
Small Residues Inhibit Homo-Dimerization of the Human Carbonic Anhydrase XII Transmembrane Domain



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Plasmid Construction
2.2. GALLEX Assay
2.3. Software
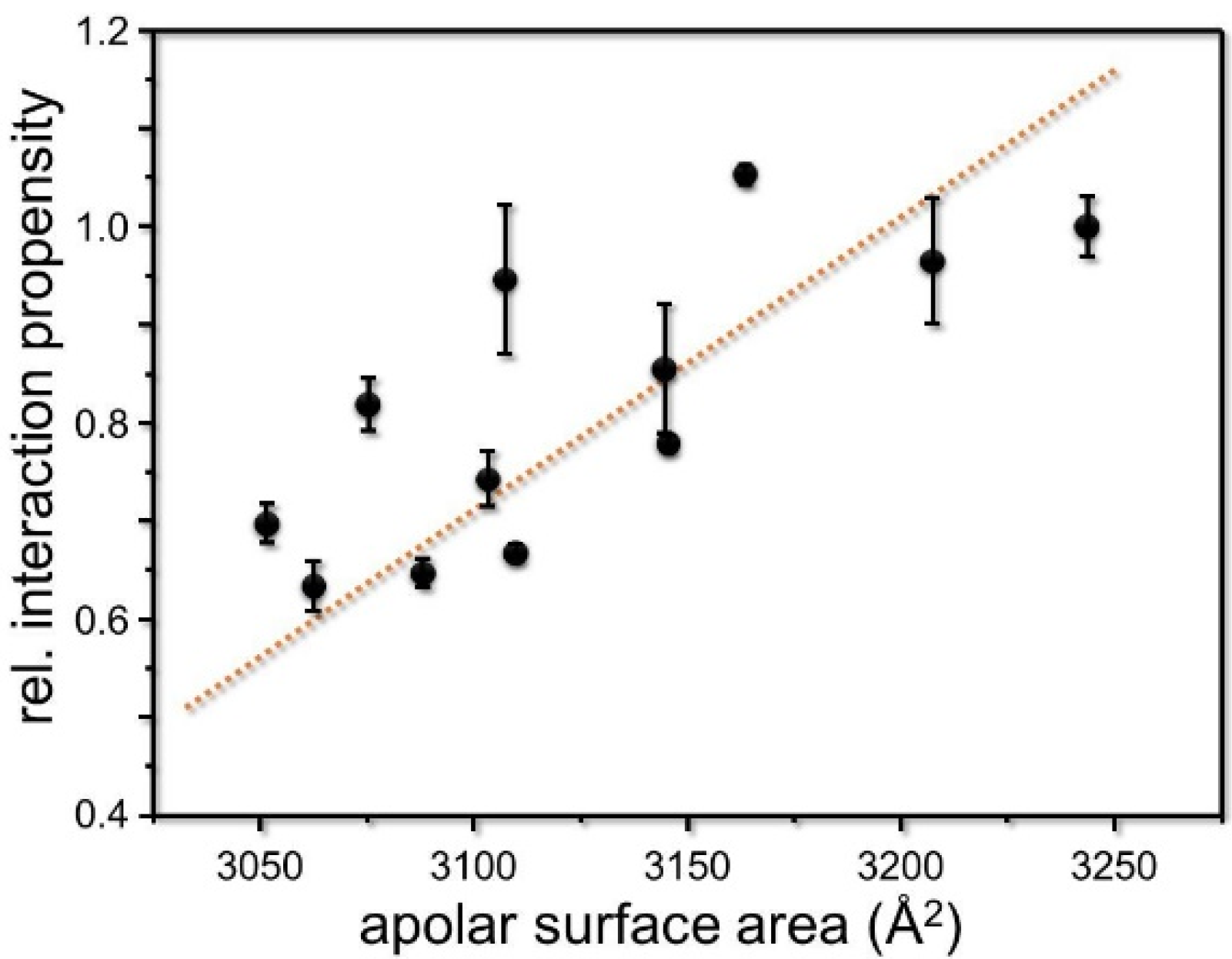
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lemmon, M.A.; Flanagan, J.M.; Treutlein, H.R.; Zhang, J.; Engelman, D.M. Sequence specificity in the dimerization of transmembrane alpha-helices. Biochemistry 1992, 31, 12719–12725. [Google Scholar] [CrossRef]
- MacKenzie, K.R.; Prestegard, J.H.; Engelman, D.M. A transmembrane helix dimer: Structure and implications. Science 1997, 276, 131–133. [Google Scholar] [CrossRef]
- Brosig, B.; Langosch, D. The dimerization motif of the glycophorin a transmembrane segment in membranes: Importance of glycine residues. Protein. Sci. 1998, 7, 1052–1056. [Google Scholar] [CrossRef] [PubMed]
- Russ, W.P.; Engelman, D.M. The GxxxG motif: A framework for transmembrane helix-helix association. J. Mol. Biol. 2000, 296, 911–919. [Google Scholar] [CrossRef] [PubMed]
- Senes, A.; Gerstein, M.; Engelman, D.M. Statistical analysis of amino acid patterns in transmembrane helices: The GxxxG motif occurs frequently and in association with beta-branched residues at neighboring positions. J. Mol. Biol. 2000, 296, 921–936. [Google Scholar] [CrossRef] [PubMed]
- Cymer, F.; Veerappan, A.; Schneider, D. Transmembrane helix-helix interactions are modulated by the sequence context and by lipid bilayer properties. Biochim. Biophys. Acta 2012, 1818, 963–973. [Google Scholar] [CrossRef]
- Teese, M.G.; Langosch, D. Role of GxxxG Motifs in Transmembrane Domain Interactions. Biochemistry 2015, 54, 5125–5135. [Google Scholar] [CrossRef]
- Schneider, D. Rendezvous in a membrane: Close packing, hydrogen bonding, and the formation of transmembrane helix oligomers. FEBS Lett. 2004, 577, 5–8. [Google Scholar] [CrossRef]
- Senes, A.; Engel, D.E.; DeGrado, W.F. Folding of helical membrane proteins: The role of polar, GxxxG-like and proline motifs. Curr. Opin. Struct. Biol. 2004, 14, 465–479. [Google Scholar] [CrossRef]
- Schneider, D.; Engelman, D.M. Motifs of Two Small Residues can ASSIST but are not sufficient to Mediate Transmembrane Helix Interactions. J. Mol. Biol. 2004, 343, 799–804. [Google Scholar] [CrossRef]
- Senes, A.; Ubarretxena-Belandia, I.; Engelman, D.M. The Calpha—H...O hydrogen bond: A determinant of stability and specificity in transmembrane helix interactions. Proc. Natl. Acad. Sci. USA 2001, 98, 9056–9061. [Google Scholar] [CrossRef] [PubMed]
- Arbely, E.; Arkin, I.T. Experimental measurement of the strength of a C alpha—H...O bond in a lipid bilayer. J. Am. Chem. Soc. 2004, 126, 5362–5363. [Google Scholar] [CrossRef]
- Walters, R.F.S.; DeGrado, W.F. Helix-packing motifs in membrane proteins. Proc. Natl. Acad. Sci. USA 2006, 103, 13658–13663. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Jeon, T.J.; Oberai, A.; Yang, D.; Schmidt, J.J.; Bowie, J.U. Transmembrane glycine zippers: Physiological and pathological roles in membrane proteins. Proc. Natl. Acad. Sci. USA 2005, 102, 14278–14283. [Google Scholar] [CrossRef]
- Herrmann, J.R.; Fuchs, A.; Panitz, J.C.; Eckert, T.; Unterreitmeier, S.; Frishman, D.; Langosch, D. Ionic Interactions Promote Transmembrane Helix-Helix Association Depending on Sequence Context. J. Mol. Biol. 2010, 396, 452–461. [Google Scholar] [CrossRef]
- Herrmann, J.R.; Panitz, J.C.; Unterreitmeier, S.; Fuchs, A.; Frishman, D.; Langosch, D. Complex Patterns of Histidine, Hydroxylated Amino Acids and the GxxxG Motif Mediate High-affinity Transmembrane Domain Interactions. J. Mol. Biol. 2008, 385, 912–923. [Google Scholar] [CrossRef] [PubMed]
- Unterreitmeier, S.; Fuchs, A.; Schaffler, T.; Heym, R.G.; Frishman, D.; Langosch, D. Phenylalanine promotes interaction of transmembrane domains via GxxxG motifs. J. Mol. Biol. 2007, 374, 705–718. [Google Scholar] [CrossRef]
- Melnyk, R.A.; Kim, S.; Curran, A.R.; Engelman, D.M.; Bowie, J.U.; Deber, C.M. The affinity of GXXXG motifs in transmembrane helix-helix interactions is modulated by long-range communication. J. Biol. Chem. 2004, 279, 16591–16597. [Google Scholar] [CrossRef]
- Gurezka, R.; Langosch, D. In vitro selection of membrane-spanning leucine zipper protein-protein interaction motifs using POSSYCCAT. J. Biol. Chem. 2001, 276, 45580–45587. [Google Scholar] [CrossRef]
- Dawson, J.P.; Weinger, J.S.; Engelman, D.M. Motifs of serine and threonine can drive association of transmembrane helices. J. Mol. Biol. 2002, 316, 799–805. [Google Scholar] [CrossRef]
- Zhou, F.X.; Merianos, H.J.; Brunger, A.T.; Engelman, D.M. Polar residues drive association of polyleucine transmembrane helices. Proc. Natl. Acad. Sci. USA 2001, 98, 2250–2255. [Google Scholar] [CrossRef] [PubMed]
- Gratkowski, H.; Lear, J.D.; De Grado, W.F. Polar side chains drive the association of model transmembrane peptides. Proc. Natl. Acad. Sci. USA 2001, 98, 880–885. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.M.; Hecht, K.; Deber, C.M. Aromatic and cation-pi interactions enhance helix-helix association in a membrane environment. Biochemistry 2007, 46, 9208–9214. [Google Scholar] [CrossRef] [PubMed]
- Sal-Man, N.; Gerber, D.; Bloch, I.; Shai, Y. Specificity in transmembrane helix-helix interactions mediated by aromatic residues. J. Biol. Chem. 2007, 282, 19753–19761. [Google Scholar] [CrossRef]
- Sal-Man, N.; Gerber, D.; Shai, Y. The composition rather than position of polar residues (QxxS) drives aspartate receptor transmembrane domain dimerization in vivo. Biochemistry 2004, 43, 2309–2313. [Google Scholar] [CrossRef]
- Steindorf, D.; Schneider, D. In vivo selection of heterotypically interacting transmembrane helices: Complementary helix surfaces, rather than conserved interaction motifs, drive formation of transmembrane hetero-dimers. Biochim. Et Biophys. Acta Biomembr. 2017, 1859, 245–256. [Google Scholar] [CrossRef]
- Hubert, P.; Sawma, P.; Duneau, J.-P.; Khao, J.; Henin, J.; Bagnard, D.; Sturgis, J. Single-spanning transmembrane domains in cell growth and cell-cell interactions. Cell Adhes. Migr. 2010, 4, 313–324. [Google Scholar] [CrossRef]
- Kirrbach, J.; Krugliak, M.; Ried, C.L.; Pagel, P.; Arkin, I.T.; Langosch, D. Self-interaction of transmembrane helices representing pre-clusters from the human single-span membrane proteins. Bioinformatics 2013, 29, 1623–1630. [Google Scholar] [CrossRef][Green Version]
- Ried, C.L.; Kube, S.; Kirrbach, J.; Langosch, D. Homotypic interaction and amino acid distribution of unilaterally conserved transmembrane helices. J. Mol. Biol. 2012, 420, 251–257. [Google Scholar] [CrossRef]
- Langosch, D.; Arkin, I.T. Interaction and conformational dynamics of membrane-spanning protein helices. Protein Sci. 2009, 18, 1343–1358. [Google Scholar] [CrossRef]
- Cymer, F.; Sanders, C.R.; Schneider, D. Analyzing oligomerization of individual transmembrane helices and of entire membrane proteins in E. coli: A hitchhiker’s guide to GALLEX. Methods Mol. Biol. 2013, 932, 259–276. [Google Scholar] [PubMed]
- Schneider, D.; Engelman, D.M. GALLEX, a Measurement of Heterologous Association of Transmembrane Helices in a Biological Membrane. J. Biol. Chem. 2003, 278, 3105–3111. [Google Scholar] [CrossRef] [PubMed]
- Treptow, N.A.; Shuman, H.A. Genetic evidence for substrate and periplasmic-binding-protein recognition by the MalF and MalG proteins, cytoplasmic membrane components of the Escherichia coli maltose transport system. J. Bacteriol. 1985, 163, 654–660. [Google Scholar] [CrossRef] [PubMed]
- Polyansky, A.A.; Chugunov, A.O.; Volynsky, P.E.; Krylov, N.A.; Nolde, D.E.; Efremov, R.G. PREDDIMER: A web server for prediction of transmembrane helical dimers. Bioinformatics 2014, 30, 889–890. [Google Scholar] [CrossRef]
- Notredame, C.; Higgins, D.G.; Heringa, J. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 2000, 302, 205–217. [Google Scholar] [CrossRef]
- Gasteiger, E.; Gattiker, A.; Hoogland, C.; Ivanyi, I.; Appel, R.D.; Bairoch, A. ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 2003, 31, 3784–3788. [Google Scholar] [CrossRef]
- Fraczkiewicz, R.; Braun, W. Exact and efficient analytical calculation of the accessible surface areas and their gradients for macromolecules. J. Comp. Chem. 1998, 19, 319–333. [Google Scholar] [CrossRef]
- Cunningham, F.; Poulsen, B.E.; Ip, W.; Deber, C.M. Beta-branched residues adjacent to GG4 motifs promote the efficient association of glycophorin A transmembrane helices. Biopolymers 2011, 96, 340–347. [Google Scholar] [CrossRef]
- Whittington, D.A.; Waheed, A.; Ulmasov, B.; Shah, G.N.; Grubb, J.H.; Sly, W.S.; Christianson, D.W. Crystal structure of the dimeric extracellular domain of human carbonic anhydrase XII, a bitopic membrane protein overexpressed in certain cancer tumor cells. Proc. Natl. Acad. Sci. USA 2001, 98, 9545–9550. [Google Scholar] [CrossRef]
- Alterio, V.; Hilvo, M.; di Fiore, A.; Supuran, C.T.; Pan, P.; Parkkila, S.; Scaloni, A.; Pastorek, J.; Pastorekova, S.; Pedone, C.; et al. Crystal structure of the catalytic domain of the tumor-associated human carbonic anhydrase IX. Proc. Natl. Acad. Sci. USA 2009, 106, 16233–16238. [Google Scholar] [CrossRef]
- Benej, M.; Pastorekova, S.; Pastorek, J. Carbonic anhydrase IX: Regulation and role in cancer. Subcell Biochem. 2014, 75, 199–219. [Google Scholar] [PubMed]
- Waheed, A.; Sly, W.S. Carbonic anhydrase XII functions in health and disease. Gene 2017, 623, 33–40. [Google Scholar] [CrossRef]
- Escher, C.; Cymer, F.; Schneider, D. Two GxxxG-like motifs facilitate promiscuous interactions of the human ErbB transmembrane domains. J. Mol. Biol. 2009, 389, 10–16. [Google Scholar] [CrossRef]
- Anderson, S.M.; Mueller, B.K.; Lange, E.J.; Senes, A. Combination of Calpha-H Hydrogen Bonds and van der Waals Packing Modulates the Stability of GxxxG-Mediated Dimers in Membranes. J. Am. Chem. Soc. 2017, 139, 15774–15783. [Google Scholar] [CrossRef]
- Xiao, Y.; Zeng, B.; Berner, N.; Frishman, D.; Langosch, D.; Teese, M.G. Experimental determination and data-driven prediction of homotypic transmembrane domain interfaces. Comput. Struct. Biotechnol. J. 2020, 18, 3230–3242. [Google Scholar] [CrossRef]
- MacKenzie, K.R.; Engelman, D.M. Structure-based prediction of the stability of transmembrane helix-helix interactions: The sequence dependence of glycophorin a dimerization. Proc. Natl. Acad. Sci. USA 1998, 95, 3583–3590. [Google Scholar] [CrossRef] [PubMed]
- Li, E.; Wimley, W.C.; Hristova, K. Transmembrane helix dimerization: Beyond the search for sequence motifs. Biochim. Biophys. Acta 2012, 1818, 183–193. [Google Scholar] [CrossRef]
- Hogel, P.; Gotz, A.; Kuhne, F.; Ebert, M.; Stelzer, W.; Rand, K.D.; Scharnagl, C.; Langosch, D. Glycine Perturbs Local and Global Conformational Flexibility of a Transmembrane Helix. Biochemistry 2018, 57, 1326–1337. [Google Scholar] [CrossRef] [PubMed]
- Li, S.C.; Deber, C.M. A measure of helical propensity for amino acids in membrane environments. Nat. Struct. Biol. 1994, 1, 558. [Google Scholar] [CrossRef]
- Padmanabhan, S.; Baldwin, R.L. Tests for helix-stabilizing interactions between various nonpolar side chains in alanine-based peptides. Protein. Sci. 1994, 3, 1992–1997. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cymer, F.; Schneider, D. Small Residues Inhibit Homo-Dimerization of the Human Carbonic Anhydrase XII Transmembrane Domain. Membranes 2021, 11, 512. https://doi.org/10.3390/membranes11070512
Cymer F, Schneider D. Small Residues Inhibit Homo-Dimerization of the Human Carbonic Anhydrase XII Transmembrane Domain. Membranes. 2021; 11(7):512. https://doi.org/10.3390/membranes11070512
Chicago/Turabian StyleCymer, Florian, and Dirk Schneider. 2021. "Small Residues Inhibit Homo-Dimerization of the Human Carbonic Anhydrase XII Transmembrane Domain" Membranes 11, no. 7: 512. https://doi.org/10.3390/membranes11070512
APA StyleCymer, F., & Schneider, D. (2021). Small Residues Inhibit Homo-Dimerization of the Human Carbonic Anhydrase XII Transmembrane Domain. Membranes, 11(7), 512. https://doi.org/10.3390/membranes11070512