): Acquisition, analysis, and applications. Methods Membr. Lipids 2015, 1232, 255–271. [Google Scholar] [CrossRef]
- Braga, J.; Desterro, J.M.; Carmo-Fonseca, M. Intracellular macromolecular mobility measured by fluorescence recovery after photobleaching with confocal laser scanning microscopes. Mol. Biol. Cell 2004, 15, 4749–4760. [Google Scholar] [CrossRef]
- Kang, M.; Day, C.A.; Drake, K.; Kenworthy, A.K.; DiBenedetto, E. A generalization of theory for two-dimensional fluorescence recovery after photobleaching applicable to confocal laser scanning microscopes. Biophys. J. 2009, 97, 1501–1511. [Google Scholar] [CrossRef] [PubMed]
- Axelrod, D.; Koppel, D.E.; Schlessinger, J.; Elson, E.; Webb, W.W. Mobility measurement by analysis of fluorescence photobleaching recovery kinetics. Biophys. J. 1976, 16, 1055–1069. [Google Scholar] [CrossRef] [PubMed]
- Kang, M. Regression Analysis of Confocal FRAP and its Application to Diffusion in Membranes. J. Fluoresc. 2022, 32, 1031–1038. [Google Scholar] [CrossRef] [PubMed]
- Soumpasis, D.M. Theoretical analysis of fluorescence photobleaching recovery experiments. Biophys. J. 1983, 41, 95–97. [Google Scholar] [CrossRef] [PubMed]
- Pinholt, H.D.; Bohr, S.S.; Iversen, J.F.; Boomsma, W.; Hatzakis, N.S. Single-particle diffusional fingerprinting: A machine-learning framework for quantitative analysis of heterogeneous diffusion. Proc. Natl. Acad. Sci. USA 2021, 118, e2104624118. [Google Scholar] [CrossRef]
- Dosset, P.; Rassam, P.; Fernandez, L.; Espenel, C.; Rubinstein, E.; Margeat, E.; Milhiet, P.E. Automatic detection of diffusion modes within biological membranes using back-propagation neural network. BMC Bioinform. 2016, 17, 197. [Google Scholar] [CrossRef]
- Feder, T.J.; Brust-Mascher, I.; Slattery, J.P.; Baird, B.; Webb, W.W. Constrained diffusion or immobile fraction on cell surfaces: A new interpretation. Biophys. J. 1996, 70, 2767–2773. [Google Scholar] [CrossRef]
- Peters, R.; Peters, J.; Tews, K.H.; Bähr, W. A microfluorimetric study of translational diffusion in erythrocyte membranes. Biochim. Biophys. Acta Biomembr. 1974, 367, 282–294. [Google Scholar] [CrossRef]
- Liebman, P.A.; Entine, G. Lateral diffusion of visual pigment in photorecptor disk membranes. Science 1974, 185, 457–459. [Google Scholar] [CrossRef]
- Poo, M.; Cone, R.A. Lateral diffusion of rhodopsin in the photoreceptor membrane. Nature 1974, 247, 438–441. [Google Scholar] [CrossRef]
- Kang, M.; Day, C.A.; Kenworthy, A.K. A novel computational framework for D(t) from Fluorescence Recovery after Photobleaching data reveals various anomalous diffusion types in live cell membranes. Traffic 2019, 20, 867–880. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.; Day, C.A.; Kenworthy, A.K.; DiBenedetto, E. Simplified equation to extract diffusion coefficients from confocal FRAP data. Traffic 2012, 13, 1589–1600. [Google Scholar] [CrossRef] [PubMed]
- Mueller, F.; Mazza, D.; Stasevich, T.J.; McNally, J.G. FRAP and kinetic modeling in the analysis of nuclear protein dynamics: What do we really know? Curr. Opin. Cell Biol. 2010, 22, 403–411. [Google Scholar] [CrossRef]
- Yechiel, E.; Edidin, M. Micrometer-scale domains in fibroblast plasma membranes. J. Cell. Biol. 1987, 105, 755–760. [Google Scholar] [CrossRef] [PubMed]
- Edidin, M.; Stroynowski, I. Differences between the lateral organization of conventional and inositol phospholipid-anchored membrane proteins. A further definition of micrometer scale membrane domains. J. Cell. Biol. 1991, 112, 1143–1150. [Google Scholar] [CrossRef]
- Schram, V.; Tocanne, J.F.; Lopez, A. Influence of obstacles on lipid lateral diffusion: Computer simulation of FRAP experiments and application to proteoliposomes and biomembranes. Eur. Biophys. J. 1994, 23, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Salomé, L.; Cazeils, J.L.; Lopez, A.; Tocanne, J.F. Characterization of membrane domains by FRAP experiments at variable observation areas. Eur. Biophys. J. 1998, 27, 391–402. [Google Scholar] [CrossRef] [PubMed]
- Cézanne, L.; Lecat, S.; Lagane, B.; Millot, C.; Vollmer, J.Y.; Matthes, H.; Galzi, J.L.; Lopez, A. Dynamic confinement of NK2 receptors in the plasma membrane. Improved FRAP analysis and biological relevance. J. Biol. Chem. 2004, 279, 45057–45067. [Google Scholar] [CrossRef]
- Pucadyil, T.J.; Chattopadhyay, A. Cholesterol depletion induces dynamic confinement of the G-protein coupled serotonin(1A) receptor in the plasma membrane of living cells. Biochim. Biophys. Acta Biomembr. 2007, 1768, 655–668. [Google Scholar] [CrossRef]
- Gelles, J.; Schnapp, B.J.; Sheetz, M.P. Tracking kinesin-driven movements with nanometre-scale precision. Nature 1988, 331, 450–453. [Google Scholar] [CrossRef]
- Sheetz, M.P.; Turney, S.; Qian, H.; Elson, E.L. Nanometre-level analysis demonstrates that lipid flow does not drive membrane glycoprotein movements. Nature 1989, 340, 284–288. [Google Scholar] [CrossRef] [PubMed]
- Geerts, H.; De Brabander, M.; Nuydens, R.; Geuens, S.; Moeremans, M.; De Mey, J.; Hollenbeck, P. Nanovid tracking: A new automatic method for the study of mobility in living cells based on colloidal gold and video microscopy. Biophys. J. 1987, 52, 775–782. [Google Scholar] [CrossRef] [PubMed]
- Clarke, D.T.; Martin-Fernandez, M.L. A Brief History of Single-Particle Tracking of the Epidermal Growth Factor Receptor. Methods Protoc. 2019, 2, 12. [Google Scholar] [CrossRef] [PubMed]
- Arnspang, E.C.; Sengupta, P.; Mortensen, K.I.; Jensen, H.H.; Hahn, U.; Jensen, E.B.V.; Lippincott-Schwartz, J.; Nejsum, L.N. Regulation of Plasma Membrane Nanodomains of the Water Channel Aquaporin-3 Revealed by Fixed and Live Photoactivated Localization Microscopy. Nano Lett. 2019, 19, 699–707. [Google Scholar] [CrossRef]
- Ries, J.; Chiantia, S.; Schwille, P. Accurate determination of membrane dynamics with line-scan FCS. Biophys. J. 2009, 96, 1999–2008. [Google Scholar] [CrossRef]
- Wawrezinieck, L.; Rigneault, H.; Marguet, D.; Lenne, P.F. Fluorescence correlation spectroscopy diffusion laws to probe the submicron cell membrane organization. Biophys. J. 2005, 89, 4029–4042. [Google Scholar] [CrossRef]
- Lenne, P.F.; Wawrezinieck, L.; Conchonaud, F.; Wurtz, O.; Boned, A.; Guo, X.J.; Rigneault, H.; He, H.T.; Marguet, D. Dynamic molecular confinement in the plasma membrane by microdomains and the cytoskeleton meshwork. EMBO J. 2006, 25, 3245–3256. [Google Scholar] [CrossRef]
- Eggeling, C.; Ringemann, C.; Medda, R.; Schwarzmann, G.; Sandhoff, K.; Polyakova, S.; Belov, V.N.; Hein, B.; von Middendorff, C.; Schönle, A.; et al. Direct observation of the nanoscale dynamics of membrane lipids in a living cell. Nature 2009, 457, 1159–1162. [Google Scholar] [CrossRef]
- Heinze, K.G.; Koltermann, A.; Schwille, P. Simultaneous two-photon excitation of distinct labels for dual-color fluorescence cross-correlation analysis. Proc. Natl. Acad. Sci. USA 2000, 97, 10377–10382. [Google Scholar] [CrossRef]
- Larson, D.R.; Gosse, J.A.; Holowka, D.A.; Baird, B.A.; Webb, W.W. Temporally resolved interactions between antigen-stimulated IgE receptors and Lyn kinase on living cells. J. Cell Biol. 2005, 171, 527–536. [Google Scholar] [CrossRef]
- Roullier, V.; Clarke, S.; You, C.; Pinaud, F.; Gouzer, G.G.; Schaible, D.; Marchi-Artzner, V.; Piehler, J.; Dahan, M. High-affinity labeling and tracking of individual histidine-tagged proteins in live cells using Ni2+ tris-nitrilotriacetic acid quantum dot conjugates. Nano Lett. 2009, 9, 1228–1234. [Google Scholar] [CrossRef] [PubMed]
- Picard, D.; Suslova, E.; Briand, P.A. 2-color photobleaching experiments reveal distinct intracellular dynamics of two components of the Hsp90 complex. Exp. Cell Res. 2006, 312, 3949–3958. [Google Scholar] [CrossRef] [PubMed]
- Kino, T.; Liou, S.H.; Charmandari, E.; Chrousos, G.P. Glucocorticoid receptor mutants demonstrate increased motility inside the nucleus of living cells: Time of fluorescence recovery after photobleaching (FRAP) is an integrated measure of receptor function. Mol. Med. 2004, 10, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Drake, K.R.; Kang, M.; Kenworthy, A.K. Nucleocytoplasmic distribution and dynamics of the autophagosome marker EGFP-LC3. PLoS ONE 2010, 5, e9806. [Google Scholar] [CrossRef]
- Howell, J.L.; Truant, R. Live-cell nucleocytoplasmic protein shuttle assay utilizing laser confocal microscopy and FRAP. Biotechniques 2002, 32, 84, 86–87. [Google Scholar] [CrossRef] [PubMed]
- Cardarelli, F.; Tosti, L.; Serresi, M.; Beltram, F.; Bizzarri, R. Fluorescent recovery after photobleaching (FRAP) analysis of nuclear export rates identifies intrinsic features of nucleocytoplasmic transport. J. Biol. Chem. 2012, 287, 5554–5561. [Google Scholar] [CrossRef]
- Goodwin, J.S.; Drake, K.R.; Rogers, C.; Wright, L.; Lippincott-Schwartz, J.; Philips, M.R.; Kenworthy, A.K. Depalmitoylated Ras traffics to and from the Golgi complex via a nonvesicular pathway. J. Cell. Biol. 2005, 170, 261–272. [Google Scholar] [CrossRef]
- Taylor, N.O.; Wei, M.T.; Stone, H.A.; Brangwynne, C.P. Quantifying Dynamics in Phase-Separated Condensates Using Fluorescence Recovery after Photobleaching. Biophys. J. 2019, 117, 1285–1300. [Google Scholar] [CrossRef]
- Simons, K.; Ikonen, E. Functional rafts in cell membranes. Nature 1997, 387, 569–572. [Google Scholar] [CrossRef]
- Kenworthy, A.K.; Nichols, B.J.; Remmert, C.L.; Hendrix, G.M.; Kumar, M.; Zimmerberg, J.; Lippincott-Schwartz, J. Dynamics of putative raft-associated proteins at the cell surface. J. Cell Biol. 2004, 165, 735–746. [Google Scholar] [CrossRef]
- Day, C.A.; Kenworthy, A.K. Tracking microdomain dynamics in cell membranes. Biochim. Biophys. Acta Biomembr. 2009, 1788, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Shvartsman, D.E.; Gutman, O.; Tietz, A.; Henis, Y.I. Cyclodextrins but not compactin inhibit the lateral diffusion of membrane proteins independent of cholesterol. Traffic 2006, 7, 917–926. [Google Scholar] [CrossRef] [PubMed]
- Shvartsman, D.E.; Kotler, M.; Tall, R.D.; Roth, M.G.; Henis, Y.I. Differently anchored influenza hemagglutinin mutants display distinct interaction dynamics with mutual rafts. J. Cell Biol. 2003, 163, 879–888. [Google Scholar] [CrossRef] [PubMed]
- Meder, D.; Moreno, M.J.; Verkade, P.; Vaz, W.L.; Simons, K. Phase coexistence and connectivity in the apical membrane of polarized epithelial cells. Proc. Natl. Acad. Sci. USA 2006, 103, 329–334. [Google Scholar] [CrossRef]
- Day, C.A.; Kenworthy, A.K. Mechanisms underlying the confined diffusion of cholera toxin B-subunit in intact cell membranes. PLoS ONE 2012, 7, e34923. [Google Scholar] [CrossRef] [PubMed]
- Simons, K.; van Meer, G. Lipid sorting in epithelial cells. Biochemistry 1988, 27, 6197–6202. [Google Scholar] [CrossRef]
- Bastiani, M.; Liu, L.; Hill, M.M.; Jedrychowski, M.P.; Nixon, S.J.; Lo, H.P.; Abankwa, D.; Luetterforst, R.; Fernandez-Rojo, M.; Breen, M.R.; et al. MURC/Cavin-4 and cavin family members form tissue-specific caveolar complexes. J. Cell Biol. 2009, 185, 1259–1273. [Google Scholar] [CrossRef]
- Rothberg, K.G.; Heuser, J.E.; Donzell, W.C.; Ying, Y.S.; Glenney, J.R.; Anderson, R.G. Caveolin, a protein component of caveolae membrane coats. Cell 1992, 68, 673–682. [Google Scholar] [CrossRef]
- Hansen, C.G.; Nichols, B.J. Exploring the caves: Cavins, caveolins and caveolae. Trends Cell Biol. 2010, 20, 177–186. [Google Scholar] [CrossRef]
- Scherer, P.E.; Okamoto, T.; Chun, M.; Nishimoto, I.; Lodish, H.F.; Lisanti, M.P. Identification, sequence, and expression of caveolin-2 defines a caveolin gene family. Proc. Natl. Acad. Sci. USA 1996, 93, 131–135. [Google Scholar] [CrossRef]
- Hill, M.M.; Bastiani, M.; Luetterforst, R.; Kirkham, M.; Kirkham, A.; Nixon, S.J.; Walser, P.; Abankwa, D.; Oorschot, V.M.; Martin, S.; et al. PTRF-Cavin, a conserved cytoplasmic protein required for caveola formation and function. Cell 2008, 132, 113–124. [Google Scholar] [CrossRef]
- Parton, R.G.; del Pozo, M.A. Caveolae as plasma membrane sensors, protectors and organizers. Nat. Rev. Mol. Cell Biol. 2013, 14, 98–112. [Google Scholar] [CrossRef]
- Breen, M.R.; Camps, M.; Carvalho-Simoes, F.; Zorzano, A.; Pilch, P.F. Cholesterol depletion in adipocytes causes caveolae collapse concomitant with proteosomal degradation of cavin-2 in a switch-like fashion. PLoS ONE 2012, 7, e34516. [Google Scholar] [CrossRef]
- Ortegren, U.; Karlsson, M.; Blazic, N.; Blomqvist, M.; Nystrom, F.H.; Gustavsson, J.; Fredman, P.; Strålfors, P. Lipids and glycosphingolipids in caveolae and surrounding plasma membrane of primary rat adipocytes. Eur. J. Biochem. 2004, 271, 2028–2036. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, P.; Roepstorff, K.; Stahlhut, M.; van Deurs, B. Caveolae are highly immobile plasma membrane microdomains, which are not involved in constitutive endocytic trafficking. Mol. Biol. Cell 2002, 13, 238–250. [Google Scholar] [CrossRef]
- Pelkmans, L.; Kartenbeck, J.; Helenius, A. Caveolar endocytosis of simian virus 40 reveals a new two-step vesicular-transport pathway to the ER. Nat. Cell Biol. 2001, 3, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Fire, E.; Zwart, D.E.; Roth, M.G.; Henis, Y.I. Evidence from lateral mobility studies for dynamic interactions of a mutant influenza hemagglutinin with coated pits. J. Cell Biol. 1991, 115, 1585–1594. [Google Scholar] [CrossRef]
- Ktistakis, N.T.; Thomas, D.; Roth, M.G. Characteristics of the tyrosine recognition signal for internalization of transmembrane surface glycoproteins. J. Cell Biol. 1990, 111, 1393–1407. [Google Scholar] [CrossRef]
- Lazarovits, J.; Roth, M. A single amino acid change in the cytoplasmic domain allows the influenza virus hemagglutinin to be endocytosed through coated pits. Cell 1988, 53, 743–752. [Google Scholar] [CrossRef] [PubMed]
- Fire, E.; Gutman, O.; Roth, M.G.; Henis, Y.I. Dynamic or stable interactions of influenza hemagglutinin mutants with coated pits. Dependence on the internalization signal but not on aggregation. J. Biol. Chem. 1995, 270, 21075–21081. [Google Scholar] [CrossRef]
- Wu, E.S.; Tank, D.W.; Webb, W.W. Unconstrained lateral diffusion of concanavalin A receptors on bulbous lymphocytes. Proc. Natl. Acad. Sci. USA 1982, 79, 4962–4966. [Google Scholar] [CrossRef] [PubMed]
- Sheetz, M.P.; Schindler, M.; Koppel, D.E. Lateral mobility of integral membrane proteins is increased in spherocytic erythrocytes. Nature 1980, 285, 510–511. [Google Scholar] [CrossRef]
- Umemura, Y.M.; Vrljic, M.; Nishimura, S.Y.; Fujiwara, T.K.; Suzuki, K.G.; Kusumi, A. Both MHC class II and its GPI-anchored form undergo hop diffusion as observed by single-molecule tracking. Biophys. J. 2008, 95, 435–450. [Google Scholar] [CrossRef]
- Fujiwara, T.; Ritchie, K.; Murakoshi, H.; Jacobson, K.; Kusumi, A. Phospholipids undergo hop diffusion in compartmentalized cell membrane. J. Cell Biol. 2002, 157, 1071–1081. [Google Scholar] [CrossRef]
- Gauthier, N.C.; Fardin, M.A.; Roca-Cusachs, P.; Sheetz, M.P. Temporary increase in plasma membrane tension coordinates the activation of exocytosis and contraction during cell spreading. Proc. Natl. Acad. Sci. USA 2011, 108, 14467–14472. [Google Scholar] [CrossRef]
- Boulant, S.; Kural, C.; Zeeh, J.C.; Ubelmann, F.; Kirchhausen, T. Actin dynamics counteract membrane tension during clathrin-mediated endocytosis. Nat. Cell Biol. 2011, 13, 1124–1131. [Google Scholar] [CrossRef] [PubMed]
- Römer, W.; Berland, L.; Chambon, V.; Gaus, K.; Windschiegl, B.; Tenza, D.; Aly, M.R.; Fraisier, V.; Florent, J.C.; Perrais, D.; et al. Shiga toxin induces tubular membrane invaginations for its uptake into cells. Nature 2007, 450, 670–675. [Google Scholar] [CrossRef] [PubMed]
- Houk, A.R.; Jilkine, A.; Mejean, C.O.; Boltyanskiy, R.; Dufresne, E.R.; Angenent, S.B.; Altschuler, S.J.; Wu, L.F.; Weiner, O.D. Membrane tension maintains cell polarity by confining signals to the leading edge during neutrophil migration. Cell 2012, 148, 175–188. [Google Scholar] [CrossRef]
- Stewart, M.P.; Helenius, J.; Toyoda, Y.; Ramanathan, S.P.; Muller, D.J.; Hyman, A.A. Hydrostatic pressure and the actomyosin cortex drive mitotic cell rounding. Nature 2011, 469, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Basu, R.; Whitlock, B.M.; Husson, J.; Le Floc’h, A.; Jin, W.; Oyler-Yaniv, A.; Dotiwala, F.; Giannone, G.; Hivroz, C.; Biais, N.; et al. Cytotoxic T Cells Use Mechanical Force to Potentiate Target Cell Killing. Cell 2016, 165, 100–110. [Google Scholar] [CrossRef]
- Shi, Z.; Graber, Z.T.; Baumgart, T.; Stone, H.A.; Cohen, A.E. Cell Membranes Resist Flow. Cell 2018, 175, 1769–1779.e13. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Baumgart, T. Membrane tension and peripheral protein density mediate membrane shape transitions. Nat. Commun. 2015, 6, 5974. [Google Scholar] [CrossRef] [PubMed]
- Frick, M.; Schmidt, K.; Nichols, B.J. Modulation of lateral diffusion in the plasma membrane by protein density. Curr. Biol. 2007, 17, 462–467. [Google Scholar] [CrossRef] [PubMed]
- Oda, K.; Hirose, S.; Takami, N.; Misumi, Y.; Takatsuki, A.; Ikehara, Y. Brefeldin A arrests the intracellular transport of a precursor of complement C3 before its conversion site in rat hepatocytes. FEBS Lett. 1987, 214, 135–138. [Google Scholar] [CrossRef] [PubMed]
- Takatsuki, A.; Tamura, G. Brefeldin A, a Specific Inhibitor of Intracellular Translocation of Vesicular Stomatitis Virus G Protein: Intracellular Accumulation of High-mannose Type G Protein and Inhibition of Its Cell Surface Expression. Agric. Biol. Chem. 1985, 49, 899–902. [Google Scholar]
- Misumi, Y.; Miki, K.; Takatsuki, A.; Tamura, G.; Ikehara, Y. Novel blockade by brefeldin A of intracellular transport of secretory proteins in cultured rat hepatocytes. J. Biol. Chem. 1986, 261, 11398–11403. [Google Scholar] [CrossRef]
- Kallen, K.J.; Quinn, P.; Allan, D. Effects of brefeldin A on sphingomyelin transport and lipid synthesis in BHK21 cells. Biochem. J. 1993, 289 Pt 1, 307–312. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Day, C.A.; Kang, M. The Utility of Fluorescence Recovery after Photobleaching (FRAP) to Study the Plasma Membrane. Membranes 2023, 13, 492. https://doi.org/10.3390/membranes13050492
Day CA, Kang M. The Utility of Fluorescence Recovery after Photobleaching (FRAP) to Study the Plasma Membrane. Membranes. 2023; 13(5):492. https://doi.org/10.3390/membranes13050492
Chicago/Turabian StyleDay, Charles A., and Minchul Kang. 2023. "The Utility of Fluorescence Recovery after Photobleaching (FRAP) to Study the Plasma Membrane" Membranes 13, no. 5: 492. https://doi.org/10.3390/membranes13050492
APA StyleDay, C. A., & Kang, M. (2023). The Utility of Fluorescence Recovery after Photobleaching (FRAP) to Study the Plasma Membrane. Membranes, 13(5), 492. https://doi.org/10.3390/membranes13050492