

Biocatalytic Membranes for Carbon Capture and Utilization


Abstract
1. Introduction
1.1. Enzymes for CO2 Capture and Utilization
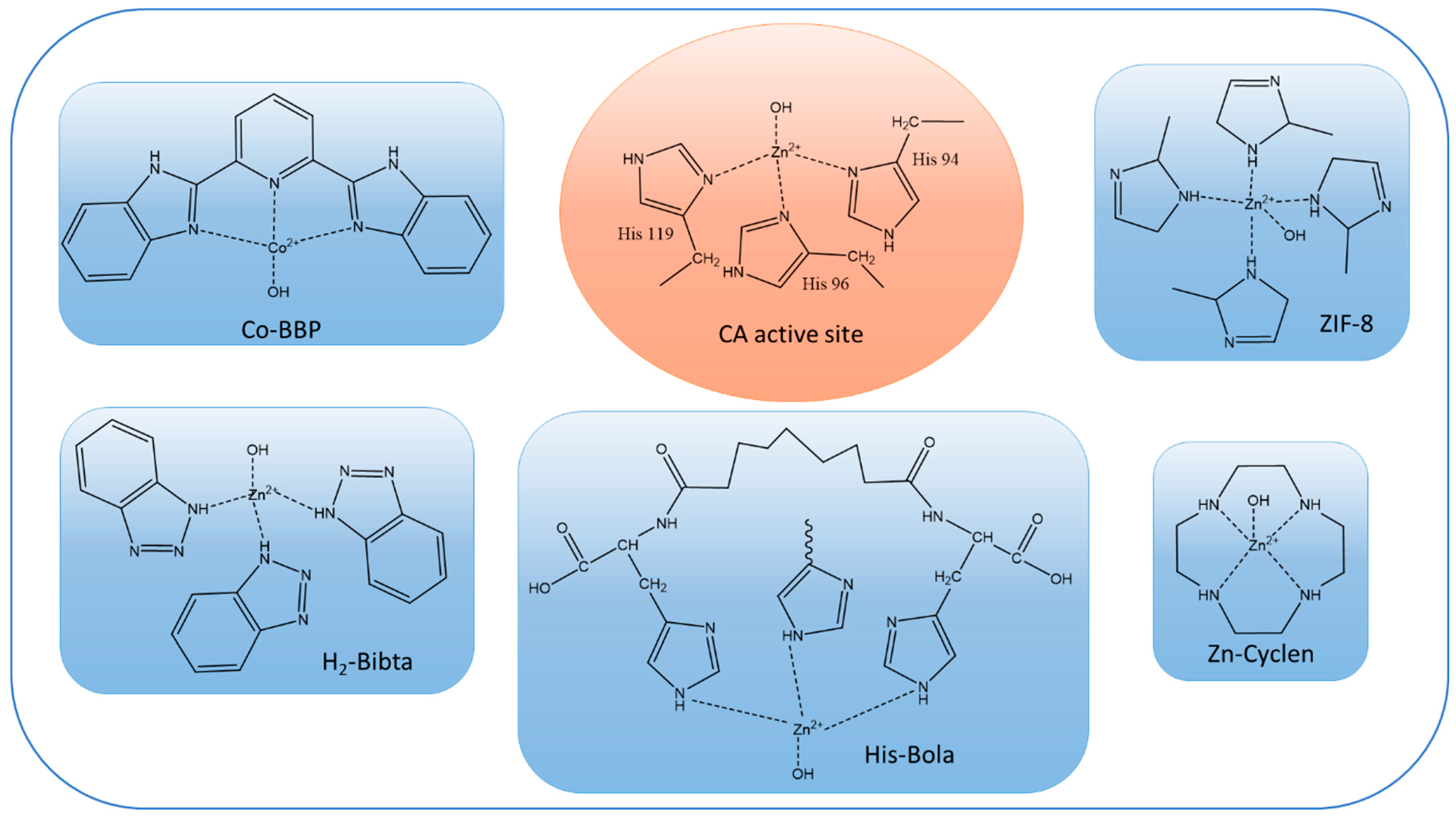
1.1.1. Carbonic Anhydrases
1.1.2. Formate Dehydrogenases
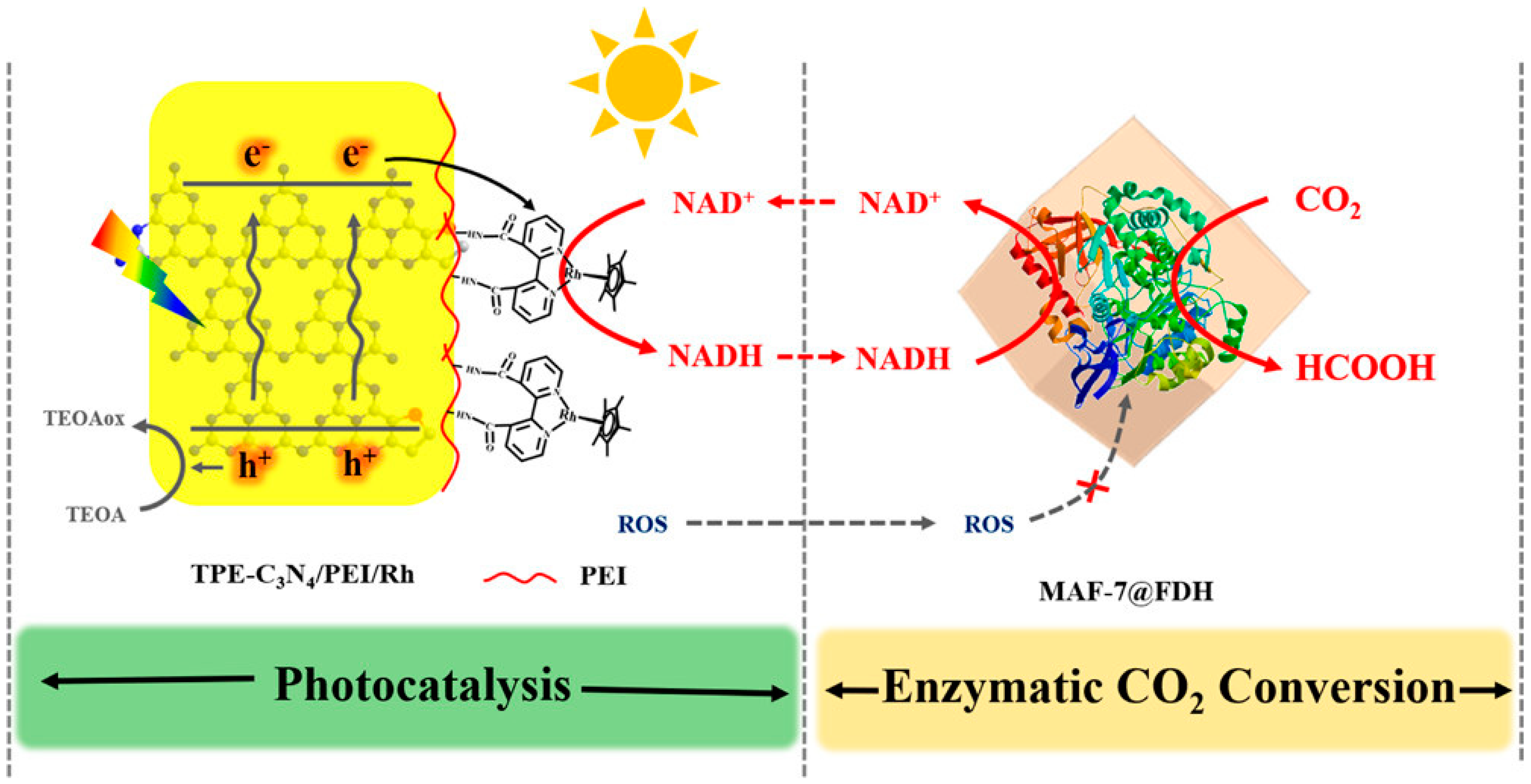
1.1.3. Enzyme Cascade with Other Oxidoreductases
1.1.4. Enzyme Immobilization
1.1.5. Comparisons of Biocatalysts with Electrocatalysts for CO2 Reduction Reaction
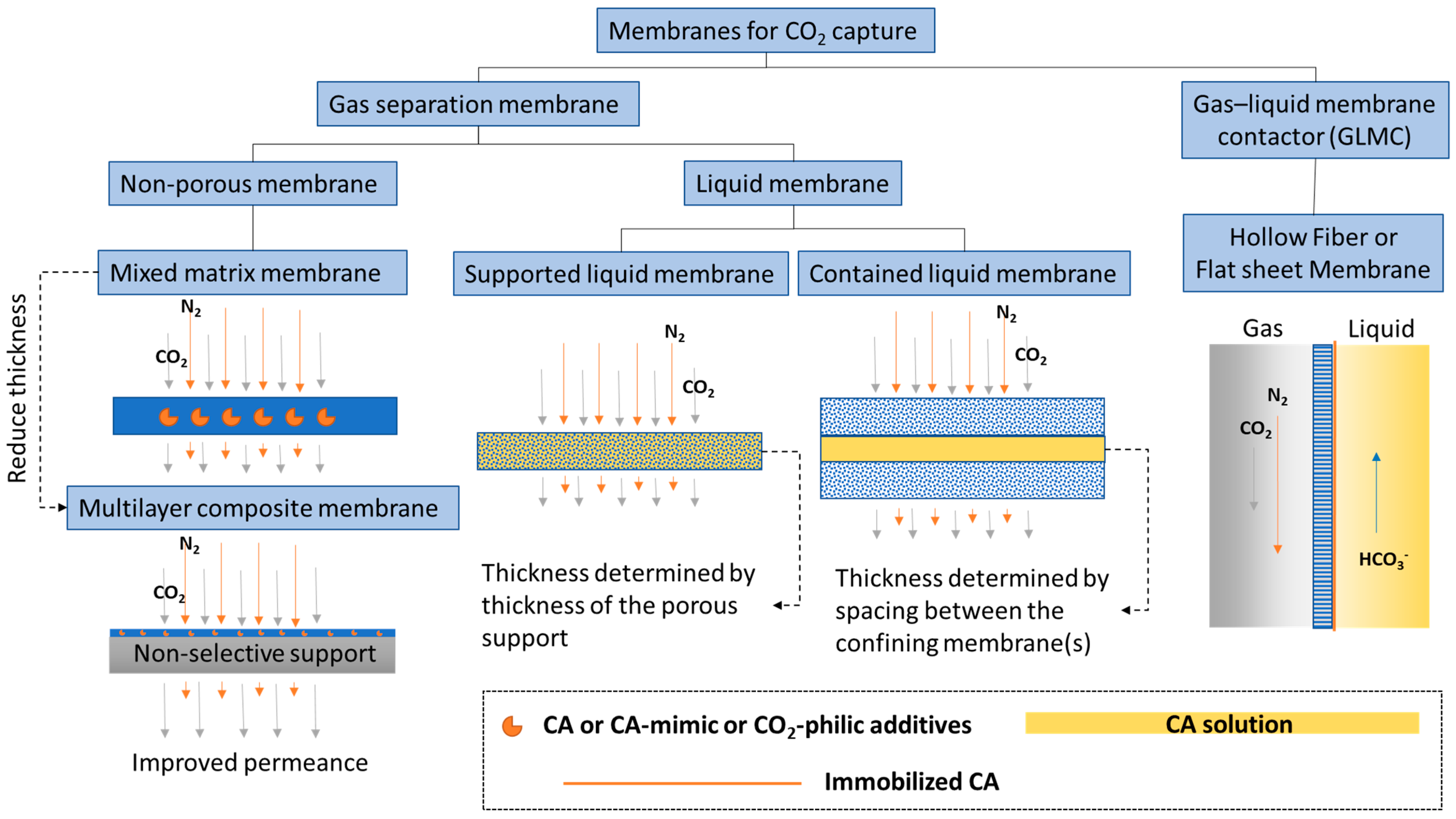
1.2. Types of Membranes for CO2 Capture and Utilization
1.2.1. CO2 Separation Membrane
1.2.2. CO2 Liquid Contactor Membrane
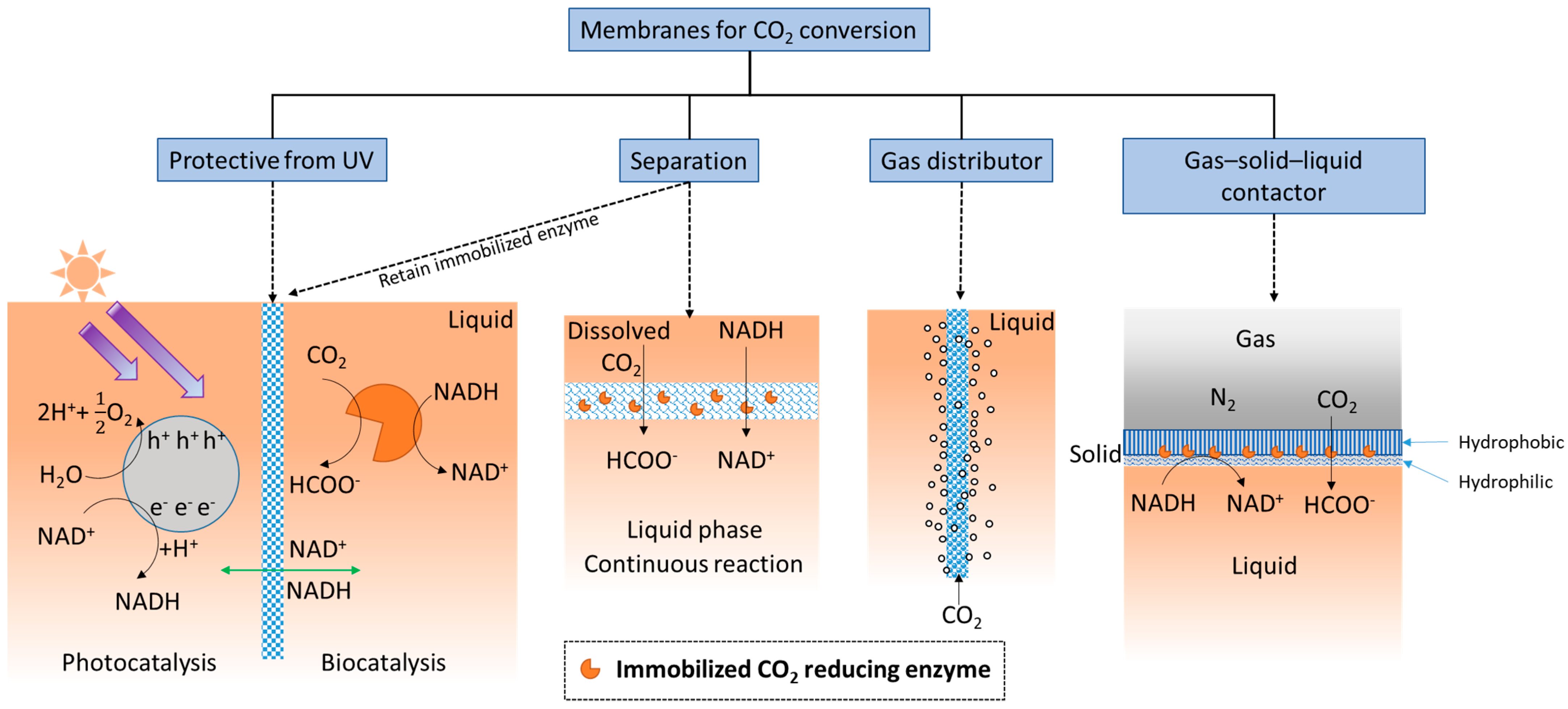
1.2.3. Other Membrane Structure Functions
2. Facilitated Transport Membranes
2.1. CA vs. CA-Mimic
2.2. Membrane Structures
2.3. Humidity
3. Liquid Membranes
3.1. Early Developments in CA-Promoted Supported Liquid Membrane (SLM)
3.2. SLM with Non-Volatile Liquids
3.3. CA-Promoted Contained Liquid Membrane (CLM)
3.4. Liquid Membrane Thickness
4. Gas–Liquid Membrane Contactor
4.1. Advantages Compared with Conventional Gas Separation Membrane and Chemical Absorption
4.2. Developments in CA-Promoted GLMC
4.3. Materials and Surface Modifications
4.4. Enzyme Immobilization
4.5. Solvents and Form of Substrate
5. Enzyme Membrane Reactor
5.1. Location of the Immobilized Enzymes
5.2. Roles of Membrane in Substrate Uptake
5.3. Cofactor Regeneration
5.4. Long-Term Stability of EMR
6. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Friedlingstein, P.; O’Sullivan, M.; Jones, M.W.; Andrew, R.M.; Hauck, J.; Olsen, A.; Peters, G.P.; Peters, W.; Pongratz, J.; Sitch, S.; et al. Global Carbon Budget 2020. Earth Syst. Sci. Data 2020, 12, 3269–3340. [Google Scholar] [CrossRef]
- Shakun, J.D.; Clark, P.U.; He, F.; Marcott, S.A.; Mix, A.C.; Liu, Z.; Otto-Bliesner, B.; Schmittner, A.; Bard, E. Global Warming Preceded by Increasing Carbon Dioxide Concentrations during the Last Deglaciation. Nature 2012, 484, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Lindsey, R. Climate Change: Atmospheric Carbon Dioxide. Available online: https://www.climate.gov/news-features/understanding-climate/climate-change-atmospheric-carbon-dioxide (accessed on 18 July 2022).
- Lenton, T.; Rockström, J.; Gaffney, O.; Rahmstorf, S.; Richardson, K.; Steffen, W.; Shellnhuber, H.J. Climate Tipping Points—Too Risky to Bet Against. Nature 2019, 575, 592–595. [Google Scholar] [CrossRef]
- Lebling, K.; Leslie-Bole, H.; Psarras, P.; Bridgwater, E.; Byrum, Z.; Pilorgé, H. Direct Air Capture: Assessing Impacts to Enable Responsible Scaling. World Resour. Inst. 2022, 1–28. [Google Scholar] [CrossRef]
- Peplow, M. The Race to Upcycle CO2 into Fuels, Concrete and More. Nature 2022, 603, 780–783. [Google Scholar] [CrossRef]
- Mondal, M.K.; Balsora, H.K.; Varshney, P. Progress and Trends in CO2 Capture/Separation Technologies: A Review. Energy 2012, 46, 431–441. [Google Scholar] [CrossRef]
- Merkel, T.C.; Lin, H.; Wei, X.; Baker, R. Power Plant Post-Combustion Carbon Dioxide Capture: An Opportunity for Membranes. J. Memb. Sci. 2010, 359, 126–139. [Google Scholar] [CrossRef]
- Witek-Krowiak, A.; Dawiec, A.; Modelski, S.; Podstawczyk, D. Carbon Dioxide Removal in a Membrane Contactor—Selection of Absorptive Liquid/Membrane System. Int. J. Chem. Eng. Appl. 2012, 3, 391–395. [Google Scholar] [CrossRef]
- Figueroa, J.D.; Fout, T.; Plasynski, S.; McIlvried, H.; Srivastava, R.D. Advances in CO2 Capture Technology—The U.S. Department of Energy’s Carbon Sequestration Program. Int. J. Greenh. Gas Control 2008, 2, 9–20. [Google Scholar] [CrossRef]
- Khalifah, R.G. The Carbon Dioxide Hydration Activity of Carbonic Anhydrase. I. Stop-Flow Kinetic Studies on the Native Human Isoenzymes B and C. J. Biol. Chem. 1971, 246, 2561–2573. [Google Scholar] [CrossRef]
- Ward, W.J.; Robb, W.L. Carbon Dioxide-Oxygen Separation: Facilitated Transport of Carbon Dioxide across a Liquid Film. Science 1967, 156, 1481–1484. [Google Scholar] [CrossRef] [PubMed]
- Kaar, J.L.; Oh, H.I.; Russell, A.J.; Federspiel, W.J. Towards Improved Artificial Lungs through Biocatalysis. Biomaterials 2007, 28, 3131–3139. [Google Scholar] [CrossRef] [PubMed]
- Salmon, S.; House, A. Enzyme-Catalyzed Solvents for CO2 Separation. In Novel Materials for Carbon Dioxide Mitigation Technology; Novel Materials for Carbon Dioxide Mitigation Technology; Elsevier: Amsterdam, The Netherlands, 2015; pp. 23–86. [Google Scholar]
- Qi, G.; Liu, K.; House, A.; Salmon, S.; Ambedkar, B.; Frimpong, R.A.; Remias, J.E.; Liu, K. Laboratory to Bench-Scale Evaluation of an Integrated CO2 Capture System Using a Thermostable Carbonic Anhydrase Promoted K2CO3 Solvent with Low Temperature Vacuum Stripping. Appl. Energy 2018, 209, 180–189. [Google Scholar] [CrossRef]
- Shen, J.; Yuan, Y.; Salmon, S. Carbonic Anhydrase Immobilized on Textile Structured Packing Using Chitosan Entrapment for CO2 Capture. ACS Sustain. Chem. Eng. 2022, 10, 7772–7785. [Google Scholar] [CrossRef]
- Shen, J.; Yuan, Y.; Salmon, S. Durable and Versatile Immobilized Carbonic Anhydrase on Textile Structured Packing for CO2 Capture. Catalysts 2022, 12, 1108. [Google Scholar] [CrossRef]
- Niazi, M.B.K.; Jahan, Z.; Ahmed, A.; Rafiq, S.; Jamil, F.; Gregersen, Ø.W. Effect of Zn-Cyclen Mimic Enzyme on Mechanical, Thermal and Swelling Properties of Cellulose Nanocrystals/PVA Nanocomposite Membranes. J. Polym. Environ. 2020, 28, 1921–1933. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, H.; Zhou, S.; Wang, J.; He, X.; Liu, J.; Zhang, Y. Biomimetic Material Functionalized Mixed Matrix Membranes for Enhanced Carbon Dioxide Capture. J. Mater. Chem. A 2018, 6, 15585–15592. [Google Scholar] [CrossRef]
- Zheng, W.; Tian, Z.; Wang, Z.; Peng, D.; Zhang, Y.; Wang, J.; Zhang, Y. Dual-Function Biomimetic Carrier Based Facilitated Transport Mixed Matrix Membranes with High Stability for Efficient CO2/N2 Separation. Sep. Purif. Technol. 2022, 285, 120371. [Google Scholar] [CrossRef]
- Alvizo, O.; Nguyen, L.J.; Savile, C.K.; Bresson, J.A.; Lakhapatri, S.L.; Solis, E.O.P.; Fox, R.J.; Broering, J.M.; Benoit, M.R.; Zimmerman, S.A.; et al. Directed Evolution of an Ultrastable Carbonic Anhydrase for Highly Efficient Carbon Capture from Flue Gas. Proc. Natl. Acad. Sci. USA 2014, 111, 16436–16441. [Google Scholar] [CrossRef]
- Reardon, J.; Bucholz, T.; Hulvey, M.; Tuttle, J.; Shaffer, A.; Pulvirenti, D.; Weber, L.; Killian, K.; Zaks, A. Low Energy CO2 Capture Enabled by Biocatalyst Delivery System. Energy Procedia 2014, 63, 301–321. [Google Scholar] [CrossRef]
- Smit, B.; Park, A.H.A.; Gadikota, G. The Grand Challenges in Carbon Capture, Utilization, and Storage. Front. Energy Res. 2014, 2, 2013–2015. [Google Scholar] [CrossRef]
- Bulushev, D.A.; Ross, J.R.H. Towards Sustainable Production of Formic Acid. ChemSusChem 2018, 11, 821–836. [Google Scholar] [CrossRef] [PubMed]
- Grubel, K.; Jeong, H.; Yoon, C.W.; Autrey, T. Challenges and Opportunities for Using Formate to Store, Transport, and Use Hydrogen. J. Energy Chem. 2020, 41, 216–224. [Google Scholar] [CrossRef]
- Bar-Even, A. Formate Assimilation: The Metabolic Architecture of Natural and Synthetic Pathways. Biochemistry 2016, 55, 3851–3863. [Google Scholar] [CrossRef] [PubMed]
- Mao, W.; Yuan, Q.; Qi, H.; Wang, Z.; Ma, H.; Chen, T. Recent Progress in Metabolic Engineering of Microbial Formate Assimilation. Appl. Microbiol. Biotechnol. 2020, 104, 6905–6917. [Google Scholar] [CrossRef]
- Marpani, F.; Pinelo, M.; Meyer, A.S. Enzymatic Conversion of CO2 to CH3OH via Reverse Dehydrogenase Cascade Biocatalysis: Quantitative Comparison of Efficiencies of Immobilized Enzyme Systems. Biochem. Eng. J. 2017, 127, 217–228. [Google Scholar] [CrossRef]
- Song, H.; Ma, C.; Liu, P.; You, C.; Lin, J.; Zhu, Z. A Hybrid CO2 Electroreduction System Mediated by Enzyme-Cofactor Conjugates Coupled with Cu Nanoparticle-Catalyzed Cofactor Regeneration. J. CO2 Util. 2019, 34, 568–575. [Google Scholar] [CrossRef]
- Chen, Y.; Li, P.; Noh, H.; Kung, C.; Buru, C.T.; Wang, X.; Zhang, X.; Farha, O.K. Stabilization of Formate Dehydrogenase in a Metal–Organic Framework for Bioelectrocatalytic Reduction of CO2. Angew. Chem. 2019, 131, 7764–7768. [Google Scholar] [CrossRef]
- Liao, Q.; Liu, W.; Meng, Z. Strategies for Overcoming the Limitations of Enzymatic Carbon Dioxide Reduction. Biotechnol. Adv. 2022, 60, 108024. [Google Scholar] [CrossRef]
- Luo, J.; Song, S.; Zhang, H.; Zhang, H.; Zhang, J.; Wan, Y. Biocatalytic Membrane: Go Far beyond Enzyme Immobilization. Eng. Life Sci. 2020, 20, 441–450. [Google Scholar] [CrossRef]
- Lim, F.N.A.R.; Marpani, F.; Dilol, V.E.A.; Pauzi, S.M.; Othman, N.H.; Alias, N.H.; Him, N.R.N.; Luo, J.; Rahman, N.A. A Review on the Design and Performance of Enzyme-Aided Catalysis of Carbon Dioxide in Membrane, Electrochemical Cell and Photocatalytic Reactors. Membranes 2022, 12, 28. [Google Scholar] [CrossRef]
- Shen, J.; Zhang, S.; Fang, X.; Salmon, S. Advances in 3D Gel Printing for Enzyme Immobilization. Gels 2022, 8, 460. [Google Scholar] [CrossRef]
- Sun, J.; Wei, L.; Wang, Y.; Zhao, Z.; Liu, W. Immobilization of Carbonic Anhydrase on Polyvinylidene Fluoride Membranes. Biotechnol. Appl. Biochem. 2018, 65, 362–371. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Wang, C.; Wang, Y.; Ji, S.; Liu, W. Immobilization of Carbonic Anhydrase on Polyethylenimine/Dopamine Codeposited Membranes. J. Appl. Polym. Sci. 2019, 136, 1–9. [Google Scholar] [CrossRef]
- Luo, J.; Meyer, A.S.; Mateiu, R.V.; Pinelo, M. Cascade Catalysis in Membranes with Enzyme Immobilization for Multi-Enzymatic Conversion of CO2 to Methanol. N. Biotechnol. 2015, 32, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Ren, S.; Li, C.; Tan, Z.; Hou, Y.; Jia, S.; Cui, J. Carbonic Anhydrase@ZIF-8 Hydrogel Composite Membrane with Improved Recycling and Stability for Efficient CO2 Capture. J. Agric. Food Chem. 2019, 67, 3372–3379. [Google Scholar] [CrossRef]
- Wen, H.; Zhang, L.; Du, Y.; Wang, Z.; Jiang, Y.; Bian, H.; Cui, J.; Jia, S. Bimetal Based Inorganic-Carbonic Anhydrase Hybrid Hydrogel Membrane for CO2 Capture. J. CO2 Util. 2020, 39, 101171. [Google Scholar] [CrossRef]
- Moehlenbrock, M.J.; Minteer, S.D. Introduction to the Field of Enzyme Immobilization and Stabilization. In Enzyme Stabilization and Immobilization: Methods and Protocols; Minteer, S.D., Ed.; Humana Press: New York, NY, USA, 2017; Volume 1504, pp. 1–7. ISBN 978-1-60761-894-2. [Google Scholar]
- Guisan, J.M. New Opportunities for Immobilization of Enzymes. In Immobilization of Enzymes and Cells: Third Edition; Guisan, J.M., Ed.; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2013; Volume 1051, pp. 1–13. ISBN 978-1-62703-549-1. [Google Scholar]
- Rasouli, H.; Nguyen, K.; Iliuta, M.C. Recent Advancements in Carbonic Anhydrase Immobilization and Its Implementation in CO2 Capture Technologies: A Review. Sep. Purif. Technol. 2022, 296, 121299. [Google Scholar] [CrossRef]
- Molina-Fernández, C.; Luis, P. Immobilization of Carbonic Anhydrase for CO2 Capture and Its Industrial Implementation: A Review. J. CO2 Util. 2021, 47, 101475. [Google Scholar] [CrossRef]
- Russo, M.E.; Capasso, C.; Marzocchella, A.; Salatino, P. Immobilization of Carbonic Anhydrase for CO2 Capture and Utilization. Appl. Microbiol. Biotechnol. 2022, 106, 3419–3430. [Google Scholar] [CrossRef]
- Hedstrom, L. Enzyme Specificity and Selectivity. In Encyclopedia of Life Sciences; Wiley: Chichester, UK, 2010; pp. 1–8. ISBN 9780470015902. [Google Scholar] [CrossRef]
- Bender, D.A. Tricarboxylic Acid Cycle. In Encyclopedia of Food Sciences and Nutrition; Elsevier: Amsterdam, The Netherlands, 2003; pp. 5851–5856. ISBN 9780123786319. [Google Scholar]
- Yates, N.D.J.; Fascione, M.A.; Parkin, A. Methodologies for “Wiring” Redox Proteins/Enzymes to Electrode Surfaces. Chem.—A Eur. J. 2018, 24, 12164–12182. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, F.; Zhang, X.; Ji, X. Recent Progress on Electrochemical Reduction of CO2 to Methanol. Curr. Opin. Green Sustain. Chem. 2020, 23, 10–17. [Google Scholar] [CrossRef]
- Saxena, A.; Liyanage, W.; Masud, J.; Kapila, S.; Nath, M. Selective Electroreduction of CO2 to Carbon-Rich Products with a Simple Binary Copper Selenide Electrocatalyst. J. Mater. Chem. A 2021, 9, 7150–7161. [Google Scholar] [CrossRef]
- Saxena, A.; Singh, H.; Nath, M. Cobalt Telluride Electrocatalyst for Selective Electroreduction of CO2 to Value-Added Chemicals. Mater. Renew. Sustain. Energy 2022, 11, 115–129. [Google Scholar] [CrossRef]
- Saxena, A.; Liyanage, W.P.R.; Kapila, S.; Nath, M. Nickel Selenide as an Efficient Electrocatalyst for Selective Reduction of Carbon Dioxide to Carbon-Rich Products. Catal. Sci. Technol. 2022, 12, 4727–4739. [Google Scholar] [CrossRef]
- Saxena, A.; Kapila, S.; Medvedeva, J.E.; Nath, M. Copper Cobalt Selenide as a Bifunctional Electrocatalyst for the Selective Reduction of CO2 to Carbon-Rich Products and Alcohol Oxidation. ACS Appl. Mater. Interfaces 2023, 15, 14433–14446. [Google Scholar] [CrossRef]
- Schlager, S.; Dumitru, L.M.; Haberbauer, M.; Fuchsbauer, A.; Neugebauer, H.; Hiemetsberger, D.; Wagner, A.; Portenkirchner, E.; Sariciftci, N.S. Electrochemical Reduction of Carbon Dioxide to Methanol by Direct Injection of Electrons into Immobilized Enzymes on a Modified Electrode. ChemSusChem 2016, 9, 631–635. [Google Scholar] [CrossRef]
- He, X. A Review of Material Development in the Field of Carbon Capture and the Application of Membrane-Based Processes in Power Plants and Energy-Intensive Industries. Energy. Sustain. Soc. 2018, 8, 34. [Google Scholar] [CrossRef]
- Bernardo, P.; Drioli, E.; Golemme, G. Membrane Gas Separation: A Review/State of the Art. Ind. Eng. Chem. Res. 2009, 48, 4638–4663. [Google Scholar] [CrossRef]
- He, X.; Lindbråthen, A.; Kim, T.J.; Hägg, M.B. Pilot Testing on Fixed-Site-Carrier Membranes for CO2 Capture from Flue Gas. Int. J. Greenh. Gas Control 2017, 64, 323–332. [Google Scholar] [CrossRef]
- Merkel, T.; Kniep, J.; Wei, X.; Carlisle, T.; White, S.; Pande, S.; Fulton, D.; Watson, R.; Hoffman, T.; Freeman, B.; et al. Pilot Testing of a Membrane System for Postcombustion CO2 Capture; National Energy Technology Laboratory: Pittsburgh, PA, USA; Morgantown, WV, USA, 2015. [Google Scholar]
- Trachtenberg, M.C.; Cowan, R.M.; Smith, D.A.; Horazak, D.A.; Jensen, M.D.; Laumb, J.D.; Vucelic, A.P.; Chen, H.; Wang, L.; Wu, X. Membrane-Based, Enzyme-Facilitated, Efficient Carbon Dioxide Capture. Energy Procedia 2009, 1, 353–360. [Google Scholar] [CrossRef]
- Zhou, F.; Tien, H.N.; Xu, W.L.; Chen, J.T.; Liu, Q.; Hicks, E.; Fathizadeh, M.; Li, S.; Yu, M. Ultrathin Graphene Oxide-Based Hollow Fiber Membranes with Brush-like CO2-Philic Agent for Highly Efficient CO2 Capture. Nat. Commun. 2017, 8, 2107. [Google Scholar] [CrossRef] [PubMed]
- McKeown, N.B. Polymers of Intrinsic Microporosity (PIMs). Polymer 2020, 202, 122736. [Google Scholar] [CrossRef]
- Comesaña-Gándara, B.; Chen, J.; Bezzu, C.G.; Carta, M.; Rose, I.; Ferrari, M.C.; Esposito, E.; Fuoco, A.; Jansen, J.C.; McKeown, N.B. Redefining the Robeson Upper Bounds for CO2/CH4 and CO2/N2 Separations Using a Series of Ultrapermeable Benzotriptycene-Based Polymers of Intrinsic Microporosity. Energy Environ. Sci. 2019, 12, 2733–2740. [Google Scholar] [CrossRef]
- Lai, H.W.H.; Benedetti, F.M.; Ahn, J.M.; Robinson, A.M.; Wang, Y.; Pinnau, I.; Smith, Z.P.; Xia, Y. Hydrocarbon Ladder Polymers with Ultrahigh Permselectivity for Membrane Gas Separations. Science 2022, 375, 1390–1392. [Google Scholar] [CrossRef] [PubMed]
- Robeson, L.M. The Upper Bound Revisited. J. Memb. Sci. 2008, 320, 390–400. [Google Scholar] [CrossRef]
- Scholes, C.A.; Kanehashi, S. Polymer of Intrinsic Microporosity (PIM-1) Membranes Treated with Supercritical CO2. Membranes 2019, 9, 41. [Google Scholar] [CrossRef]
- Tan, X.; Robijns, S.; Thür, R.; Ke, Q.; De Witte, N.; Lamaire, A.; Li, Y.; Aslam, I.; Van Havere, D.; Donckels, T.; et al. Truly Combining the Advantages of Polymeric and Zeolite Membranes for Gas Separations. Science 2022, 378, 1189–1194. [Google Scholar] [CrossRef]
- Torre-Celeizabal, A.; Casado-Coterillo, C.; Garea, A. Biopolymer-Based Mixed Matrix Membranes (MMMs) for CO2/CH4 Separation: Experimental and Modeling Evaluation. Membranes 2022, 12, 561. [Google Scholar] [CrossRef]
- Casado-Coterillo, C.; Fernández-Barquín, A.; Zornoza, B.; Téllez, C.; Coronas, J.; Irabien, Á. Synthesis and Characterisation of MOF/Ionic Liquid/Chitosan Mixed Matrix Membranes for CO2/N2 Separation. RSC Adv. 2015, 5, 102350–102361. [Google Scholar] [CrossRef]
- Borgohain, R.; Jain, N.; Prasad, B.; Mandal, B.; Su, B. Carboxymethyl Chitosan/Carbon Nanotubes Mixed Matrix Membranes for CO2 Separation. React. Funct. Polym. 2019, 143, 104331. [Google Scholar] [CrossRef]
- Casado-Coterillo, C.; Fernández-Barquín, A.; Irabien, A. Effect of Humidity on CO2/N2 and CO2/CH4 Separation Using Novel Robust Mixed Matrix Composite Hollow Fiber Membranes: Experimental and Model Evaluation. Membranes 2019, 10, 6. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Nada, A.A.; Abou-Zeid, N.Y.; Hudson, S.M. Synthesis of Chitosan Iodoacetamides via Carbodiimide Coupling Reaction: Effect of Degree of Substitution on the Hemostatic Properties. Carbohydr. Polym. 2020, 229, 115522. [Google Scholar] [CrossRef]
- Nada, A.A.; Ali, E.A.; Soliman, A.A.F.; Shen, J.; Abou-Zeid, N.Y.; Hudson, S.M. Multi-Layer Dressing Made of Laminated Electrospun Nanowebs and Cellulose-Based Adhesive for Comprehensive Wound Care. Int. J. Biol. Macromol. 2020, 162, 629–644. [Google Scholar] [CrossRef]
- Nada, A.A.; Abdellatif, F.H.H.; Soliman, A.A.F.; Shen, J.; Hudson, S.M.; Abou-Zeid, N.Y. Fabrication and Bioevaluation of a Medicated Electrospun Mat Based on Azido-Cellulose Acetate via Click Chemistry. Cellulose 2019, 26, 9721–9736. [Google Scholar] [CrossRef]
- Ji, Y.; Zhang, M.; Guan, K.; Zhao, J.; Liu, G.; Jin, W. High-Performance CO2 Capture through Polymer-Based Ultrathin Membranes. Adv. Funct. Mater. 2019, 29, 1–9. [Google Scholar] [CrossRef]
- Sandru, M.; Sandru, E.M.; Ingram, W.F.; Deng, J.; Stenstad, P.M.; Deng, L.; Spontak, R.J. An Integrated Materials Approach to Ultrapermeable and Ultraselective CO2 Polymer Membranes. Science 2022, 376, 90–94. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, H.; Liu, J.; Hou, J.; Zhang, Y. Enzyme-Embedded Metal-Organic Framework Membranes on Polymeric Substrates for Efficient CO2 Capture. J. Mater. Chem. A 2017, 5, 19954–19962. [Google Scholar] [CrossRef]
- Saeed, M.; Deng, L. CO2 Facilitated Transport Membrane Promoted by Mimic Enzyme. J. Memb. Sci. 2015, 494, 196–204. [Google Scholar] [CrossRef]
- Gilassi, S.; Taghavi, S.M.; Rodrigue, D.; Kaliaguine, S. Techno-Economic Evaluation of Membrane and Enzymatic-Absorption Processes for CO2 Capture from Flue-Gas. Sep. Purif. Technol. 2020, 248, 116941. [Google Scholar] [CrossRef]
- Bao, L.; Trachtenberg, M.C. Facilitated Transport of CO2 across a Liquid Membrane: Comparing Enzyme, Amine, and Alkaline. J. Memb. Sci. 2006, 280, 330–334. [Google Scholar] [CrossRef]
- Cheng, L.H.; Zhang, L.; Chen, H.L.; Gao, C.J. Hollow Fiber Contained Hydrogel-CA Membrane Contactor for Carbon Dioxide Removal from the Enclosed Spaces. J. Memb. Sci. 2008, 324, 33–43. [Google Scholar] [CrossRef]
- Bednár, A.; Nemestóthy, N.; Bakonyi, P.; Fülöp, L.; Zhen, G.; Lu, X.; Kobayashi, T.; Kumar, G.; Xu, K.; Bélafi-Bakó, K. Enzymatically-Boosted Ionic Liquid Gas Separation Membranes Using Carbonic Anhydrase of Biomass Origin. Chem. Eng. J. 2016, 303, 621–626. [Google Scholar] [CrossRef]
- De Castro, A.M.; Prasavath, D.; Bevilaqua, J.V.; Portugal, C.A.M.; Neves, L.A.; Crespo, J.G. Role of Water on Deep Eutectic Solvents (DES) Properties and Gas Transport Performance in Biocatalytic Supported DES Membranes. Sep. Purif. Technol. 2021, 255, 117763. [Google Scholar] [CrossRef]
- Nemestóthy, N.; Bakonyi, P.; Németh, Z.; Bélafi-Bakó, K. Evaluation of Pectin-Reinforced Supported Liquid Membranes Containing Carbonic Anhydrase: The Role of Ionic Liquid on Enzyme Stability and CO2 Separation Performance. J. CO2 Util. 2018, 24, 59–63. [Google Scholar] [CrossRef]
- Rivero, J.R.; Panagakos, G.; Lieber, A.; Hornbostel, K. Hollow Fiber Membrane Contactors for Post-Combustion Carbon Capture: A Review of Modeling Approaches. Membranes 2020, 10, 382. [Google Scholar] [CrossRef]
- Vadillo, J.M.; Gómez-Coma, L.; Garea, A.; Irabien, A. Hollow Fiber Membrane Contactors in CO2 Desorption: A Review. Energy Fuels 2021, 35, 111–136. [Google Scholar] [CrossRef]
- Porcheron, F.; Ferré, D.; Favre, E.; Nguyen, P.T.; Lorain, O.; Mercier, R.; Rougeau, L. Hollow Fiber Membrane Contactors for CO2 Capture: From Lab-Scale Screening to Pilot-Plant Module Conception. Energy Procedia 2011, 4, 763–770. [Google Scholar] [CrossRef]
- Ibrahim, M.H.; El-Naas, M.H.; Zhang, Z.; Van Der Bruggen, B. CO2 Capture Using Hollow Fiber Membranes: A Review of Membrane Wetting. Energy Fuels 2018, 32, 963–978. [Google Scholar] [CrossRef]
- Zare, A.; Perna, L.; Nogalska, A.; Ambrogi, V.; Cerruti, P.; Tylkowski, B.; García-Valls, R.; Giamberini, M. Polymer Blends for Improved CO2 Capture Membranes. Polymers 2019, 11, 1662. [Google Scholar] [CrossRef]
- Yong, J.K.J.; Cui, J.; Cho, K.L.; Stevens, G.W.; Caruso, F.; Kentish, S.E. Surface Engineering of Polypropylene Membranes with Carbonic Anhydrase-Loaded Mesoporous Silica Nanoparticles for Improved Carbon Dioxide Hydration. Langmuir 2015, 31, 6211–6219. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.J.; Lang, A.; Chikukwa, A.; Sheridan, E.; Dahl, P.I.; Leimbrink, M.; Skiborowski, M.; Roubroeks, J. Enzyme Carbonic Anhydrase Accelerated CO2 Absorption in Membrane Contactor. Energy Procedia 2017, 114, 17–24. [Google Scholar] [CrossRef]
- RASOULI, H.; ILIUTA, I.; BOUGIE, F.; GARNIER, A.; ILIUTA, M.C. Hybrid Enzymatic CO2 Capture Process in Intensified Flat Sheet Membrane Contactors with Immobilized Carbonic Anhydrase. Sep. Purif. Technol. 2022, 287, 120505. [Google Scholar] [CrossRef]
- Nguyen, K.; Iliuta, I.; Bougie, F.; Pasquier, L.C.; Iliuta, M.C. Techno-Economic Assessment of Enzymatic CO2 Capture in Hollow Fiber Membrane Contactors with Immobilized Carbonic Anhydrase. Sep. Purif. Technol. 2023, 307, 122702. [Google Scholar] [CrossRef]
- Kurayama, F.; Matsuyama, T.; Yamamoto, H. Kinetic Study of a New Photosynthesis Bioreactor Design Using TiO2 Particles Combined with Enzymes. Adv. Powder Technol. 2005, 16, 517–533. [Google Scholar] [CrossRef]
- Gundersen, M.T.; Gladis, A.; Fosbøl, P.L.; Von Solms, N.; Woodley, J.M. Operating Considerations of Ultrafiltration in Enzyme Enhanced Carbon Capture. Energy Procedia 2017, 114, 735–743. [Google Scholar] [CrossRef][Green Version]
- Guo, M.; Gu, F.; Meng, L.; Liao, Q.; Meng, Z.; Liu, W. Synthesis of Formaldehyde from CO2 Catalyzed by the Coupled Photo-Enzyme System. Sep. Purif. Technol. 2022, 286, 120480. [Google Scholar] [CrossRef]
- Wang, Y.Z.; Zhao, Z.P.; Li, M.F.; Chen, Y.Z.; Liu, W. fang Development of a Hollow Fiber Membrane Micro-Reactor for Biocatalytic Production of Formate from CO2. J. Memb. Sci. 2016, 514, 44–52. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, J. Bioinspired Photocatalytic NADH Regeneration by Covalently Metalated Carbon Nitride for Enhanced CO2 Reduction. Chem.–Eur. J. 2022, 28, e202201430. [Google Scholar] [CrossRef]
- Nilouyal, S.; Karahan, H.E.; Isfahani, A.P.; Yamaguchi, D.; Gibbons, A.H.; Ito, M.M.M.; Sivaniah, E.; Ghalei, B. Carbonic Anhydrase-Mimicking Supramolecular Nanoassemblies for Developing Carbon Capture Membranes. ACS Appl. Mater. Interfaces 2022, 14, 37595–37607. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, Y.; Wang, J.; Zhang, Y. Bioinspired Porous Organic Polymer-Functionalized Membranes for Efficient CO2 Capture. Sustain. Energy Fuels 2020, 4, 1191–1198. [Google Scholar] [CrossRef]
- Jahan, Z.; Niazi, M.B.K.; Gul, S.; Sher, F.; Kakar, S.J.; Hägg, M.B.; Gregersen, Ø.W. Mimic Enzyme Based Cellulose Nanocrystals/PVA Nanocomposite Membranes for Enrichment of Biogas as a Natural Gas Substitute. J. Polym. Environ. 2021, 29, 2598–2608. [Google Scholar] [CrossRef]
- Saeed, M.; Deng, L. Carbon Nanotube Enhanced PVA-Mimic Enzyme Membrane for Post-Combustion CO2 Capture. Int. J. Greenh. Gas Control 2016, 53, 254–262. [Google Scholar] [CrossRef]
- Duan, S.; Kai, T.; Nakao, S.I. Effect of Carbonic Anhydrase on CO2 Separation Performance of Thin Poly(Amidoamine) Dendrimer/Poly(Ethylene Glycol) Hybrid Membranes. Membranes 2019, 9, 167. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.H.; Liu, N.; Yu, M.R.; Yang, S.T.; Chen, H.L. Cell Surface Display of Carbonic Anhydrase on Escherichia Coli Using Ice Nucleation Protein for CO2 Sequestration. Biotechnol. Bioeng. 2011, 108, 2853–2864. [Google Scholar] [CrossRef]
- Favre, N.; Pierre, A.C. Synthesis and Behaviour of Hybrid Polymer-Silica Membranes Made by Sol Gel Process with Adsorbed Carbonic Anhydrase Enzyme, in the Capture of CO2. J. Sol-Gel Sci. Technol. 2011, 60, 177–188. [Google Scholar] [CrossRef]
- Neves, L.A.; Afonso, C.; Coelhoso, I.M.; Crespo, J.G. Integrated CO2 Capture and Enzymatic Bioconversion in Supported Ionic Liquid Membranes. Sep. Purif. Technol. 2012, 97, 34–41. [Google Scholar] [CrossRef]
- Fu, Y.; Jiang, Y.B.; Dunphy, D.; Xiong, H.; Coker, E.; Chou, S.; Zhang, H.; Vanegas, J.M.; Croissant, J.G.; Cecchi, J.L.; et al. Ultra-Thin Enzymatic Liquid Membrane for CO2 Separation and Capture. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef]
- Craveiro, R.; Neves, L.A.; Duarte, A.R.C.; Paiva, A. Supported Liquid Membranes Based on Deep Eutectic Solvents for Gas Separation Processes. Sep. Purif. Technol. 2021, 254, 117593. [Google Scholar] [CrossRef]
- Mondal, S.; Alke, B.; de Castro, A.M.; Ortiz-Albo, P.; Syed, U.T.; Crespo, J.G.; Brazinha, C. Design of Enzyme Loaded W/O Emulsions by Direct Membrane Emulsification for CO2 Capture. Membranes 2022, 12, 797. [Google Scholar] [CrossRef]
- De Castro, A.M.; Neves, L.A.; Corvo, M.C.; Cabrita, E.J.; Crespo, J.G. Effect of Carbonic Anhydrase on CO2 Absorption Promoted by Choline Hydroxide Using Supported Liquid Membranes. Sep. Purif. Technol. 2022, 280, 119921. [Google Scholar] [CrossRef]
- Enns, T. Facilitation by Carbonic Anhydrase of Carbon Dioxide Transport. Science. 1967, 155, 44–47. [Google Scholar] [CrossRef] [PubMed]
- Abdelrahim, M.Y.M.; Martins, C.F.; Neves, L.A.; Capasso, C.; Supuran, C.T.; Coelhoso, I.M.; Crespo, J.G.; Barboiu, M. Supported Ionic Liquid Membranes Immobilized with Carbonic Anhydrases for CO2 Transport at High Temperatures. J. Memb. Sci. 2017, 528, 225–230. [Google Scholar] [CrossRef]
- Zhang, Y.-T.; Zhang, L.; Chen, H.-L.; Zhang, H.-M. Selective Separation of Low Concentration CO2 Using Hydrogel Immobilized CA Enzyme Based Hollow Fiber Membrane Reactors. Chem. Eng. Sci. 2010, 65, 3199–3207. [Google Scholar] [CrossRef]
- COWAN, R.M.; GE, J.-J.; QIN, Y.-J.; McGREGOR, M.L.; TRACHTENBERG, M.C. CO2 Capture by Means of an Enzyme-Based Reactor. Ann. N. Y. Acad. Sci. 2003, 984, 453–469. [Google Scholar] [CrossRef]
- Amann, J.M.G.; Bouallou, C. CO2 Capture from Power Stations Running with Natural Gas (NGCC) and Pulverized Coal (PC): Assessment of a New Chemical Solvent Based on Aqueous Solutions of N-Methyldiethanolamine + Triethylene Tetramine. Energy Procedia 2009, 1, 909–916. [Google Scholar] [CrossRef]
- Liu, S.; Gao, H.; He, C.; Liang, Z. Experimental Evaluation of Highly Efficient Primary and Secondary Amines with Lower Energy by a Novel Method for Post-Combustion CO2 Capture. Appl. Energy 2019, 233–234, 443–452. [Google Scholar] [CrossRef]
- Vega, F.; Baena-Moreno, F.M.; Gallego Fernández, L.M.; Portillo, E.; Navarrete, B.; Zhang, Z. Current Status of CO2 Chemical Absorption Research Applied to CCS: Towards Full Deployment at Industrial Scale. Appl. Energy 2020, 260, 114313. [Google Scholar] [CrossRef]
- N.Borhani, T.; Wang, M. Role of Solvents in CO2 Capture Processes: The Review of Selection and Design Methods. Renew. Sustain. Energy Rev. 2019, 114, 109299. [Google Scholar] [CrossRef]
- Zhang, S.; Lu, Y. Kinetic Performance of CO2 Absorption into a Potassium Carbonate Solution Promoted with the Enzyme Carbonic Anhydrase: Comparison with a Monoethanolamine Solution. Chem. Eng. J. 2015, 279, 335–343. [Google Scholar] [CrossRef]
- Leimbrink, M.; Neumann, K.; Kupitz, K.; Górak, A.; Skiborowski, M. Enzyme Accelerated Carbon Capture in Different Contacting Equipment—A Comparative Study. Energy Procedia 2017, 114, 795–812. [Google Scholar] [CrossRef]
- Zhao, S.; Feron, P.H.M.; Deng, L.; Favre, E.; Chabanon, E.; Yan, S.; Hou, J.; Chen, V.; Qi, H. Status and Progress of Membrane Contactors in Post-Combustion Carbon Capture: A State-of-the-Art Review of New Developments. J. Memb. Sci. 2016, 511, 180–206. [Google Scholar] [CrossRef]
- Arazawa, D.T.; Oh, H.I.; Ye, S.H.; Johnson, C.A.; Woolley, J.R.; Wagner, W.R.; Federspiel, W.J. Immobilized Carbonic Anhydrase on Hollow Fiber Membranes Accelerates CO2 Removal from Blood. J. Memb. Sci. 2012, 403–404, 25–31. [Google Scholar] [CrossRef]
- Kimmel, J.D.; Arazawa, D.T.; Ye, S.H.; Shankarraman, V.; Wagner, W.R.; Federspiel, W.J. Carbonic Anhydrase Immobilized on Hollow Fiber Membranes Using Glutaraldehyde Activated Chitosan for Artificial Lung Applications. J. Mater. Sci. Mater. Med. 2013, 24, 2611–2621. [Google Scholar] [CrossRef]
- Arazawa, D.T.; Kimmel, J.D.; Finn, M.C.; Federspiel, W.J. Acidic Sweep Gas with Carbonic Anhydrase Coated Hollow Fiber Membranes Synergistically Accelerates CO2 Removal from Blood. Acta Biomater. 2015, 25, 143–149. [Google Scholar] [CrossRef]
- Arazawa, D.T.; Kimmel, J.D.; Federspiel, W.J. Kinetics of CO2 Exchange with Carbonic Anhydrase Immobilized on Fiber Membranes in Artificial Lungs. J. Mater. Sci. Mater. Med. 2015, 26, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Koonaphapdeelert, S.; Wu, Z.; Li, K. Carbon Dioxide Stripping in Ceramic Hollow Fibre Membrane Contactors. Chem. Eng. Sci. 2009, 64, 1–8. [Google Scholar] [CrossRef]
- Kosaraju, P.; Kovvali, A.S.; Korikov, A.; Sirkar, K.K. Hollow Fiber Membrane Contactor Based CO2 Absorption-Stripping Using Novel Solvents and Membranes. Ind. Eng. Chem. Res. 2005, 44, 1250–1258. [Google Scholar] [CrossRef]
- Saunders, P.; Salmon, S.; Borchert, M.; Lessard, L.P. Modular Membrane Reactor and Process for Carbon Dioxide Extraction. US Patent 20110223650 A1, 15 September 2011. [Google Scholar]
- Hou, J.; Dong, G.; Xiao, B.; Malassigne, C.; Chen, V. Preparation of Titania Based Biocatalytic Nanoparticles and Membranes for CO2 Conversion. J. Mater. Chem. A 2015, 3, 3332–3342. [Google Scholar] [CrossRef]
- Hou, J.; Ji, C.; Dong, G.; Xiao, B.; Ye, Y.; Chen, V. Biocatalytic Janus Membranes for CO2 Removal Utilizing Carbonic Anhydrase. J. Mater. Chem. A 2015, 3, 17032–17041. [Google Scholar] [CrossRef]
- Hou, J.; Zulkifli, M.Y.; Mohammad, M.; Zhang, Y.; Razmjou, A.; Chen, V. Biocatalytic Gas-Liquid Membrane Contactors for CO2 Hydration with Immobilized Carbonic Anhydrase. J. Memb. Sci. 2016, 520, 303–313. [Google Scholar] [CrossRef]
- Saeed, M.; Deng, L. Post-Combustion CO2 Membrane Absorption Promoted by Mimic Enzyme. J. Memb. Sci. 2016, 499, 36–46. [Google Scholar] [CrossRef]
- Yong, J.K.J.; Stevens, G.W.; Caruso, F.; Kentish, S.E. In Situ Layer-by-Layer Assembled Carbonic Anhydrase-Coated Hollow Fiber Membrane Contactor for Rapid CO2 Absorption. J. Memb. Sci. 2016, 514, 556–565. [Google Scholar] [CrossRef]
- Yong, J.K.J.; Stevens, G.W.; Caruso, F.; Kentish, S.E. The Resilience of Carbonic Anhydrase Enzyme for Membrane-Based Carbon Capture Applications. Int. J. Greenh. Gas Control 2017, 62, 122–129. [Google Scholar] [CrossRef]
- Malankowska, M.; Martins, C.F.; Rho, H.S.; Neves, L.A.; Tiggelaar, R.M.; Crespo, J.G.; Pina, M.P.; Mallada, R.; Gardeniers, H.; Coelhoso, I.M. Microfluidic Devices as Gas—Ionic Liquid Membrane Contactors for CO2 Removal from Anaesthesia Gases. J. Memb. Sci. 2018, 545, 107–115. [Google Scholar] [CrossRef]
- Xu, Y.; Lin, Y.; Chew, N.G.P.; Malde, C.; Wang, R. Biocatalytic PVDF Composite Hollow Fiber Membranes for CO2 Removal in Gas-Liquid Membrane Contactor. J. Memb. Sci. 2019, 572, 532–544. [Google Scholar] [CrossRef]
- Kim, H.S.; Hong, S.G.; Yang, J.; Ju, Y.; Ok, J.; Kwon, S.J.; Yeon, K.M.; Dordick, J.S.; Kim, J. 3D-Printed Interfacial Devices for Biocatalytic CO2 Conversion at Gas-Liquid Interface. J. CO2 Util. 2020, 38, 291–298. [Google Scholar] [CrossRef]
- Liu, Q.; Bai, X.; Pham, H.; Hu, J.; Dinu, C.Z. Active Nanointerfaces Based on Enzyme Carbonic Anhydrase and Metal–Organic Framework for Carbon Dioxide Reduction. Nanomaterials 2021, 11, 1008. [Google Scholar] [CrossRef]
- Rasouli, H.; Iliuta, I.; Bougie, F.; Garnier, A.; Iliuta, M.C. Enzyme-Immobilized Flat-Sheet Membrane Contactor for Green Carbon Capture. Chem. Eng. J. 2021, 421, 129587. [Google Scholar] [CrossRef]
- Iliuta, I.; Iliuta, M.C. Investigation of CO2 Removal by Immobilized Carbonic Anhydrase Enzyme in a Hollow-Fiber Membrane Bioreactor. AIChE J. 2017, 63, 2996–3007. [Google Scholar] [CrossRef]
- Wang, X.; Zhou, D.; Sinniah, K.; Clarke, C.; Birch, L.; Li, H.; Rayment, T.; Abell, C. Electrostatic Orientation of Enzymes on Surfaces for Ligand Screening Probed by Force Spectroscopy. Langmuir 2006, 22, 887–892. [Google Scholar] [CrossRef] [PubMed]
- Zeuner, B.; Ma, N.; Berendt, K.; Meyer, A.S.; Andric, P.; Jørgensen, J.H.; Pinelo, M. Immobilization of Alcohol Dehydrogenase on Ceramic Silicon Carbide Membranes for Enzymatic CH3OH Production. J. Chem. Technol. Biotechnol. 2018, 93, 2952–2961. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, Y.; Wang, C.; Sun, J.; Zhao, Z.; Liu, W. Polyethylenimine-Modified Membranes for CO2 Capture and in Situ Hydrogenation. ACS Appl. Mater. Interfaces 2018, 10, 29003–29009. [Google Scholar] [CrossRef] [PubMed]
- Barin, R.; Biria, D.; Rashid-Nadimi, S.; Asadollahi, M.A. Enzymatic CO2 Reduction to Formate by Formate Dehydrogenase from Candida Boidinii Coupling with Direct Electrochemical Regeneration of NADH. J. CO2 Util. 2018, 28, 117–125. [Google Scholar] [CrossRef]
- Zhu, D.; Ao, S.; Deng, H.; Wang, M.; Qin, C.; Zhang, J.; Jia, Y.; Ye, P.; Ni, H. Ordered Coimmobilization of a Multienzyme Cascade System with a Metal Organic Framework in a Membrane: Reduction of CO2 to Methanol. ACS Appl. Mater. Interfaces 2019, 11, 33581–33588. [Google Scholar] [CrossRef] [PubMed]
- Gu, F.J.; Wang, Y.Z.; Meng, Z.H.; Liu, W.F.; Qiu, L.Y. A Coupled Photocatalytic/Enzymatic System for Sustainable Conversion of CO2 to Formate. Catal. Commun. 2020, 136, 105903. [Google Scholar] [CrossRef]
- Guo, M.Y.; Zhai, T.T.; Wang, C.H.; Meng, Z.H.; Liu, W.F. Immobilization of Formate Dehydrogenase on Polyethyleneimine Modified Carriers for the Enhancement of Catalytic Performance. Catal. Commun. 2021, 149, 106259. [Google Scholar] [CrossRef]
- Chai, M.; Razmjou, A.; Chen, V. Metal-Organic-Framework Protected Multi-Enzyme Thin-Film for the Cascade Reduction of CO2 in a Gas-Liquid Membrane Contactor. J. Memb. Sci. 2021, 623, 118986. [Google Scholar] [CrossRef]
- Lin, G.; Zhang, Y.; Hua, Y.; Zhang, C.; Jia, C.; Ju, D.; Yu, C.; Li, P.; Liu, J. Bioinspired Metalation of the Metal-Organic Framework MIL-125-NH2 for Photocatalytic NADH Regeneration and Gas-Liquid-Solid Three-Phase Enzymatic CO2 Reduction. Angew. Chemie Int. Ed. 2022, 61, e202206283. [Google Scholar] [CrossRef]
- Tian, Y.; Zhou, Y.; Zong, Y.; Li, J.; Yang, N.; Zhang, M.; Guo, Z.; Song, H. Construction of Functionally Compartmental Inorganic Photocatalyst-Enzyme System via Imitating Chloroplast for Efficient Photoreduction of CO2 to Formic Acid. ACS Appl. Mater. Interfaces 2020, 12, 34795–34805. [Google Scholar] [CrossRef]
- Hollmann, F.; Witholt, B.; Schmid, A. [Cp*Rh(Bpy)(H2O)]2+: A Versatile Tool for Efficient and Non-Enzymatic Regeneration of Nicotinamide and Flavin Coenzymes. J. Mol. Catal. B Enzym. 2002, 19–20, 167–176. [Google Scholar] [CrossRef]
- Yuan, M.; Sahin, S.; Cai, R.; Abdellaoui, S.; Hickey, D.P.; Minteer, S.D.; Milton, R.D. Creating a Low-Potential Redox Polymer for Efficient Electroenzymatic CO2 Reduction. Angew. Chem. -Int. Ed. 2018, 57, 6582–6586. [Google Scholar] [CrossRef] [PubMed]
- Sahin, S.; Cai, R.; Milton, R.D.; Abdellaoui, S.; Macazo, F.C.; Minteer, S.D. Molybdenum-Dependent Formate Dehydrogenase for Formate Bioelectrocatalysis in a Formate/O2 Enzymatic Fuel Cell. J. Electrochem. Soc. 2018, 165, H109–H113. [Google Scholar] [CrossRef]
- Minteer, S.D.; Liaw, B.Y.; Cooney, M.J. Enzyme-Based Biofuel Cells. Curr. Opin. Biotechnol. 2007, 18, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Moore, C.M.; Akers, N.L.; Hill, A.D.; Johnson, Z.C.; Minteer, S.D. Improving the Environment for Immobilized Dehydrogenase Enzymes by Modifying Nafion with Tetraalkylammonium Bromides. Biomacromolecules 2004, 5, 1241–1247. [Google Scholar] [CrossRef]
- Moehlenbrock, M.J.; Minteer, S.D. Extended Lifetime Biofuel Cells. Chem. Soc. Rev. 2008, 37, 1188–1196. [Google Scholar] [CrossRef]
- Arechederra, R.L.; Treu, B.L.; Minteer, S.D. Development of Glycerol/O2 Biofuel Cell. J. Power Sources 2007, 173, 156–161. [Google Scholar] [CrossRef]
- Rubenwolf, S.; Sané, S.; Hussein, L.; Kestel, J.; Von Stetten, F.; Urban, G.; Krueger, M.; Zengerle, R.; Kerzenmacher, S. Prolongation of Electrode Lifetime in Biofuel Cells by Periodic Enzyme Renewal. Appl. Microbiol. Biotechnol. 2012, 96, 841–849. [Google Scholar] [CrossRef]
- Herkendell, K.; Stemmer, A.; Tel-Vered, R. Extending the Operational Lifetimes of All-Direct Electron Transfer Enzymatic Biofuel Cells by Magnetically Assembling and Exchanging the Active Biocatalyst Layers on Stationary Electrodes. Nano Res. 2019, 12, 767–775. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Application | Membrane Configuration | Enzyme, Concentration | Performance | Year, 1st Author, Ref |
|---|---|---|---|---|
| CO2 separation from N2 | PVA selective layer containing enzyme mimic supported by PSf ultrafiltration membrane | 5 μmol/g (Zn–cyclen /PVA) with 1 wt% CNT | CO2 permeance: 256–363 GPU CO2/N2 selectivity: 107–120 | 2015–2016 Saeed [76,100] |
| CO2 separation from N2 | Biocatalytic composite membranes HNTs/MOF/CA selective layer supported by PAN membrane | 24.2 wt% CA in MOF | CO2 permeance: 24.2 GPU CO2/N2 selectivity: 165.5 | 2017 Zhang [75] |
| CO2 separation from N2 | MMM with Cobalt-based CA-mimic CoBBP dispersed in Pebax-1657 (PEO:PA6 polyamide 60:40 wt%) | 1.33 wt% CoBBP in Pebax | CO2 permeability: 675.5 Barrer CO2 permeance: 9 GPU (75 μm thickness) CO2/N2 selectivity: 62 | 2018 Zhang [19] |
| CO2 separation from H2 | PAMAM/PEGDMA/GMA hybrid membrane supported on PES porous support | 1 wt% CA loading spray-coated on the hybrid membrane | CO2 permeance: 14.4 GPU CO2 permeability: 216 Barrer (15 μm thickness) CO2/He selectivity: 28.7 | 2019 Duan [101] |
| CO2 separation from N2 | MMM with CoBBP CA-mimic loaded on POP (CoBBP@POP) and together both were loaded in Pebax-1657 matrix | 28.5 wt% CoBBP in POP. 5 wt% CoBBP@POP composite in Pebax matrix. | CO2 permeability: 1620 Barrer CO2 permeance: 32.4 GPU (50 μm thickness) CO2/N2 selectivity: 102 | 2020 Wang [98] |
| CO2 separation from CH4 | PVA selective layer containing CA-mimic supported by PSf ultrafiltration membrane | 5 μmol/g (Zn–cyclen /PVA with 1 wt% CNC) | CO2 permeance: 126 GPU CO2/CH4 selectivity: 42 | 2021 Jahan [99] |
| CO2 separation from N2 | MMM with His-NPs CA-mimic loaded in Pebax-1657 matrix | 0–9 wt% His-NPs in Pebax-1657 matrix | CO2 permeability: 188.4 Barrer CO2 permeance: 2.7 GPU (70 μm thickness) CO2/N2 selectivity: 158.2 | 2022 Nilouyal [97] |
| CO2 separation from N2 | MMM with Zinc-coordinated MOF CA-mimic loaded in Pebax-1657 matrix | 3% MOF CA-mimic in Pebax-1657 | CO2 permeability: 869 (dry) Barrer 1409 (humid) Barrer CO2 permeance: 28.2 GPU (50 μm thickness) CO2/N2 selectivity: 88.6 (dry) 83 (humid) | 2022 Zheng [20] |
| Application | Membrane Configuration | Enzyme, Concentration | Performance | Year, 1st Author, Ref |
|---|---|---|---|---|
| CO2 separation from N2 | Microporous PP HFCLM mat with heat exchanger type design (mutually orthogonal fiber orientation) | 3 mg/mL CA in 1.0 M NaHCO3 | At 10% CO2 CO2 permeance: 90 GPU CO2/N2 selectivity: 234 | 2006 Bao [78] |
| CO2 separation from air | Microporous PP HFCLM bundle with feed and sweep fibers intimately commingled | 10 mg/L CA in poly(acrylic acid-co-acrylamide) hydrogel | Able to reduce CO2 from 0.52% to 0.09% | 2008 Cheng [79] |
| CO2 separation from air | Microporous PVDF HFCLM bundle with feed and sweep fibers intimately commingled | 121.8 W-A U/L CA displayed on the surface of E. coli suspended in water | 40% increase in CO2 removal rate, 2 times more stable than free CA | 2011 Fan [102] |
| CO2 separation from N2 | SLM with enzyme solution impregnating hydrophilic PVDF membrane; hybrid nylon-silica CLM sandwiched between two hydrophobic PVDF membranes | 0.2 mg/mL CA in 1 M NaHCO3 pH~8 | CO2 permeance: 108 GPU, silica xerogel provides additional catalytic benefit | 2011 Favre [103] |
| CO2 separation from N2 | SILM with porous hydrophobic PVDF membrane | 0.01 wt% CA in hydrophobic [C4MIM][Tf2N] ionic liquid or PEG 300 | Max CO2/N2 selectivity: 48; enzyme enhancement is more profound at higher water content | 2012 Neves [104] |
| CO2 separation from N2, H2, CH4 | SILM with hydrophobic PVDF microfiltration membrane | 5 mg/mL in hydrophobic [C4MIM][Tf2N] ionic liquid | Selectivity: CO2/N2:30.3 CO2/CH4:19.9 CO2/H2:11.2 | 2016 Bednar [80] |
| CO2 separation from N2 | SLM with porous hydrophilic cellulose acetate membrane reinforced by pectin | 2 mg CA/mL in Tris buffer (20Mm, pH 8.3) | CO2 permeability: 93 Barrer CO2 permeance: 0.75 GPU (120 μm thickness) CO2/N2 selectivity: 54 | 2018 Nemestóthy [82] |
| CO2 separation from N2 | ILM within 8 nm hydrophilic silica mesopores and thickness of 18 nm | 2 CA per nanopore; effective conc. of 100 mg CA mL–1 | CO2 permeance: 2600 GPU Selectivity: CO2/N2:788; CO2/H2:1500 | 2018 Fu [105] |
| CO2 separation from N2 | SLM with DES filling hydrophilic PTFE microfiltration membrane | 0.5 mg CA/g DES (choline chloride and levulinic acid) | CO2 permeability: 78 Barrer CO2/N2 selectivity: 32 Adding CA failed to enhance selectivity | 2021 de Castro [81] |
| CO2 separation from N2 and CH4 | SLM with DES filling hydrophilic PTFE microfiltration membrane | 0.1 mg CA/mL DES (choline chloride and urea) | CO2 permeability: 140 Barrer (w/CA) Selectivity: CO2/N2: below RUB CO2/CH4: above (w/o CA) and on (w/CA) RUB | 2021 Craveiro [106] |
| CO2 separation from CH4 | SLM with water-in-oil emulsion filling porous hydrophobic PVDF membrane | 1 wt% disperse phase (0.5 g CA/L K2CO3 pH 11, 5% PEG 300) in corn oil with 2 wt% Tween 80 | Permeability of CO2 increased by ~15% and CO2/CH4 selectivity increased by 2.9-fold with CA | 2022 Mondal [107] |
| CO2 separation from N2 and CH4 | SLM with hydrophilic PTFE microfiltration membrane | 0.5 mg CA/g solvent (12.5/14.5/75.0 wt% ChOH/water/glycerol) | CO2 permeability: 81 Barrer Selectivity: CO2/N2: 90.5 | 2022 Castro [108] |
| Application | Membrane Configuration | Enzyme, Concentration | Performance | Year, 1st Author, Ref |
|---|---|---|---|---|
| Artificial lungs CO2 desorption | PMP HFGLMC | Immobilized CA up to 88% theoretical monolayer coverage, 0.3 U esterase activity | Rates of CO2 exchange from buffer increased by 75% with immobilized CA | 2007 Kaar [13] |
| Artificial lungs CO2 desorption | PMP and PP HFGLMC | Immobilized CA 0.99–8.8 U esterase activity | CO2 removal rate increased by 115% and 37% from buffer and from blood, respectively | 2012–2015 Arazawa [120,121,122,123] |
| CO2 absorption | PP flat sheet membrane with LbL polyelectrolytes PEI/PSS/PAH/MSNP | 440 μg CA cm−2 per layer tested up to 3 layers | CO2 hydration rate of 19 ± 4 μmol cm−2 min−1 per layer tested up to 3 layers using CO2-saturated buffer | 2015 Yong [88] |
| CO2 absorption | Hydrophobic PVDF flat sheet membrane with TiO2 coating | 700 μg CA cm−2 | CO2 hydration rate of 140 μmol cm−2 min−1 nominal membrane area using CO2-saturated buffer | 2015 Hou [127] |
| CO2 absorption | PVDF flat sheet Janus membrane with fluorosilane-treated superhydrophobic and CNT-coated hydrophilic sides | 165 ± 22 μg CA cm−2 | CO2 hydration rate of 1.32 μmol cm−2 min−1 from 100% CO2 gas to pure water | 2015 Hou [128] |
| CO2 absorption | PP- or TiO2-coated superhydrophobic PP HFGLMC | 200 μg immobilized CA (on TiO2 NP)/mL suspended in absorption buffer | CO2 hydration rate of 0.96 μmol cm−2 min−1 from 20% CO2 gas mixture to buffer | 2016 Hou [129] |
| CO2 absorption | GLMC with a tubular porous glass membrane with hydrophobic coating on the outer skin | 10 mM enzyme mimic in 0.5 M K2CO3 | 10.8 μmol cm−2 min−1 from 10% CO2 gas mixture to 1 M NaOH; 10-fold increase in rate constant using 10 mM enzyme mimic in 0.5 M K2CO3 | 2016 Saeed [130] |
| CO2 absorption | HFGLMC with porous PP or non-porous Psf with PDMS coating; LbL polyelectrolytes PEI/PSS/PAH for enzyme adsorption | Three tri-layers (PSS/PAH/CA) and about 0.15 mg CA in HFGLMC. | 3-fold improvement in CO2 absorption rate in 30 wt% K2CO3 with immobilized CA which retained > 80% activity after exposure to common contaminant from flue gas but did not tolerate high pH combined with high temperature | 2016–2017 Yong [131,132] |
| CO2 absorption | HFGLMC based on hydrophobic porous PP (bulk and pores) and a dense hydrophilic PVDF layer and PIL coating on flue gas side | 1 wt% CA concentrate dissolved in 30% MDEA or MEA. | 2.2- and 1.7-fold enzyme enhancement in 30% MDEA with (CO2 flux 0.41 μmol cm−2 min−1 from 15% CO2) or without PIL coating, respectively; negative effect for adding CA in 30% MEA | 2017 Kim [89] |
| CO2 absorption from CO2/Xe mixture | Dense flat sheet PDMS GLMC separating gas and liquid phase microfluidic channels with alveolar design | 0.1 mg CA/g of CP ionic liquid with water activity of 0.753 | Enzyme has no effect on Xe transport but has 1.9-fold enhancement for CO2 absorption | 2018 Malankowska [133] |
| CO2 absorption | PVDF HFGLMC with co-deposited PDA/PEI for enzyme immobilization | 498 U esterase activity per m2 membrane | 15 μmol cm−2 min−1, 150% higher than non-biocatalytic membrane | 2019 Xu [134] |
| CO2 absorption | Electrospun PSMA nanofiber membrane as enzyme carrier and gas–liquid contacting surface positioned by flotation device | 10 mg CA/mg nanofiber membrane | CO2 hydration rate 8.9 μmol cm−2 min−1 from 100% CO2 gas | 2020 Kim [135] |
| CO2 absorption | MOF grown on Al2O3 membrane filter for enzyme adsorption | 0.1 mg CA/membrane or 75 μg CA/cm2 nominal area | CO2 hydration rate 108 μmol cm−2 min−1 from 5% CO2 gas into water | 2021 Liu [136] |
| CO2 absorption | Flat sheet PP GLMC with co-deposited PEI/PDA for enzyme immobilization | 94.3 µg CA/cm2 | CO2 hydration rate 1.74 μmol cm−2 min−1 from 15% CO2 into 100 mM Tris buffer | 2021 Rasouli [137] |
| CO2 absorption | Biocatalytic Flat sheet PP GLMC and MNP both were co-deposited with PEI/PDA and used for enzyme immobilization | 6.49–65.44 mg CA/Lreactor | CO2 hydration rate 1.7 μmol cm−2 min−1 from 15% CO2 into 100 mM Tris buffer | 2022 Rasouli [90] |
| Application | Membrane Configuration | Enzyme, Concentration | Cofactor Regeneration Electron Transfer System | Year, 1st Author, Ref |
|---|---|---|---|---|
| CO2 conversion to formic acid | Ceramic tubular membrane as UV-light blocker | FDH; DAH | UV > TiO2 (EtOH as hole quencher) > MV > DAH > NADH > FDH | 2005 Kurayama [92] |
| CO2 conversion to methanol | Flat sheet polymeric membranes with immobilized enzymes by direct membrane fouling | FDH; FaldDH; ADH | NADH > FDH; FaldDH; ADH | 2015 Luo [37] |
| CO2 conversion to formic acid | Hydrophobic HFM as gas distributor and PAA-grafted PE HFM as enzyme carrier | FDH | NADH > FDH | 2016 Wang [95] |
| Formaldehyde conversion to methanol | Hydrophilic flat sheet macroporous (200 nm) SiC membrane pretreated with NaOH and surface functionalized with PEI or APTES as enzyme carrier | ADH | NADH > ADH | 2018 Zeuner [140] |
| CO2 conversion to formic acid | PAA-grafted PE HFM modified by PEI through electrostatic interaction as CO2-philic surface | FDH | NADH > FDH | 2018 Wang [141] |
| CO2 conversion to formic acid | Electrospun PS nanofiber membrane surface modified by acid treatment, APTES, and GA activation as enzyme carrier | FDH | Cu foam electrode > NADH > FDH | 2018 Barin [142] |
| CO2 conversion to methanol | PVDF porous membrane functionalized by dead-end filtration of MOFs containing enzymes and cofactor | FDH; FaldDH; ADH; GDH | L-glutamate > GDH > NADH > FDH; FaldDH; ADH | 2019 Zhu [143] |
| CO2 conversion to formic acid | Porous HFM used as both gas distributor and enzyme carrier | FDH | UV > TiO2 (EDTA as hole quencher) > [Cp⁎Rh(bpy) (H2O)]2+ > NADH > FDH | 2020 Gu [144] |
| CO2 conversion to formic acid | PAA-grafted PE HFM modified by PEI compared with PEI/PDA co-deposited SiO2 microsphere as enzyme carriers | FDH | NADH > FDH | 2021 Guo [145] |
| CO2 absorption and conversion to formic acid | PP or ceramic GLMC modified by PEI/PDA and in situ grown MOFs encapsulating enzymes | CA; FDH | NADH > FDH | 2021 Chai [146] |
| CO2 conversion to formic acid | Ultrafiltration membrane with hydrophobic PP support layer and hydrophilic regenerated cellulose skin layer for enzyme immobilization | FDH | UV > MIL-125-Py-Rh (TEOA as hole quencher) > NADH > FDH | 2022 Lin [147] |
| CO2 conversion to formic acid | Ultrafiltration membrane with hydrophobic PP support layer and hydrophilic regenerated cellulose skin layer for enzyme immobilization | FDH | UV > Rhm3-N-PCN (TEOA as hole quencher) > NADH > FDH | 2022 Zhang [96] |
| CO2 conversion to formaldehyde | PE hollow fiber membrane was used as the enzyme-bearing reactor and gas distributor | FDH; FaldDH | UV > TiO2 (EDTA or H2O as hole quencher) > [Cp⁎Rh(bpy) (H2O)]2+ > NADH > FDH; FaldDH | 2022 Guo [94] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shen, J.; Salmon, S. Biocatalytic Membranes for Carbon Capture and Utilization. Membranes 2023, 13, 367. https://doi.org/10.3390/membranes13040367
Shen J, Salmon S. Biocatalytic Membranes for Carbon Capture and Utilization. Membranes. 2023; 13(4):367. https://doi.org/10.3390/membranes13040367
Chicago/Turabian StyleShen, Jialong, and Sonja Salmon. 2023. "Biocatalytic Membranes for Carbon Capture and Utilization" Membranes 13, no. 4: 367. https://doi.org/10.3390/membranes13040367
APA StyleShen, J., & Salmon, S. (2023). Biocatalytic Membranes for Carbon Capture and Utilization. Membranes, 13(4), 367. https://doi.org/10.3390/membranes13040367